

Summary
Quorum sensing, a process of bacterial cell-cell communication, relies on production, detection, and response to autoinducer signaling molecules. Here we focus on LuxN, a nine transmembrane domain protein from Vibrio harveyi, and the founding example of membrane-bound receptors for acyl-homoserine lactone (AHL) autoinducers. Previously, nothing was known about signal recognition by membrane-bound AHL receptors. We used mutagenesis and suppressor analyses to identify the AHL-binding domain of LuxN, and discovered LuxN mutants that confer decreased and increased AHL sensitivity. Our analysis of dose-response curves of multiple LuxN mutants pins these inverse phenotypes on quantifiable opposing shifts in the free-energy bias of LuxN for its kinase and phosphatase states. To extract signaling parameters, we exploited a strong LuxN antagonist, one of fifteen small-molecule antagonists we identified. We find that quorum-sensing-mediated communication can be manipulated positively and negatively to control bacterial behavior, and that signaling parameters can be deduced from in vivo data.
Keywords: Quorum Sensing, Autoinducer, Receptor, Kinase, Phosphatase, Free-Energy Bias
Introduction
Quorum sensing is a process of bacterial cell-cell communication that involves production and detection of secreted signaling molecules called autoinducers (Waters and Bassler, 2005). Quorum sensing allows bacteria to collectively regulate gene expression and thereby function as multi-cellular organisms. The bioluminescent Gram-negative quorum-sensing bacterium Vibrio harveyi integrates information from three different diffusible autoinducers that together enable intra-and inter-species communication (Henke and Bassler, 2004b). The three V. harveyi autoinducers are AI-1 (3-hydroxybutanoyl homoserine lactone), AI-2 ((2S,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran-borate), and CAI-1 ((S)-3-hydroxytridecan-4-one) (Bassler et al., 1993; Cao and Meighen, 1989; Chen et al., 2002; Higgins et al., 2007; Surette et al., 1999). These signals are detected by the sensor-kinase proteins, LuxN, LuxQ, and CqsS, respectively (Figure 1A) ((Henke and Bassler, 2004b). At low cell density, (i.e., in the absence of autoinducers), these sensor kinases autophosphorylate and transfer phosphate to the shared phospho-transfer protein, LuxU (Freeman and Bassler, 1999a, b). LuxU transfers the phosphoryl-group to the DNA-binding response regulator, LuxO, which activates transcription of genes encoding five redundant small regulatory RNAs called the quorum regulatory RNAs (Qrrs) (Figure 1A) (Lenz et al., 2004; Tu and Bassler, 2007). The Qrrs destabilize the mRNA transcript encoding the master quorum-sensing regulator, LuxR (Martin et al., 1989; Showalter et al., 1990; Svenningsen et al., 2008). Therefore, under low-cell-density conditions, the bacteria do not display quorum-sensing behaviors. In contrast, at high cell density the three autoinducers accumulate and bind to their cognate receptors. These binding events switch the receptors to phosphatases, resulting in dephosphorylation of LuxO and termination of Qrr production. The luxR transcript is stabilized, leading to LuxR protein production (Figure 1A and Tu and Bassler, 2007). LuxR controls the genes in the quorum sensing, including those required for bioluminescence, siderophore production, type III secretion, and metalloprotease production (Fuqua et al., 1996; Hammer and Bassler, 2003; Henke and Bassler, 2004a; McFall-Ngai and Ruby, 2000; Miller and Bassler, 2001; Waters and Bassler, 2005).
Figure 1. The V. harveyi Quorum-Sensing Circuit and the LuxN Trans-Membrane Domain.
