

Abstract
Oxidative cyclizations of a variety of heteroatom nucleophiles onto unactivated olefins are catalyzed by palladium(II) and pyridine in the presence of molecular oxygen as the sole stoichiometric oxidant in a nonpolar solvent (toluene). Reactivity studies of a number of N-ligated palladium complexes show that chelating ligands slow the reaction. Nearly identical conditions are applicable to five different types of nucleophiles: phenols, primary alcohols, carboxylic acids, a vinylogous acid, and amides. Electron-rich phenols are excellent substrates, and multiple olefin substitution patterns are tolerated. Primary alcohols undergo oxidative cyclization without significant oxidation to the aldehyde, a fact that illustrates the range of reactivity available from various Pd(II) salts under differing conditions. Alcohols can form both fused and spirocyclic ring systems, depending on the position of the olefin relative to the tethered alcohol; the same is true of the acid derivatives. The racemic conditions served as a platform for the development of an enantioselective reaction. Experiments with stereospecifically deuterated primary alcohol substrates rule out a “Wacker-type” mechanism involving anti oxypalladation and suggest that the reaction proceeds by syn oxypalladation for both mono- and bidentate ligands. In contrast, cyclizations of deuterium-labeled carboxylic acid substrates undergo anti oxypalladation.
INTRODUCTION
Oxidation is one of the most fundamental and important processes in nature. It has also become one of the most effective ways for chemists to induce asymmetry in organic transformations for the production of enantioenriched materials.1 Most enantioselective oxidations involve the transfer of a heteroatom, commonly oxygen, to a substrate in a manner analogous to that of oxygenase metalloenzymes. In contrast, there is a significant lack of asymmetric two-electron oxidations that do not involve heteroatom transfer. This second mode of oxidation is similar to oxidase-type enzymatic reactions for which oxygen atoms instead act as electron and proton acceptors in substrate oxidation.
The ability of palladium (Pd) to serve as both a nucleophile (Pd(0)) and an electrophile (Pd(II)) has led to an explosion in Pd(0)-catalyzed reactions that have become indispensable to organic chemistry.2 This property has also led to the development of racemic oxidase-type reactions that feature electrophilic Pd(II), some examples of which are shown in Figure 1, although they are much less prevalent.3 Such reactions require the recycling of Pd(0) to Pd(II) by a stoichiometric oxidant. Ideally, such a process would take advantage of the most abundant and inexpensive oxidant, molecular oxygen (O2). In fact, one of the early systems developed for the catalytic oxidation of secondary alcohols by Pd(II) employed O2 directly as the only stoichiometric oxidant.4 The use of DMSO as solvent also has enabled racemic O2-coupled Pd(II)-catalyzed oxidative cyclizations of olefin-appended nucleophiles such as phenols, alcohols, acids and tosylamides, as well as alcohol oxidations to ketones and aldehydes.5 Two other reoxidation systems include the traditional Wacker process that employs a copper cocatalyst in the presence of O2, and the use of the organic oxidant benzoquinone as a stoichiometric oxidant. Racemic cyclization reactions have also been developed using these reoxidants.6,7
Figure 1.
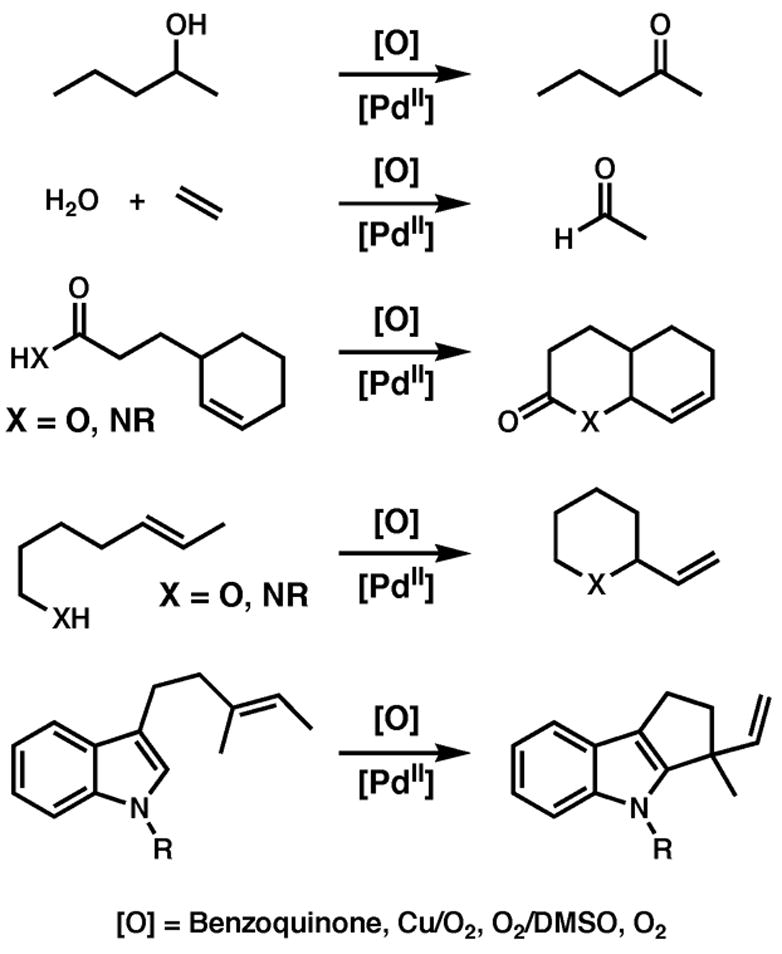
Prototypical Pd(II)-catalyzed dehyrogenation reactions.
Despite these advances in racemic Pd(II)-catalyzed oxidation reactions, many do not meet the ideal criteria for enantioselective reactions of employing only O2 as the stoichiometric oxidant in a non-coordinating solvent. Instead, the necessary use of cocatalysts, organic oxidants or DMSO has complicated the development of asymmetric oxidase-type reactions. For example, the use of copper/O2 introduces a secondary catalytic cycle and another metal that could compete with Pd for the chiral ligand. The benzoquinone system requires the removal of stoichiometric amounts of organic compounds8 and benzoquinone itself can act as a ligand.9,10 In addition, DMSO is a highly donating solvent that could interfere with the coordination of a chiral ligand to Pd. Despite the obstacles presented by the traditional oxidation systems, seminal works by Hosokawa and Murahashi,11 Hayashi,12 Sasai13 and Bäckvall9 established the potential of enantioselective Pd(II)-catalyzed oxidative cyclizations and dialkoxylations.14
With the requirement that O2 be the only stoichiometric oxidant, we began to address the lack of enantioselective oxidase-type catalysis with the development of the Pd(II)-catalyzed asymmetric dehydrogenation of secondary alcohols illustrated in Figure 2.15,16 This oxidation effects a kinetic resolution to provide enantioenriched alcohol, and was the first example of an asymmetric oxidase-type reaction in that it employs O2 as the terminal oxidant. The basis for this chemistry was another racemic Pd(II)-catalyzed alcohol oxidation reported in 1999 by Uemura and coworkers.17 Significantly, the reaction used only O2 in a non-cooridnating solvent (toluene), and was accelerated by the presence of a ligand (pyridine). We subsequently initiated an effort to apply our enantioselective alcohol oxidation to the development of asymmetric versions of reactions such as those shown in Figure 1. In line with this goal, we recently communicated the extension of Uemura’s conditions to the cyclizations of heteroatoms and electron-rich indoles onto pendant olefins, as well as an asymmetric version of the former reaction.18,19 This work established that heteroatom-olefin cyclizations that use O2 as the sole oxidant could be amenable to asymmetric catalysis. We now report the full details regarding the development of these racemic and asymmetric heterocyclizations, an expanded substrate scope for both, and deuterium-labeling studies that establish the stereochemistry of oxypalladation for two types of substrates. On the basis of our results, we offer a rationale for the difficulty in developing the asymmetric reaction in this system specifically. As recent work by Hayashi, Wolfe, Sanford, White, Stahl, Sigman and others has also shown, our studies further demonstrate that oxidase-type catalysis by Pd(II) is both advantageously versatile in terms of reactivity and frustratingly promiscuous in terms of mechanism.
Figure 2.
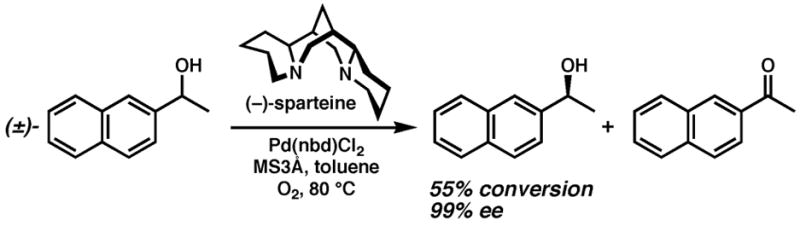
An oxidase-type kinetic resolution of secondary alcohols.