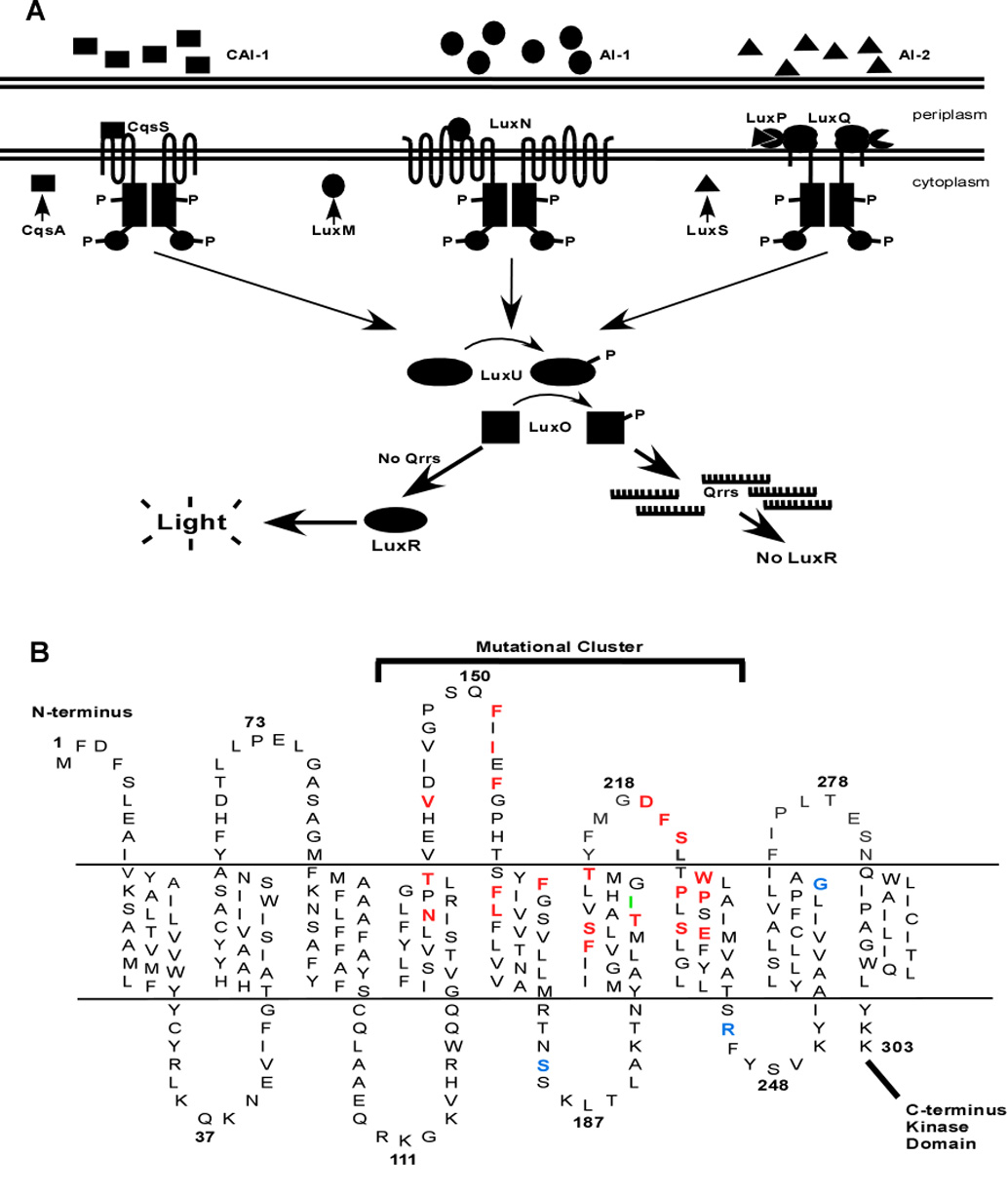
(A) The autoinducer receptor systems are CAI-1/CqsS, AI-1/LuxN, and AI-2/LuxPQ. CAI-1 is (S)-3-hydroxytridecan-4-one (squares), AI-1 is 3-hydroxybutanoyl homoserine lactone (ovals), and AI-2 is (2S,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran-borate (triangles), and they are synthesized by CqsA, LuxM, and LuxS, respectively. At low cell densities, in the absence of appreciable autoinducer, CqsS, LuxN, and LuxQ act as kinases funneling phosphate via LuxU to LuxO (arrows). Phospho-LuxO activates expression of the qrr genes; the Qrr sRNAs (comb shapes) are transcribed and they bind to and facilitate the degradation of the mRNA encoding LuxR. Without LuxR, there is no quorum sensing, and thus no light production. At high cell density, in the presence of autoinducers, the receptors act as phosphatases, draining phosphate from LuxO via LuxU. Transcription of the qrr genes is terminated, the LuxR mRNA is stabilized, and LuxR protein is produced. By activating and repressing a variety of genes, LuxR facilitates the transition of the cells into quorum-sensing mode. One operon activated by LuxR at high cell density encodes luciferase, so in the presence of autoinducers, V. harveyi produces light. (B) The cartoon depicts the putative topology of the N-terminal region of LuxN. Amino acids in red, when mutated, confer a dark phenotype. Amino acids in blue denote sites where mutations enhance sensitivity of LuxN to AI-1. The amino acid in green represents the LuxN* suppressor mutation that prevents C450-0730 antagonism.
AI-1 is an AHL and it is the strongest of the three V. harveyi signals and, thus, the major input controlling quorum-sensing-regulated behaviors (Henke and Bassler, 2004b). Typically, AHL autoinducers are detected by cytoplasmic LuxR-type transcriptional activators (note: these LuxR-type proteins are unrelated to V. harveyi LuxR, Figure 1A) (Fuqua et al., 2001; Fuqua et al., 1996; Fuqua et al., 1994) (Engebrecht and Silverman, 1984, 1987). V. harveyi is unusual because all three of its autoinducers, including AI-1, are detected by membrane-bound sensor-kinase proteins (in the case of AI-2, however, an additional periplasmic binding protein LuxP is required in conjunction with the membrane-bound two-component protein LuxQ). AI-1 is also the defining member of a growing family of recognized AHLs that interact with membrane-bound sensor-kinases like LuxN, rather than with cytosolic LuxR-type proteins (Freeman et al., 2000; Jung et al., 2007; Timmen et al., 2006). There are currently 11 LuxN homologs in the NCBI database, but nothing is known about how AHLs interact with this important class of receptors (Figure S2).
Membrane-topology analysis predicts that LuxN is bound to the bacterial inner-membrane by nine trans-membrane (TM) spanning helices (Figure 1B) (Jung et al., 2007). The N-terminus of LuxN is on the periplasmic side of the bacterial inner-membrane, while the histidine-kinase portion of LuxN resides in the cytosol as judged by reporter-protein fusion analyses (Jung et al., 2007). Therefore, LuxN contains four periplasmic loops and four cytosolic loops connecting the nine TM segments (Figure 1B). By analogy to homologous membrane-bound sensor kinases, LuxN is believed to assemble into homodimers (Park et al., 1998). To locate the AI-1 binding domain of LuxN, we performed a genetic screen to identify luxN mutants encoding proteins incapable of properly responding to AI-1. We found that the LuxN AI-1 binding domain is composed of TM helices 4, 5, 6, and 7 as well as the intervening periplasmic loops 2 and 3. We also used a high-throughput chemical screen to identify a set of small molecules that specifically antagonize the LuxN/AI-1 interaction. All of these LuxN antagonist molecules have IC50 values in the low micromolar range, and, based on competition assays and genetic evidence, the most potent LuxN antagonist competes for the AI-1 binding site. These antagonists provided a molecular tool with which to further probe the AI-1 binding pocket and characterize the signaling properties of V. harveyi LuxN. Quantitative analysis of the sensing and binding properties of our LuxN mutants suggests a two-state, kinase vs. phosphatase model for receptor function. Indeed, when signaling output (bioluminescence) was plotted as a function of the free-energy difference between kinase and phosphatase states our data collapsed to a single curve, allowing us to extract signaling parameters for both wild-type and mutant LuxN proteins.
Only through this quantitative analysis was it revealed that, unlike the paradigmatic two-state chemotaxis receptors which spend roughly equal time in the active and inactive states for maximum sensitivity to ligand the quorum-sensing receptor LuxN spends ~96% of its time in the active/kinase state and requires establishment of a threshold concentration of autoinducer to inactivate it (Sourjik, 2004; Sourjik and Berg, 2004). Remarkably, although the chemotaxis and LuxN receptors are homologous, they solve fundamentally different biological problems by operating in different regimes. Chemotaxis, a system tuned for sensitivity, allows instantaneous alterations in behavior in response to small fluctuations in signal concentration. Quorum sensing, by contrast, a system built to ignore small perturbations, initiates a slow, all-or-nothing commitment program only upon reaching a signal threshold.