RESULTS AND DISCUSSION
Reaction development
The effect of palladium X− ligand, exogenous base and nitrogen-containing ligand on reaction rate
Our initial aim was to establish Pd(II)-catalyzed conditions for racemic aerobic cyclizations to which a chiral ligand eventually could be introduced. Thus, we began our investigation of aerobic oxidative cyclizations with 2-(1,2-dimethyl-1-propenyl)phenol (1) using a variety of palladium(II) salts, pyridine, O2, and MS3Å in toluene at 80 °C (Table 1). These conditions are modeled after Uemura’s alcohol oxidation conditions,17a which, as stated above, were also employed as a starting point for our oxidative kinetic resolution chemistry.15a Surprisingly, Pd(nbd)Cl2, which is the most effective catalyst for the kinetic resolution chemistry, was ineffective for the cyclization of 1 to dihydrofuran 2 (entry 1). Treatment of 1 with a range of palladium(II) salts (entries 1–4) led to the discovery that the electron-deficient palladium(II) trifluoroacetate (Pd(TFA)2) was most effective for producing 2 in good yield after reasonable reaction time (entry 4). A number of exogenous bases were examined with the hope that proton consumption would accelerate the reaction (not shown, see Supporting Information). Sodium carbonate exerts the most positive effect to give 95% yield in under 30 minutes (entry 5). Pd(0) sources were found to be poor catalysts for the reaction: Pd2(dba)3 resulted in the formation of a small amount of product, along with palladium black (entry 6), and palladium black itself gave no reaction (entry 7). No cyclization occurred in the absence of any palladium (entry 8). O2 is necessary for good reactivity, although there appears to be a background reaction that leads to cyclization since the product is formed in greater than 5% yield (entry 9). The presence of 30 equivalents of elemental mercury slowed the cyclization, but did not prevent reaction altogether, which contraindicates colloidal Pd or Pd nanoparticles as the relevant catalytic species (entry 10).20
Table 1.
Optimization of Pd Source.
 | ||||
|---|---|---|---|---|
| entry | Pd source | additive | time | yieldb |
| 1. | Pd(nbd)Cl2 | none | 24 h | 7% |
| 2. | PdCl2 | none | 24 h | 27% |
| 3. | Pd(OAc)2 | none | 24 h | 76% |
| 4. | Pd(TFA)2 | none | 60 min | 87% |
| 5. | Pd(TFA)2 | Na2CO3 | 20 min | 95% |
| 6. | Pd(dba)3 | none | 24 h | 25% |
| 7. | Pd Black | none | 24 h | NR |
| 8. | None | none | 24 h | NR |
| 9. | Pd(TFA)2 | None, no O2 | 24 h | 24% |
| 10.c | Pd(TFA)2 | Na2CO3, Hg0 | 5 h | 84%d |
5 mol% Pd source, 20 mol% pyridine, 500 mg/mmol MS3Å, 1 atm O2, toluene (0.1M), 80 °C.
Isolated yield.
5 mol % (pyridine)2Pd(TFA)2, 10 mol% pyridine, 2 equiv Na2CO3, 30 equiv Hg0.
Conversion determined by GC.
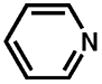
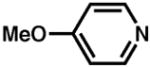
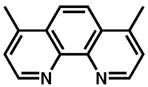
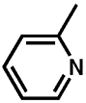
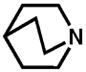
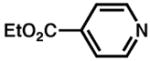
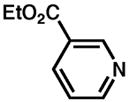
Although pyridine had been a competent ligand during our optimization studies, we carried out a small ligand screen of other nitrogen-containing ligands.21 Each LnPd(TFA)2 complex was synthesized separately and characterized, rather than generated in situ, in order to limit uncertainty regarding the catalyst or catalyst precursor. Reactions were performed with no additive, 40 mol% excess ligand, and both excess ligand and Na2CO3. As shown in Table 2, substituted pyridyls that are less coordinating than pyridine, whether due to electronic or steric reasons, result in the precipitation of Pd black. Bidentate nitrogen-containing ligands such as 2,2′-dipyridyl (entry 6), 4,7-dimethyl-1,10-phenanthroline (entry 7), TMEDA (entry 10), or TMPDA (entry 11) significantly slow the reaction. Weak alkyl amine donors result in the precipitation of Pd black (entries 8–9, 11). Although some rate enhancement was observed for the nicotinate derivatives (entries 4 and 5), pyridine offered the best combination of reactivity, catalyst stability, and availability.
Table 2.
Oxidative cyclizations with substituted pyridyl and alkyl amine ligands.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entryb | ligand | no additive | 40% ligand | 40% ligand + 40% Na2CO3 | entry | ligand | no additive | 40% ligand | 40% ligand + 40% Na2CO3 |
| 1. |
|
98%
1 h |
99% 1 h | 99%
15 h |
6.d |
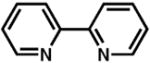
|
92%
18 h |
97%
5 h |
93%
5 h |
| 2. |
|
85%
2.5 h |
96% 9 h | 99%
2 h |
7.d |
|
94%
8 h |
96%
24 h |
99%
24 h |
| 3. |
|
50%
24 hc |
68% 24 h | 91%
5 h |
8. |
|
82%
20 h |
90%
5 hc |
92%
5 h |
| 4. |
|
84%
8 hc |
99% 40 min | 99%
15 min |
9. |
|
15%
24 h |
15%
24 hc |
10%
24 h |
| 10.d |
|
73%
12 h |
97%
8 h |
99%
8 h |
|||||
| 5. |
|
70%
30 minc |
99% 30 min | 99%
5 min |
11.d |
|
34%
20 hc |
54%
20 hc |
76%
11 h |
5 mol% LnPd(TFA)2, 500 mg/mmol MS3Å, 1 atm O2, toluene (0.1 M), 80 °C.
Conversion determined by GC.
Pd black precipitate was observed in the reaction mixture.
For entries 6, 7, 10 and 11, 20 mol% excess ligand was added.
Cyclizations of phenols and primary alcohols
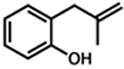
Under our optimized conditions, oxidative cyclization of a variety of substituted phenols occurs readily with 5 mol% Pd(TFA)2, 20 mol% pyridine, 2 equiv Na2CO3 and 500 mg 3Å molecular sieves/mmol substrate at 0.1 M concentration in toluene under a balloon of oxygen (Table 3). Workup involves simple filtration through a pad of silica gel. Cyclizations of electron-rich phenols are facile and provide excellent yields in under 30 min (entries 1–4, 7–10). An electron deficient phenol (9) serves as an excellent substrate as well, albeit with slower reaction time (entry 5). In contrast, a p-bromo-substituted substrate (11, entry 6) appears to undergo alternate reaction pathways leading to decomposition, possibly via oxidative addition to Pd(0). Cyclization onto a tetrasubstituted olefin (21) proceeds in good yield (entry 11), as does cyclization of a disubstituted olefin (23, entry 12). A dihydropyran (26) is available under identical conditions (entry 13). Finally, high yields and reasonable rates persist with reduced catalyst loading (2 mol%, entry 14). For terminal olefin substrate 27, reaction does not take place, most likely because exo cyclization occurs that then leaves no β-hydrogens to be eliminated from the presumed Pd-alkyl intermediate.
Table 3.
Oxidative cyclizations of phenols.a
| entry | substrate | product | time | yield | entry | substrate | product | time | yield |
|---|---|---|---|---|---|---|---|---|---|
| 1. |
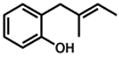
1 |
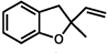
2 |
20 min | 95% | 9. |
17 |
18 |
10 min | 86% |
| 2. |
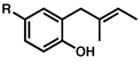
R = Me 3 |
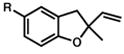
R = Me 4 |
20 min | 99% | 10. |
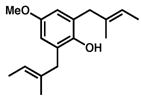
19 |
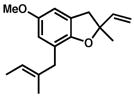
20 |
2 h | 93% |
| 3. | R = t-Bu
5 |
R = t-Bu
6 |
25 min | 90% | |||||
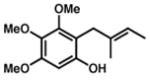
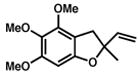
| 4. | R = OMe
7 |
R = OMe
8 |
15 min | 89% | 11. |
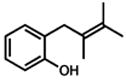
21 |
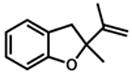
22 |
25 min | 80% |
| 5. | R = COCH3
9 |
R = COCH3
10 |
25 h | 93% | |||||
| 6. | R = Br
11 |
R = Br
12 |
24 h | 33% | 12. |
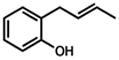
23 |
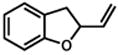
24 |
3 h | 74% |
| 7. |
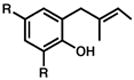
R = Me 13 |
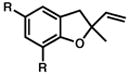
R = Me 14 |
20 min | 85% | 13.b |
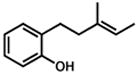
25 |
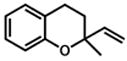
26 |
75 min | 85% |
| 8. | R = OMe
15 |
R = OMe
16 |
40 min | 80% | 14.c | 1 | 2 | 14 h | 86% |
| 15. |
27 |
NR | 24 h |
5 mol% Pd(TFA)2, 20 mol% pyridine, 2 equiv Na2CO3, 500 mg/mmol MS3Å, 1 atm O2, toluene (0.1 M), 80 °C.
The starting material was used as a 3.6:1 mixture of olefin isomers.
2 mol% Pd(TFA)2, 8 mol% pyridine.
In addition to phenols, we have investigated primary alcohol/olefin oxidative cyclizations. Remarkably, these reactions proceed to the heterocyclic ethers with, in most cases, little or no oxidation to the aldehyde under our optimized conditions (Table 4). In addition to benzyl alcohol 28, cyclopentene (30 and 32) and cyclohexene (34) derivatives provide moderate to excellent yields of spirocycles (31) and fused ring systems (33, 35). The mode of oxidative reactivity – cyclization versus alcohol oxidation – appears dependent not only on the substrate (i.e., primary vs. secondary alcohols) but also on the specific palladium source (cf. Uemura’s work17).
Table 4.
Oxidative cyclizations of primary alcohols.a
| entry | substrate | product | time | yield |
|---|---|---|---|---|
| 1.b |
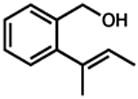
28 |
29 |
3 h | 87% |
| 2. |
30 |
31 |
10 h | 93%c |
| 3. |
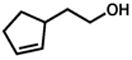
32 |
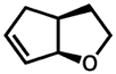
33 |
7.5 h | 69%d |
| 4. |
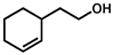
34 |
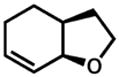
35 |
20 h | 60%e |
5 mol% Pd(TFA)2 20 mol% pyridine, 2 equiv Na2CO3, 500 mg/mmol MS3Å, 1 atm O2, toluene (0.1 M), 80 °C.
The starting material was used as a mixture of E and Z olefins.
Isolated with 7% of the aldehyde.
Isolated with 7% of an olefin isomer.
Isolated as a 5:2.3:1 mixture of 35/olefin isomer/aldehyde.
Cyclizations of carboxylic acids and derivatives
A range of carboxylic acid and carboxylic acid derivatives were synthesized and subjected to our optimized conditions. For some carboxylic acid derivatives (36, 38, 40, 44), the addition of an inorganic base was found to be unnecessary, and exposure simply to 5 mol% Pd(TFA)2, 20 mol% pyridine, 500 mg MS3Å/mmol substrate and 1 atm O2 in toluene at 80 °C led to the oxidatively cyclized products (Table 5, entries 1–3, 5). Benzoic acids (36) and amides (38, 40) are cyclized in good to excellent yields (entries 1–3). A β-keto ester (42) undergoes cyclization as a vinylogous acid to form a heterocycle (43) rather than a carbocycle (entry 4). Primary acid derivatives react to form spirocycles (45) or fused bicyclic systems (47 and 49), depending on the position of the olefin (entries 5–7). The cyclization of derivatives 46 and 48 is more facile with double the catalyst loading and is accelerated by the presence of 2 equiv Na2CO3.
Table 5.
Oxidative cyclizations of carboxylic acids and derivatives.a
| entry | substrate | product | time | yield |
|---|---|---|---|---|
| 1.b |
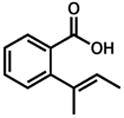
36 |
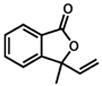
37 |
8 h | 90 % |
| 2.b |
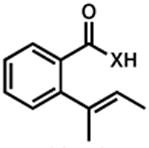
38, 40 |
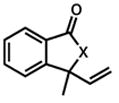
39, 41 |
8 h | 88 % |
| 3.b | 4 h | 82 % | ||
| 4.b |
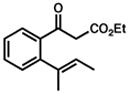
42 |
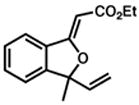
43 |
48 h | 63 %c,d |
| 5. |
44 |
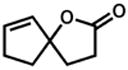
45 |
48 h | 62 %e |
| 6. |
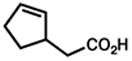
46 |
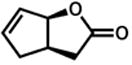
47 |
2 h | 77 %f |
| 7. |
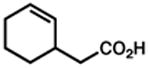
48 |
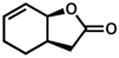
49 |
12 h | 86 %f,d |
5 mol% Pd(TFA)2, 20 mol% pyridine, MS3Å, 1 atm O2, toluene, 80 °C.
The starting material was used as a mixture of E:Z olefins.
10 mol% pyridine, 2 equiv LiOAc.
3:1 Z:E.
10 mol% Pd(TFA)2, 40 mol% pyridine.
10 mol% Pd(TFA)2, 40 mol% pyridine, 2 equiv Na2CO3.
Isolated with 6% of an olefin isomer.
Reaction scope and limitations
The high yields, usually brief reaction times, and range of substrates that are characteristic of this aerobic Pd-catalyzed oxidative cyclization demonstrate the utility of the simple reaction conditions – Pd, ligand, base, O2, and solvent. Nearly identical conditions are applicable to five different types of nucleophiles: phenols, primary alcohols, carboxylic acids, a vinylogous acid, and amides. Electron-rich phenols are excellent substrates, and multiple olefin substitution patterns are tolerated. Primary alcohols undergo oxidative cyclization without significant oxidation to the aldehyde, a fact that illustrates the range of reactivity available from various Pd(II) salts under differing conditions. In addition to the cyclization of a benzylic alcohol, non-benzylic alcohols can form both fused and spirocyclic ring systems; the same is true of the acid derivatives. Undoubtedly the range of alcohol substrates could be increased.22 While phenol/olefin, alcohol/olefin,5a,6a,23 and carboxylic acid/olefin5c,24,25 cyclizations have been achieved before under Pd(II)/oxidant catalysis, our conditions differentiate themselves by meeting the criteria for extension to an asymmetric version: simplicity, capacity to accommodate a chiral ligand, and active catalysis in a non-coordinating solvent. Without a system of this type the development of a direct aerobic asymmetric cyclization would be less feasible.
Elaboration of racemic conditions to an asymmetric version
A number of chiral ligands were screened with the conditions established for the racemic cyclizations, including typical chiral ligands such as bisoxazolines, as well as less common frameworks for catalysis such as brucine (not shown, see Supporting Information). As we observed during the development of our oxidative kinetic resolution chemistry,15a the natural product (36 h)-sparteine was by far the most successful at inducing asymmetry in the cyclization. Treatment of 1 with Pd(TFA)2 in the presence of (−)-sparteine, MS3Å and O2 in toluene led to a 72% conversion to 76% enantioenriched dihydrobenzofuran (+)-2 after 36 h (eq. 1).26
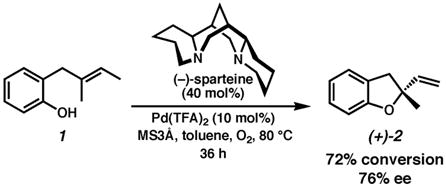 |
(1) |
Optimization of the asymmetric reaction – screen of palladium sources
With (−)-sparteine as the most enantioselective candidate to emerge from the chiral ligand screen, we set out to increase selectivity through an optimization of Pd(II) source and basic additive. This seemed essential in light of what we had observed during the development of the racemic reaction (cf. Table 1), as well as with the kinetic resolution chemistry.15a,27 Pd(II) halide sources provided product in some cases, but with degradation of enantiomeric excess (Table 6, entries 1–2, 4–5). Pd(OAc)2 is more effective at inducing asymmetry, but at the expense of conversion (entry 6). The extent of asymmetric induction varied surprisingly in the presence of different Pd(II) sources. For example, Pd(COD)Cl2 results in only 10% ee (entry 2), whereas Pd(TFA)2 provides 76% ee (entry 7). It is remarkable that this change has such a large effect on enantioselectivity.28 Pd(TFA)2 remained the optimal catalyst, and it was found that by using the complex of Pd(TFA)2 and (−)-sparteine directly rather than generating it in situ led to slightly improved and more reliable results (entry 8). Generally, more electron-deficient Pd sources were more selective in the cyclization. However, switching the anion from trifluoroacetate to triflate resulted in degradation of the catalyst (formation of Pd black). In addition to palladium source, a basic additive affects reaction rate and selectivity. Best results occur with Ca(OH)2, the presynthesized (sp)Pd(TFA)2 complex, and 1 equiv of (−)-sparteine (entry 11).
Table 6.
Optimization of Pd Source and Additive for the Asymmetric Oxidative Cyclization.
 | |||||
|---|---|---|---|---|---|
| entry | Pd source | additive | time | convb | eeb |
| 1. | Pd(nbd)Cl2 | none | 36 h | 68% | 12% |
| 2. | Pd(COD)Cl2 | none | 36 h | 30% | 10% |
| 3. | PdCl2 | none | 36 h | 2% | 12% |
| 4. | Pd(CH3CN)2Cl2 | none | 36 h | 53% | 12% |
| 5. | PdBr2 | none | 36 h | 32% | 8% |
| 6. | Pd(OAc)2 | none | 36 h | 18% | 51% |
| 7. | Pd(TFA)2 | none | 36 h | 72% | 76% |
| 8.c | (sp)Pd(TFA)2d | none | 36 h | 83% | 77% |
| 9. | Pd(OTf)2(CH3CN)4 | none | 160 h | 20% | 16% |
| 10.e | (sp)Pd(TFA)2 | Na2CO3 | 36 h | 53% | 76% |
| 11.e | (sp)Pd(TFA)2 | Ca(OH)2 | 36 h | 87% | 81% |
10 mol% Pd source, 40 mol% (−)-sparteine, 500 mg/mmol MS3Å, 1 atm O2, toluene (0.1 M), 80 °C.
Conversion measured by GC or by 1H NMR.
30 mol% (−)-sparteine.
sp = (−)-sparteine.
10 mol% (sp)Pd(TFA)2, 100 mol% (−)-sparteine.
Asymmetric oxidative cyclization of phenol substrates
Under the optimized conditions, phenol 1 was cyclized to provide dihydrobenzofuran (+)-2 in 81% ee and 87% isolated yield (Table 7, entry 1).29 Application of these conditions to other substrates that reacted successfully under the racemic conditions proved difficult.30 p-Methoxyphenol 7 is transformed with high selectivity to give (+)-8 in 90% ee and 57% yield (entry 2). t-Butylphenol 5 and p-methylphenol 3 do not react quickly but are obtained with good levels of enantiomeric excess (entries 3 and 4). p-Acylphenol 9 cyclizes, but with low %ee, perhaps indicative of a change in mechanism for this less electron-rich substrate.
Table 7.
Pd-Catalyzed asymmetric aerobic oxidative cyclization.a
| entry | substrate | product | time | yield | eec |
|---|---|---|---|---|---|
| 1. |
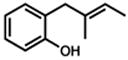
1 |
(+)-2 |
36 h | 87 % | 81 % |
| 2. |
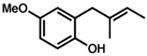
7 |
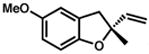
(+)-8 |
24 h
60 h @ 55°C |
64 %
57 % |
88 %
90 % |
| 3. |
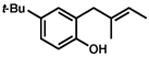
5 |
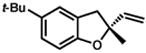
(−)-6 |
36 h | 47 % | 83 % |
| 4. |
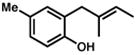
3 |
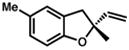
(+)-4 |
36 h | 47 % | 86 % |
| 5. |
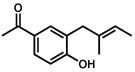
9 |
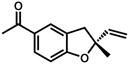
(−)-10 |
24 h | 60 % | 20 % |
10 mol% (sp)Pd(TFA)2, 100 mol% (−)-sparteine, 2 equiv Ca(OH)2, 500 mg/mmol MS3Å, 1 atm O2, toluene (0.1 M), 80 °C.
Isolated yield.
Measured by GC.
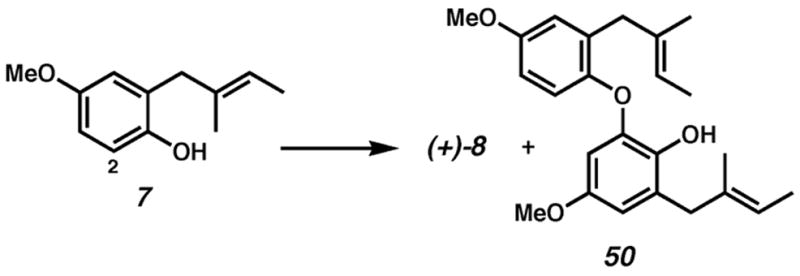
Unfortunately, the highly enantioentriched p-methoxy dihydrobenzofuran (+)-8 is produced with a dimeric aryl ether byproduct (50, Table 8). This byproduct could form via palladation ortho to the phenol, followed by coupling to another molecule of substrate. The addition of various acids suppressed the formation of the byproduct, perhaps by decomposing the postulated palladium aryl species, but also depressed the enantioselectivity of the cyclization (entry 3).
Table 8.
Aryl ether formation and attempted suppression.
 | ||||
|---|---|---|---|---|
| entry | substrate | time | (+)-8:50b | ee of (+)-8c |
| 1.a | amberlyst 15 resin (10 mol%) | 24 h | 5:3 | 87% |
| 2. | TsOH·H2O(10 mol%) | 24 h | 3:1 | 81% |
| 3. | TsOH·H2O(100 mol%) | 80 h | 6:1 | 50% |
| 4. | none | 16 h | 2:1 | 86% |
10 mol% (sp)Pd(TFA)2, 100 mol% (−)-sparteine, 500 mg/mmol MS3Å, 1 atm O2, toluene (0.1 M), 80 °C.
Measured by 1H NMR.
Measured by GC.
While the asymmetric oxidative cyclization conditions that we have developed are not yet general, we have established that it is possible to adapt a direct dioxygen-coupled racemic reaction to aerobic asymmetric catalysis, which had not been achieved previously for this class of reaction. In conjunction with our work on the oxidative kinetic resolution of secondary alcohols, the asymmetric cyclization chemistry makes significant headway into the realm of enantioselective oxidase-type chemistry.
Mechanistic investigations
Deuterium-labeling studies of primary alcohol substrates in the presence of a monodentate ligand
We hypothesized that the sluggishness of the asymmetric cyclization could be attributed to the difference in ligand denticity between pyridine and (−)-sparteine. In fact, in our screen of nitrogen-containing ligands (Table 2), the decreased reactivity of all the bidentate ligands confirmed that (−)-sparteine was not uniquely deactivating. We thus wished to gain an understanding of the mechanism of the cyclization, and whether a change from a mono- to a bidentate ligand would involve a change in mechanism. One commonly proposed mechanism for Pd(II)-catalyzed cyclizations involves activation of the olefin by Pd(II) followed by anti nucleophilic attack, i.e, anti oxypalladation; indeed, this was our original hypothesis.18,31 The resulting Pd-alkyl intermediate would then undergo β-hydrogen elimination. Such a mechanism is reminiscent of that proposed by Bäckvall, Stille and Kurosawa for the Wacker oxidation of ethylene to acetaldehyde.32 A second possible mechanism is supported by recent reports from Hayashi and Wolfe that find certain heteroatom/olefin cyclizations catalyzed by Pd(II) occur with syn oxypalladation.33 Wolfe further suggests that this involves an olefin insertion into a Pd(II)–heteroatom bond.34,35,36 This pathway is analogous to the alternative mechanism for the Wacker oxidation proposed by Henry and others that involves syn oxypalladation.37 A third mechanism entails allylic C–H activation by Pd(II) to form an intermediate π-allyl species that would then undergo reductive elimination with the heteroatom nucleophile.9,38,39
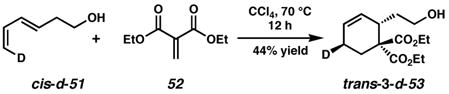
We set out to differentiate these mechanisms by synthesizing stereospecifically deuterium-labeled substrates, and observing the products of the cyclization in the presence of a mono- and bidentate ligand. Deuterium-labeled alcohols trans-3-d-53 and cis-3-d-53 were designed such that oxidative cyclization would result in retention or elimination of the label. Recently, during the course of this work, Hayashi and coworkers reported a similar study for the oxidative cyclization of olefin-appended phenols by Pd(II); in those examples syn oxypalladation occurs exclusively except in the presence of additional chloride ion.33a We chose to focus on primary alcohols because of the interesting dichotomy between oxidative cyclization and alcohol oxidation and because a study of this type had not yet been carried out for this substrate class.
 |
(2) |
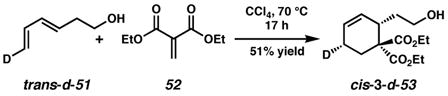 |
(3) |
Stereospecific deuterium incorporation into primary alcohol substrates could be effected with the Diels-Alder reaction shown in eqs. 2 and 3 that completed the relatively straightforward synthesis of trans- and cis-3-d-53.40 We assumed the constraints that a cis 6-5 fused ring system would form, and that β-hydrogen elimination only occur when the Pd atom and eliminated H or D atom are syn to each other.41 The presence or absence of a deuterium label in the product would then differentiate mechanistic pathways. Although a π-allyl mechanism could be difficult to unambiguously rule out with our test substrate, we hoped to at least distinguish syn from anti oxypalladation.42
Treatment of trans-3-d-53 with 10 mol% (pyridine)2Pd(TFA)2, 20 mol % pyridine, 2 equiv Na2CO3, 1 atm O2 and 500 mg/mmol MS3Å in toluene at 80 °C for 3 h provided 3-d-54 along with olefin isomer 3-d-55 in a 4:1 ratio and 95% overall yield (Scheme 1). Likewise, reaction of the cis isomer (cis-3-d-53) under the same conditions led to the formation of a 1:0.7 mixture of undeuterated 54 and cis-2-d-55 in nearly quantitative yield. Reaction of cis-3-d-53 in the absence of Na2CO3 leads to a nearly identical result.43 The stereochemistry of cis-2-d-55 was confirmed by 1H NMR homodecoupling and NOE experiments.40 Reexposure of the product mixture (54 and cis-2-d-55) to the same reaction conditions and an additional 10 mol% starting material (cis-3-d-53) for 4 h resulted in an identical ratio of products in 90% isolated yield. Comparison of the 1H and 2H NMR spectra of the products of the above reactions with those formed from undeuterated cyclohexene 53 confirmed the presence or absence of a deuterium label.40
Scheme 1.
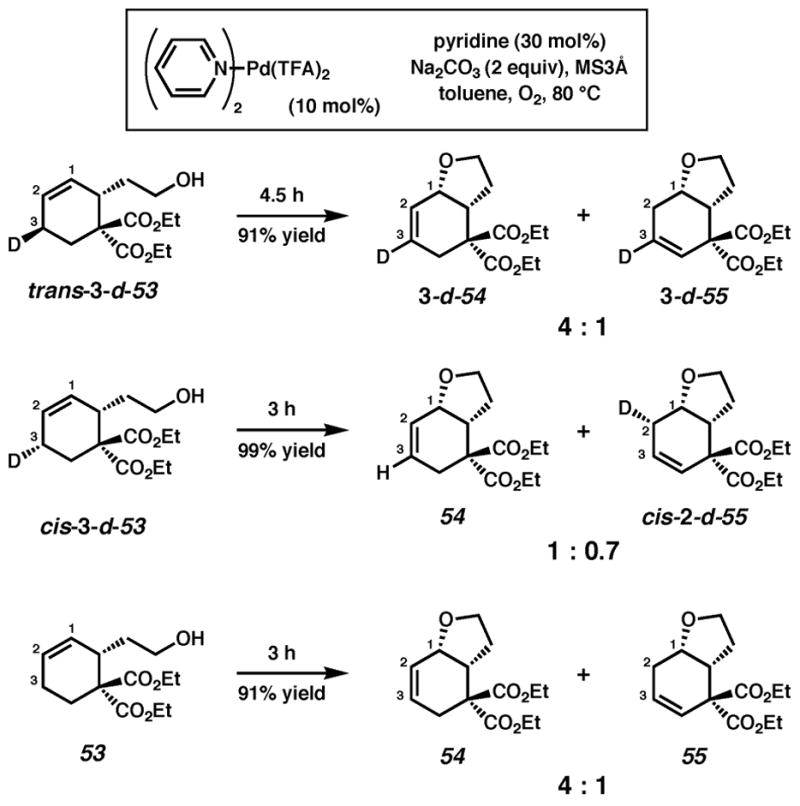
Cyclization of deuterium-labeled primary alcohol substrates with pyridine as ligand.
The mechanistic origin of the products, illustrated for the cis diastereomer (cis-3-d-53), is shown in Scheme 2. For comparison, all three possible pathways are shown. Path A involves anti nucleophilic attack of the Pd-coordinated olefin by the pendant alcohol or alkoxide. Subsequent β-hydrogen elimination would lead to the deuterium labeled product 3-d-54, but this product is not observed (vide supra, Scheme 1). In Path B, oxypalladation entails allylic C–H(D) activation and subsequent reductive elimination to Pd(0) upon formation of the C–O bond. The stereochemistry of the reductive elimination would likely be anti.44 Further, unless selective C–D activation occurs, a mixture of labeled and unlabeled products would be expected; instead a single product is observed. In Path C, a Pd-alkoxide undergoes syn oxypalladation followed by syn β-deuterium elimination to provide the observed major product, 54. Reinsertion of the Pd–D intermediate to the product olefin accounts for formation of the observed minor product, cis-2-d-55.45 The fact that an identical product mixture is obtained upon reexposure of the products to the reaction conditions supports the occurrence of reinsertion before dissociation of the product olefin from the Pd–D fragment.46 Finally, because the Pd0 intermediate in Path B cannot easily account for the formation of olefin isomer cis-2-d-55, we favor syn oxypalladation Path C.
Scheme 2.

Three oxypalladation pathways.
The remaining steps of the catalytic cycle involve reprocessing of Pd(II) by molecular oxygen. On the basis of earlier work by Murahashi and Takehira,3g,47 Uemura has proposed that this occurs by insertion of O2 directly into the [PdII]–H to form a Pd-hydroperoxide intermediate.17a Stahl has elaborated the details of a mechanism for aerobic oxidation catalysis by Pd(II) that entails reductive elimination of HX from [PdII]–H to form Pd(0) which is then oxidized by O2 in the formation of a Pd-peroxo intermediate.48,49 In any case, these steps and those outlined in Scheme 2 proceed through Pd(II) and Pd(0) intermediates; however, we cannot definitively discount a process involving Pd(IV).50
We strove to further discount the π-allyl mechanism (Path B, Scheme 2) by comparing the reactivity of two phenol substrates. As shown in Scheme 3, a phenol with a disubstituted pendant olefin (23) cyclizes in good yield to provide dihydrobenzofuran 24. The most likely π-allyl species to form from 23 would involve activation of a benzylic proton (63), but intramolecular nucleophilic attack of this intermediate (63) would not lead to 24. The π-allyl intermediate that could lead to cyclization (64) could be expected to arise from terminal olefin 62 as well (Scheme 3); both starting materials would lead to the same cyclized product (24). Instead, treatment of 62 with our standard racemic conditions leads to a complex mixture after several hours and less than 5% of 24. It may be that allylic or benzylic C–H activation does occur, but is not productive for cyclization.
Scheme 3.
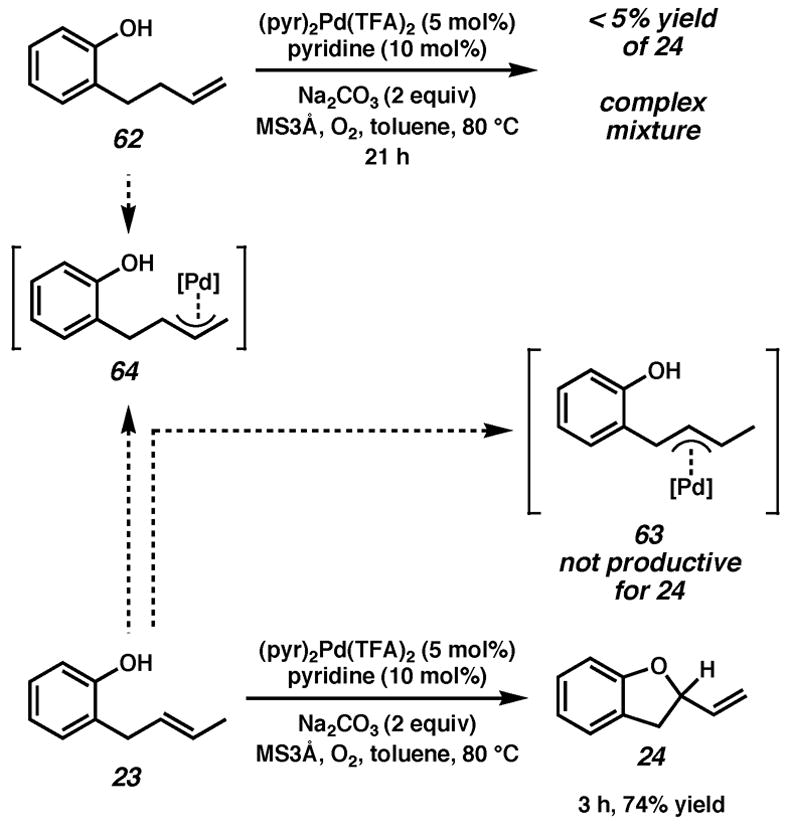
Attempted cyclization of a terminal olefin-appended phenol.
To explore whether the large effect of Pd(II) source on reactivity originated from a change in mechanism, the cyclization of cis-3-d-53 was carried out under the same reaction conditions but in the presence of (pyridine)2PdCl2 (eq. 4).51 The initial cyclization product (55) was identical to that obtained under the conditions employing (pyridine)2Pd(TFA)2; however, the olefin isomer cis-2-d-55 was now the major product. The cyclic ethers were obtained along with aldehyde cis-3-d-65 in 74% overall yield after 20 h. The formation of ether 55 and ether cis-2-d-55 implies that syn oxypalladation still occurs, contrary to what Hayashi observed for a similar phenol substrate, for which anti oxypalladation dominates upon the addition of chloride ion.33a The formation of aldehyde cis-3-d-65 likely occurs from a common alkoxide intermediate, and highlights the effect that subtle changes in reaction conditions can have on the mode of oxidation.
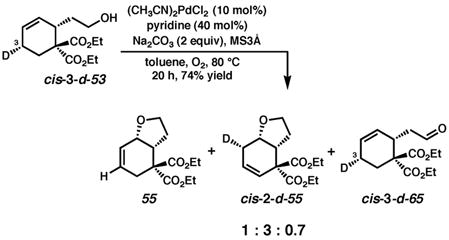 |
(4) |
Deuterium-labeling studies of primary alcohol substrates in the presence of a bidentate ligand
The deuterium labeled alcohol substrates trans-3-d-53 and cis-3-d-53 were then cyclized under conditions that employed dipyridyl as a bidentate ligand. Treatment of both trans-3-d-53 and cis-3-d-53 with 10 mol% (dipyridyl)Pd(TFA)2, Na2CO3 (2 equiv), 1 atm O2, 500 mg/mmol MS3Å in toluene at 80 °C led to the formation of the same major products (3-d-54 and 54, respectively) observed with the conditions that use pyridine (Scheme 4). None of the olefin isomer was observed; instead a small amount of aldehyde (trans-3-d-65 and cis-3-d-65) was formed for both starting material diastereomers.52 Both diastereomers were slower to cyclize in the presence of (dipyridyl)Pd(TFA)2 than in the presence of (pyridine)2Pd(TFA)2. The reaction of trans-3-d-53 under the asymmetric conditions using (−)-sparteine led only to oxidation to the aldehyde. The major product distribution from cyclization in the presence of both a mono- and bidentate ligand is the same. Presumably the aldehyde forms via β-hydrogen elimination from a common alkoxide intermediate before oxypalladation can occur.
Scheme 4.
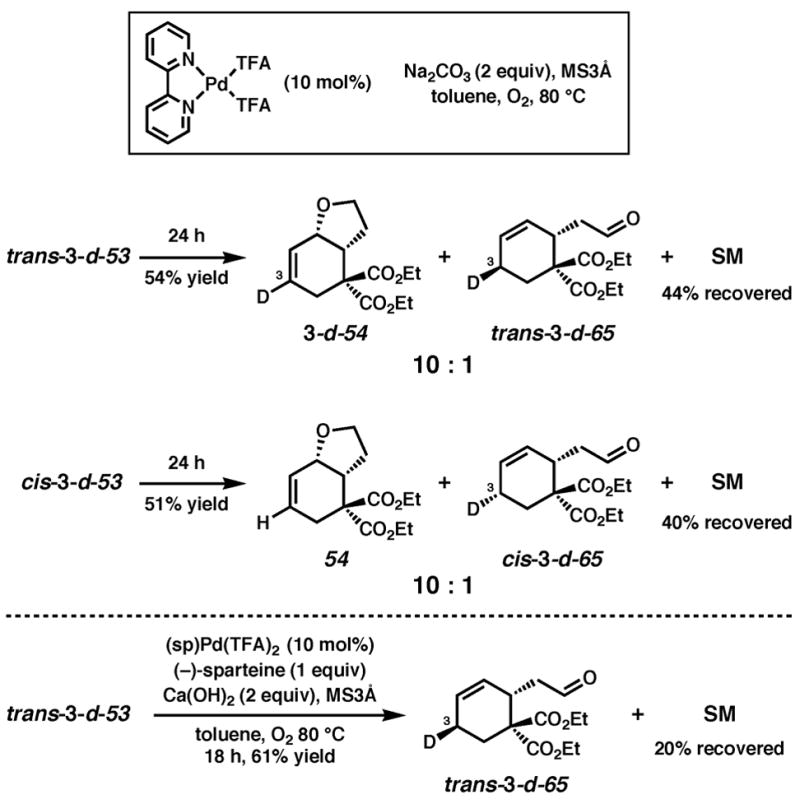
Cyclization of deuterium-labeled primary alcohol substrates with dipyridyl as ligand.
On the basis of the results outlined above, we propose that at least for the alcohol substrates, reaction under mono- or bidentate ligated conditions occurs by a similar pathway. The difference in reaction rate could arise from the degree to which each ligand can stabilize intermediates under otherwise identical reaction conditions. A neutral monodentate ligand like pyridine can dissociate when necessary to free a coordination site for the substrate while maintaining charge-neutral intermediates (67–69, Scheme 5, A). When dipyridyl is used, neutral ligand dissociation may be more difficult due to chelation, and instead an anionic ligand must dissociate, resulting in charged intermediates (71 and 73, Scheme 5, B). Such intermediates may be higher in energy under the reaction conditions, which in turn results in decreased reactivity.53 (−)-Sparteine (cf. Scheme 4) not only leads to the same limitation, but also prevents cyclization, perhaps due to steric congestion at Pd that prevents formation of the Pd–O–olefin chelate (71).
Scheme 5.

Comparison of reaction pathways with a monodentate and bidentate ligand.
Although an extension of these mechanistic conclusions to the racemic and asymmetric phenol cyclizations may be tenuous, the same scenario would account for the slowness of the reactions catalyzed by ((−)-sparteine)Pd(TFA)2 relative to (pyridine)2Pd(TFA)2. Hayashi and coworkers’ results also support this conclusion. Their system undergoes syn oxypalladation in the presence of a bidentate ligand with a dicationic Pd(II) catalyst in methanol using benzoquinone as the terminal oxidant.33a The higher dielectric solvent and dicationic catalyst may facilitate the formation of the charged intermediates shown in Scheme 5B, whereas our toluene-based conditions slow their formation. Because most of the chiral ligands that are effective in inducing asymmetry are bidentate, the mechanistic implication of the results described above is that selectivity and good reactivity will be difficult to attain in the Pd(II)/toluene/O2 system.
Deuterium-labeling studies of carboxylic acid substrates
Alcohols trans-3-d-53 and cis-3-d-53 were oxidized40 to the corresponding carboxylic acid derivatives (trans-3-d-74 and cis-3-d-74) and subjected to the cyclization conditions that employ pyridine (Scheme 6). Both were slow to cyclize and formation of Pd-black was observed; nevertheless, some product was obtained and analyzed by 1H and 2H NMR. In contrast to the alcohol substrates, the formation of unlabeled lactone 75 from trans-3-d-74 and labeled lactone 3-d-75 from cis-3-d-74 indicates that cyclization of the acid proceeds through anti oxypalladation. The olefin isomer arising from reinsertion was not observed. Cyclization of trans-3-d-74 in the absence of Na2CO3 resulted in greatly diminished yield (4% yield after 24 h) of the same product (75) but slower formation of Pd-black, which indicates that the inorganic base does not affect the stereochemistry of oxypalladation, but may play a role in catalyst decomposition.
Scheme 6.
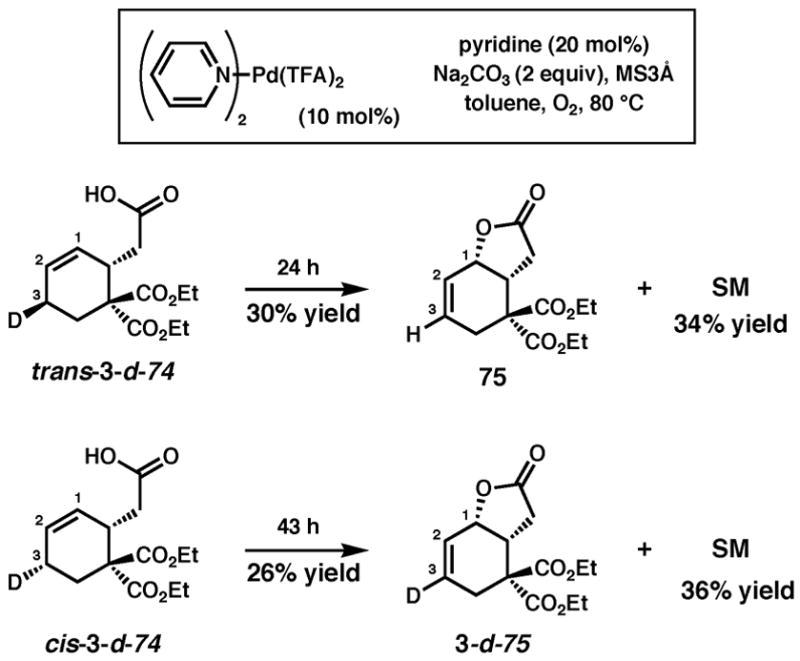
Cyclization of deuterium-labeled carboxylic acid substrates with pyridine as ligand.
Although the reasons for the change in product distribution at this point remain unclear, geometrical constraints, pKa differences, or differences in nucleophilicity between an acid and an alcohol are all possibilities. The additional unsaturation in the forming lactone could make geometrically unfavorable the Pd–O–olefin chelate that would precede syn oxypalladation. Whatever the case, these results confirm the versatility and undiscriminating reactivity of Pd(II) catalysts in oxidation reactions. As demonstrated by these results and the accumulating evidence from several groups, generalizations about reactivity and mechanism that span different substrates and different reaction conditions are difficult to make for palladium catalyzed oxidation reactions.9,32,33,37,38,50
SUMMARY AND CONCLUSION
In summary, we have demonstrated that oxidase-type cyclizations of several different nucleophiles onto pendant olefins can be achieved in excellent yields under simple conditions. Reactivity is highly dependent on Pd(II) source, basic additive, and ligand. Several different types of cyclic systems are attainable, including aryl and alkyl bicycles, as well as fused and spirocyclic motifs. These cyclizations are part of an ongoing effort to develop enantioselective oxidase-type reactions using Pd(II) catalysis and molecular oxygen. To this end, the racemic conditions we developed were suitable for extension to an enantioselective cyclization that uses the chiral ligand (−)-sparteine. While the asymmetric oxidative cyclization conditions that we have developed are not yet general, we have established that it is possible to adapt a direct dioxygen-coupled racemic reaction to aerobic asymmetric catalysis, which had not before been achieved for this class of reaction. In conjunction with our work on the oxidative kinetic resolution of secondary alcohols, the asymmetric cyclization chemistry is a significant stride into the arena of enantioselective oxidase-type chemistry.
Stereospecifically deuterated primary alcohol substrates were used to probe the stereochemistry of oxypalladation and to gain insight into the mechanism of cyclization. Contrary to the common mechanistic proposal for reactions of this type (i.e., “Wacker cyclization” or anti oxypalladation), reaction of the primary alcohol substrates appears to occur via syn oxypalladation. This is in agreement with more recent reports from Hayashi and Wolfe on related systems. Neither the presence of chloride anion, nor the use of a bidentate instead of a monodentate ligand changes the stereochemistry of oxypalladation. The implications for the asymmetric reaction are that bidentate ligands may destabilize the intermediates necessary for a syn oxypalladation pathway; thus reactivity is decreased relative to the conditions employing a monodentate ligand. On the other hand, similarly deuterated carboxylic acid substrates react in the opposite sense, i.e., via anti oxypalladation.
As contemporary work in this rapidly developing, recently reborn field continues to demonstrate, oxidase-type catalysis by Pd(II) is highly versatile and adaptable to a variety of applications.54 Our studies reported herein emphasize the subtleties of reactivity and mechanism in this field. Further development of dehydrogenation reactions using Pd(II) and molecular oxygen, racemic and asymmetric, is ongoing.
Supplementary Material
Full experimental details and characterization data for all new compounds. This material is available free of charge from the Internet at http://pubs.acs.org.
Acknowledgments
The authors wish to thank the NIH-NIGMS (R01 GM65961-01), Bristol-Myers Squibb Company and the American Chemical Society (graduate fellowship to R.M.T.), the University of California TRDRP (postdoctoral fellowship to Y.K.R.), the Dreyfus Foundation, Merck Research Laboratories, Research Corporation, Abbott Laboratories, Pfizer, Amgen, GlaxoSmithKline, Lilly, and Johnson and Johnson for generous financial support. Drs. Eric Ferreira, Jonathan Owen, and Andrew Waltman are acknowledged for helpful discussions.
REFERENCES SECTION
- 1.Johnson RA, Sharpless KB. In: Catalytic Asymmetric Synthesis. Ojima I, editor. Wiley & Sons, Inc; New York: 2000. pp. 231–280.Katsuki T. In: Catalytic Asymmetric Synthesis. Ojima I, editor. Wiley & Sons, Inc; New York: 2000. pp. 287–325.Jacobsen EN. In: Comprehensive Asymmetric 21 Catalysis. Jaobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 2. Springer; Berlin: 1999. pp. 607–618.For a recent review of advances in transition metal catalyzed oxidation, see: Punniyamurthy T, Velusamy S, Iqbal J. Chem Rev. 2005;105:2329–2364. doi: 10.1021/cr050523v.
- 2.(a) Tsuji J. Palladium Reagents and Catalysis. John Wiley & Sons, Ltd; Chichester, UK; Hoboken, NJ: 2004. [Google Scholar]; (b) Negishi E, editor. Handbook of Organopalladium Chemistry for Organic Synthesis. Wiley & Sons, Inc; New York: 2002. [Google Scholar]; (c) Tietze LF, Ila H, Bell HP. Chem Rev. 2004;104:3453–3516. doi: 10.1021/cr030700x. [DOI] [PubMed] [Google Scholar]
- 3.For reviews, see: Stoltz BM. Chem Lett. 2004;33:362–367.Stahl SS. Angew Chem, Int Ed. 2004;43:3400–3420. doi: 10.1002/anie.200300630.Sigman MS, Schultz MJ. Org Biomol Chem. 2004;2:2551–2554. doi: 10.1039/B409127M.For a recent review of Pd-catalyzed alcohol oxidation, see: Muzart J. Tetraherdon. 2003;59:5789–5816.Trost BM. Acc Chem Res. 1990;23:34–42.Hedgedus LS. Tetrahedron. 1984;40:2415–2434.Hosokawa T, Murahashi SI. Acc Chem Res. 1990;23:49–54.Semmelhack MF, Kim C, Zhang N, Bodurow C, Sanner M, Dobler W, Meier M. Pure Appl Chem. 1990;23:2035–2040.Tsuji J. Palladium Reagents and Catalysts. John Wiley & Sons, Ltd; Chichester, UK: 1995. pp. 125–527.Zeni G, Larock RC. Chem Rev. 2004;104:2285–2309. doi: 10.1021/cr020085h.Wolfe JP, Thomas JS. Curr Org Chem. 2005;9:625–655.
- 4.Blackburn TF, Schwartz J. J Chem Soc, Chem Commun. 1977:157–158. [Google Scholar]
- 5.For examples of phenol cyclizations, see: Larock RC, Wei L, Hightower T. Synlett. 1998:522–524.For examples of alcohol cyclizations, see: Rönn M, Bäckvall J-E, Andersson PG. Tetrahedron Lett. 1995;36:7749–7752.For examples of acid cyclizations, see: Larock RC, Hightower TR. J Org Chem. 1993;58:5298–5300.For examples of tosylamide cyclizations, see: Larock RC, Hightower TR, Hasvold LA, Peterson KP. J Org Chem. 1996;61:3584–3585. doi: 10.1021/jo952088i. and references therein.For examples of primary and secondary alcohol oxidation to aldehydes and ketones, see: Peterson KP, Larock RC. J Org Chem. 1998;63:3185–3189.
- 6.For examples of racemic systems that employ the copper/O2 system, see: Phenol cyclizations: Hosokawan T, Ohkata H, Moritani I. Bull Chem Soc Japan. 1975;48:1533–1536.Alcohol cyclizations: Hosokawa T, Hirata M, Murahashi S-I, Sonoda A. Tetrahedron Lett. 1976;21:1821–1824.
- 7.For examples of racemic systems that employ benzoquinone, see Aniline cyclizations: Hegedus LS, Allen GF, Bozell JJ, Waterman EL. J Am Chem Soc. 1978;100:5800–5807.For oxidative carbon-carbon bond forming aryl/olefin cyclizations: Zhang H, Ferreira EM, Stoltz BM. Angew Chem, Int Ed. 2004;43:6144–6148. doi: 10.1002/anie.200461294.
- 8.In contrast, reactions that are solely aerobic produce only H2O or H2O2 as byproducts.
- 9.(a) Thorarensen A, Palmgren A, Itami K, Bäckvall JE. Tetrahedron Lett. 1997;38:8541–8544. [Google Scholar]; (b) Itami K, Palmgren A, Thorarensen A, Bäckvall JE. J Org Chem. 1998;63:6466–6471. [Google Scholar]; (c) Cotton HK, Verboom RC, Johansson L, Plietker BJ, Bäckvall JE. Organometallics. 2002;21:3367–3375. [Google Scholar]
- 10.Chen MS, Prabagaran N, Labenz NA, White MC. J Am Chem Soc. 2005;102:6970–6971. doi: 10.1021/ja0500198. [DOI] [PubMed] [Google Scholar]
- 11.(a) Hosokawa T, Uno T, Inui S, Murahashi SI. J Am Chem Soc. 1981;103:2318–2323. [Google Scholar]; (b) Hosokawa T, Okuda C, Murahashi SI. J Org Chem. 1985;50:1282–1287. [Google Scholar]
- 12.(a) Uozumi Y, Kato K, Hayashi T. J Am Chem Soc. 1997;119:5063–5064. [Google Scholar]; (b) Uozumi Y, Kato K, Hayashi T. J Org Chem. 1998;63:5071–5075. doi: 10.1021/jo980100b. [DOI] [PubMed] [Google Scholar]; (c) Uozumi Y, Kyota H, Ogasawara M, Hayashi T. J Org Chem. 1999;64:1620–1625. doi: 10.1021/jo982104m. [DOI] [PubMed] [Google Scholar]
- 13.Arai MA, Kuraishi M, Arai T, Sasai H. J Am Chem Soc. 2001;123:2907–2908. doi: 10.1021/ja005920w. [DOI] [PubMed] [Google Scholar]
- 14.For examples of non-oxidative Pd(II)-mediated enantioselective catalysis, see: Fuiji A, Hagiwara E, Sodeoka M. J Am Chem Soc. 1999;121:5450–5458.El-Qisairi A, Hamed O, Henry PM. J Org Chem. 1998;63:2790–2791. doi: 10.1021/jo005627e.Zhang Q, Lu X. J Am Chem Soc. 2000;122:7604–7605.Overman LE, Remarchuk TP. J Am Chem Soc. 2002;124:12–13. doi: 10.1021/ja017198n.
- 15.Ferreira EM, Stoltz BM. J Am Chem Soc. 2001;123:7725–7726. doi: 10.1021/ja015791z.Bagdanoff JT, Ferreira EM, Stoltz BM. Org Lett. 2003;5:835–837. doi: 10.1021/ol027463p.Caspi DD, Ebner DC, Bagdanoff JT, Stoltz BM. Adv Synth Catal. 2004;346:185–189.For conditions employing chloroform and air, see: Bagdanoff JT, Stoltz BM. Angew Chem, Int Ed. 2004;43:353–357. doi: 10.1002/anie.200352444.Trend RM, Stoltz BM. J Am Chem Soc. 2004;126:4482–4483. doi: 10.1021/ja039551q.
- 16.For a similar system, see: Jensen DR, Pugsley JS, Sigman MS. J Am Chem Soc. 2001;123:7475–7476. doi: 10.1021/ja015827n.Mueller JA, Jensen DR, Sigman MS. J Am Chem Soc. 2002;124:8202–8203. doi: 10.1021/ja026553m.Jensen DR, Sigman MS. Org Lett. 2003;5:63–65. doi: 10.1021/ol027190y.Mandal SK, Jensen DR, Pugsley JS, Sigman MS. J Org Chem. 2003;68:4600–4603. doi: 10.1021/jo0269161.Mueller JA, Sigman MS. J Am Chem Soc. 2003;125:7005–7013. doi: 10.1021/ja034262n.Mandal SK, Sigman MS. J Org Chem. 2003;68:7535–7537. doi: 10.1021/jo034717r.
- 17.Nishimura T, Onoue T, Ohe K, Uermura S. J Org Chem. 1999;64:6750–6755. doi: 10.1021/jo9906734.Uemura has used similar conditions for the ring cleavage of tert-cyclobutanols, see: Nishimura T, Ohe K, Uemura S. J Am Chem Soc. 1999;121:2645–2646.Related heterogeneous conditions that use a hydrotalcite solid support have been developed: Kakiuchi N, Maeda Y, Nishimura T, Uemura S. J Org Chem. 2001;66:6620–6625. doi: 10.1021/jo010338r.
- 18.Trend RM, Ramtohul YK, Ferreira EM, Stoltz BM. Angew Chem, Int Ed. 2003;42:2892–2895. doi: 10.1002/anie.200351196. [DOI] [PubMed] [Google Scholar]
- 19.Ferreira EM, Stoltz BM. J Am Chem Soc. 2003;125:9578–9579. doi: 10.1021/ja035054y. [DOI] [PubMed] [Google Scholar]
- 20.(a) Anton DR, Crabtree RH. Organometallics. 1983;2:855–859. [Google Scholar]; (b) Foley P, DiCosimo R, Whitesides GM. J Am Chem Soc. 1980;102:6713–6725. [Google Scholar]
- 21.For an example of a Pd-catalyzed phenol/olefin cyclization using an N-heterocyclic carbene ligand, see: Muñiz K. Adv Synth Catal. 2004;346:1425–1428.
- 22.For a recent review of Pd-catalyzed reactions of alcohols to form ether linkages, see: Muzart J. Tetrahedron. 2005;61:5955–6008.
- 23.Semmelhack MF, Kin CR, Dobler W, Meier M. Tetrahedron Lett. 1989;30:4925–4928. [Google Scholar]
- 24.Åkermark B, Larsson EM, Oslob JD. J Org Chem. 1994;59:5729–5733. [Google Scholar]
- 25.A similar cyclization of sulfonamides was recently reported, see: Fix SR, Brice JL, Stahl SS. Angew Chem, Int Ed. 2002;41:164–166. doi: 10.1002/1521-3773(20020104)41:1<164::aid-anie164>3.0.co;2-b.
- 26.10 mol% Pd(TFA)2, 40 mol% ligand, 500 mg/mmol MS3Å, 0.41 equiv tridecane internal GC standard, Conversion and enantiomeric excess were determined by GC. 1 atm O2, toluene (0.1 M), 80 °C.
- 27.Attempted cyclization under the chloroform-based rate-enhanced conditions for oxidative kinetic resolution ((sp)PdCl2 (5 mol%), (−)-sparteine (7 mol%), Cs2CO3 (0.4 equiv), CHCl3 (0.1 M), O2 (1 atm, balloon), 25 °C) resulted in no reaction.
- 28.A similar, albeit less dramatic counterion effect was observed in the oxidative kinetic resolution of secondary alcohols; see Ref. 15a and Nielsen RJ, Keith JM, Stoltz BM, Goddard WA., III J Am Chem Soc. 2004;126:7967–7974. doi: 10.1021/ja031911m.
- 29.The absolute stereochemistry was determined by comparison of the optical rotation with that reported for (−)-2, by Uozumi and Hayashi, Ref. 12c (see Supporting Information for rotation data). We report the absolute stereochemistry of (+)-8, (−)-6, (+)-4 and (−)-10 by analogy to this substrate.
- 30.Tetrasubstituted olefin-containing phenol 21, which is Hayashi’s most selectively cyclized substrate,12a fails to cyclize under our optimized conditions.
- 31.(a) Ref. 3g and references therein. Coleman JP, Hegedus LS. Principles and Applications of Organometallic Chemistry. University Science Books; Mill Valley, CA: 1980. pp. 401–424.
- 32.(a) Bäckvall JE, Åkermark B, Ljunggren SO. J Chem Soc, Chem Commun. 1977:264–265. [Google Scholar]; (b) Bäckvall JE, Åkermark B, Ljunggren SO. J Am Chem Soc. 1979;101:2411–2416. [Google Scholar]; (c) James DE, Hines LF, Stille JK. J Am Chem Soc. 1976;98:1806–1809. [Google Scholar]; (d) Stille JK, Divakaruni R. J Am Chem Soc. 1978;100:1303–1304. [Google Scholar]; (e) Majima T, Kurosawa H. J Chem Soc, Chem Commun. 1977:610–611. [Google Scholar]
- 33.For systems containing oxygen nucleophiles, see: Hayashi T, Yamasaki K, Mimura M, Uozumi Y. J Am Chem Soc. 2004;126:3036–3037. doi: 10.1021/ja031946m.Hay MB, Hardin AR, Wolfe JP. J Org Chem. 2005;70:3099–3107. doi: 10.1021/jo050022+.For systems containing nitrogen nucleophiles, see: Ney JE, Wolfe JP. Angew Chem, Int Ed. 2004;43:3605–3608. doi: 10.1002/anie.200460060.Ney JE, Wolfe JP. J Am Chem Soc. 2005;127:8644–8657. doi: 10.1021/ja0430346.
- 34.Bäckvall and coworkers have characterized computationally the reactivity of nucleophiles toward cis migration, or olefin insertion, in (π-olefin)Pd(II) complexes and found that the HOMO-LUMO gap between the π* olefin orbital and a Pd–OH nucleophile is too large for migration to be frontier controlled. Rather, the process is charge controlled, and likely does not occur through a concerted, 4-center transition state as in true olefin insertion reactions of Pd–CH3 or Pd–H nucleophiles. See: Bäckvall JE, Björkman EE, Pettersson L, Siegbahn P. J Am Chem Soc. 1984;106:4369–4373.Bäckvall JE, Björkman EE, Pettersson L, Siegbahn P. J Am Chem Soc. 1985;107:7265–7267.
- 35.For experimental evidence of insertion of tetrafluoroethylene into a Pt–O bond, see: Bryndza HE. Organometallics. 1985;4:406–408.
- 36.For a recent example of olefin insertion into a rhodium amide, see: Zhao P, Krug C, Hartwig JF. J Am Chem Soc. 2005;127:12066–12073. doi: 10.1021/ja052473h.
- 37.(a) Francis JW, Henry PM. Organometallics. 1991;10:3498–3503. [Google Scholar]; (b) Zaw K, Henry PM. Organometallics. 1992;11:2832–2836. [Google Scholar]; (c) Francis JW, Henry PM. Organometallics. 1992;11:2008–2015. [Google Scholar]; (d) Hamed O, Henry PM. Organometallics. 1997;16:4903–4909. [Google Scholar]; (e) Hamed O, Thompson C, Hernry PM. J Org Chem. 1997;62:7082–7083. doi: 10.1021/jo971051q. [DOI] [PubMed] [Google Scholar]; (f) Hamed O, Henry PM, Thompson C. J Org Chem. 1999;64:7745–7750. [Google Scholar]; (g) ten Brink G-J, Arends IWCW, Papadogianakis G, Sheldon RA. Appl Catal, A. 2000;194–195:435–442. [Google Scholar]; (h) Nelson DJ, Li R, Brammer C. J Am Chem Soc. 2001;123:1564–1568. doi: 10.1021/ja002190j. [DOI] [PubMed] [Google Scholar]
- 38.For evidence supporting this mechanism in intermolecular examples, see: Grennberg H, Simon V, Bäckvall JE. J Chem Soc, Chem Commun. 1994:265–266.Grennberg H, Bäckvall JE. Chem Eur J. 1998;4:1083–1089.
- 39.Trost has shown that Pd(TFA)2 will form π-allyl complexes by C–H activation of olefins in acetone, see: Trost BM, Metzner PJ. J Am Chem Soc. 1980;102:3572–3577.
- 40.See Supporting Information for details.
- 41.Examples of anti β-hydrogen elimination under various conditions have been reported, but involve aromatization or the formation of highly conjugated systems; see: Toyota M, Ilangovan A, Okamoto R, Masaki T, Arakawa M, Ihara M. Org Lett. 2002;4:4293–4296. doi: 10.1021/ol020187u.Lautens M, Fang YQ. Org Lett. 2003;5:3679–3682. doi: 10.1021/ol035354k.Hughes CC, Trauner D. Angew Chem Int Ed. 2002;41:1569–1572. doi: 10.1002/1521-3773(20020503)41:9<1569::aid-anie1569>3.0.co;2-8.Buchwald SL, Hennessy EJ. J Am Chem Soc. 2003;125:12084–12084. doi: 10.1021/ja037546g.
- 42.For a study that attempts to differentiate between oxypalladation and a π-allyl route for a Pd(II)-catalyzed cyclization, see: Zanoni G, Porta A, Meriggi A, Franzini M, Vidari G. J Org Chem. 2002;67:6064–6069. doi: 10.1021/jo025722i.
- 43.After 5.5 h, 89% yield of a 1:0.75 mixture of 54 and cis-2-d-55 was obtained.
- 44.For a Pd(II)-π-allyl electrophile, a primary alcohol (or alkoxide) would fall into the class of “soft” nucleophiles, the conjugate acids of which as defined by Trost have a pKa < 25, see: Trost BM, Van Vranken DL. Chem Rev. 1996;96:395–422. doi: 10.1021/cr9409804.
- 45.The difference in the product ratios formed from the trans and cis isomers could be accounted for by an isotope effect in the reinsertion step. Possibilities include: slower reinsertion for the [Pd]–H bound olefin than for the [Pd]–D bound olefin, faster dissociation for the [Pd]–H bound olefin, faster reductive elimination of HX before dissociation or reinsertion, or faster coordination and insertion of O2 into the [Pd]–H bond, if turnover of the catalyst occurs by that mechanism. At this time we cannot rule out any of or distinguish among these possibilities.
- 46.Because both [Pd]–D and [Pd]–H are formed, reinsertion after [Pd] dissociates from product 54 would lead to scrambling of the isotopic label.
- 47.Takehira K, Hayakawa T, Orita H. Chem Lett. 1985:1835–1838. and references therein. [Google Scholar]
- 48.Stahl SS, Thorman JL, Nelson RC, Kozee MA. J Am Chem Soc. 2001;123:7188–7189. doi: 10.1021/ja015683c.See also: Steinhoff BA, Stahl SS. Org Lett. 2002;4:4179–4181. doi: 10.1021/ol026988e.
- 49.Keith JM, Nielsen RJ, Oxgaard J, Goddard WA., III J Am Chem Soc. 2005;127:13172–13179. doi: 10.1021/ja043094b. [DOI] [PubMed] [Google Scholar]
- 50.In one scenario, oxidative activation of an allylic C–H bond could lead to a Pd(IV)-alkyl intermediate for the formation of a Pd(II) π-allyl species. Or, reductive elimination upon formation of the C–O bond could lead to a Pd(II) intermediate. In another scenario, Pd(II) could undergo oxidative activation of an O–H bond to form a Pd(IV)-alkoxide, although this is unlikely in the presence of base. Although such pathways cannot be ruled out, the recently reported Pd oxidation reactions proposed to involve Pd(IV) also involve strong oxidizing agents such as PhI(OAc)2 and I2. For recent articles invoking Pd(IV) in Pd-catalyzed oxidative processes, see: Dick AR, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m.Dick AR, Kampf JW, Sanford MS. Organometallics. 2005;24:482–485.Alexanian EJ, Lee C, Sorensen EJ. J Am Chem Soc. 2005;127:7690–7691. doi: 10.1021/ja051406k.Yoneyama T, Crabtree RH. J Mol Catal A. 1996;108:35–40.For a related Pd-catalyzed iodination system, see: Giri R, Chen X, Yu JQ. Angew Chem, Int Ed. 2005;44:2112–2115. doi: 10.1002/anie.200462884.
- 51.The catalyst was generated in situ by heating (CH3CN)2PdCl2 and all other reagents except the substrate for 15 min before addition of the substrate.
- 52.The identity of the aldehyde was confirmed by oxidation of the alcohol by another method, see Supporting Information.
- 53.We cannot rule out the possibility that the reaction with dipyridyl as ligand proceeds through similar intermediates with slow, partial dissociation of one of the dipyridyl nitrogen atoms. The reaction with (−)-sparteine as ligand is much less likely to undergo partial dissociation due to its structural rigidity.
- 54.(a) Sohn JH, Waizumi N, Zhong HM, Rawal VH. J Am Chem Soc. 2005;127:7290–7291. doi: 10.1021/ja050728l. [DOI] [PubMed] [Google Scholar]; (b) Garg NK, Caspi DD, Stoltz BM. J Am Chem Soc. 2005;127:5970–5978. doi: 10.1021/ja050586v. [DOI] [PubMed] [Google Scholar]; (c) Tietze LF, Sommer KM, Zinngrebe J, Stecker F. Angew Chem, Int Ed. 2004;44:257–259. doi: 10.1002/anie.200461629. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full experimental details and characterization data for all new compounds. This material is available free of charge from the Internet at http://pubs.acs.org.