Results
Identification of LuxN mutants with defective responses to AI-1
The aim of this study was to determine how LuxN and AI-1 interact in order to understand how trans-membrane receptors couple AHL signaling to changes in gene expression. However, as is the case for most histidine sensor kinases, the complex trans-membrane topology of LuxN makes direct structural analysis extremely difficult. Therefore, to pinpoint the AI-1 binding site in the periplasmic domain of LuxN, directed mutagenesis of the 1 kb region of luxN encoding the membrane-binding domain was performed using error-prone PCR. The library of luxN mutants generated by this approach was cloned into a version of the luxN gene lacking this region to regenerate full-length luxN. The mutant library was introduced into the double sensor mutant JMH625 (luxN luxQ), which has a bright phenotype because there is no flow of phosphate to LuxO (Figure 1A). We note that the CAI-1-CqsS system is intact in the strain used for this screen. Because saturating levels of CAI-1 are always present in our experiments, CqsS exists as a phosphatase and thus does not contribute in funneling phosphate to LuxO. We reasoned that when a wild-type copy of luxN is introduced into this strain in the presence of AI-1, it would remain bright because binding of AI-1 to LuxN induces phosphatase activity. However, if a mutant luxN allele encoding a LuxN protein that is incapable of binding or responding to AI-1 is introduced, it will confer a dark phenotype due to high levels of LuxN auto-phosphorylation and phospho-transfer to LuxO (Figure 1A).
Approximately 30,000 luxN mutants were screened for those alleles causing a reduction in bioluminescence. Ten alleles were confirmed to produce dark phenotypes. These luxN genes were sequenced to identify the mutations (Table S1, Figure 1B). Several candidates contained multiple mutations, and these mutations were uncoupled by site-directed mutagenesis to produce genes encoding LuxN proteins with single amino-acid substitutions (Table S1). Interestingly, in the case of LRS6 two of the uncoupled mutations independently caused dark phenotypes (Table S1). Further analyses were carried out on LuxN mutants containing only single amino-acid changes.
The mutations conferring dark phenotypes cluster to the periplasmic region of TMs 4, 5, 6, and 7 and periplasmic loops 2 and 3 (Figure 1B in red) suggesting that the AI-1 binding site resides there. To explore this hypothesis further, we compared the 11 available LuxN homologs and replaced every 100% conserved amino acid as well as the other most highly conserved amino acids within this region with alanine, and screened them as above. This analysis produced an additional 20 mutants defective in response to AI-1. (Table 1, Figure 1B).
Table 1.
LuxN Mutant Phenotypes
| Allele | Lux Phenotpye | AI-1 EC50 (M) | Fold change in EC50a | Location |
|---|---|---|---|---|
| Wild type | WT | 2.3×10−8 | ||
| H46Y | WT | NM | TM2 | |
| S54P | WT | NM | TM2 | |
| A77D | WT | NM | PL1 | |
| H155Q | WT | NM | CL1 | |
| N133Ab | Dark | 8.2×10−8 | 3.6 | TM4 |
| L138A | WT | 3.0×10−8 | 1.3 | TM4 |
| T139A | WT | 1.4×10−8 | 0.6 | TM4 |
| T139I | Dark | 7.4×10−8 | 3.2 | TM4 |
| V140A | WT | NM | PL2 | |
| V143A | Dark | 9.9×10−8 | 4.3 | PL2 |
| I145A | WT | NM | PL2 | |
| P148A | WT | NM | PL2 | |
| S149A | WT | 6.1×10−8 | 2.7 | PL2 |
| F151A | Dark | 6.9×10−5 | 3000 | PL2 |
| I153A | Dark | 1.2×10−6 | 52.2 | PL2 |
| I153F | Dark | 1.3×10−7 | 5.7 | PL2 |
| I153L | WT | 6.6×10−8 | 2.9 | PL2 |
| E154Q | WT | 5.5×10−8 | 2.4 | PL2 |
| E154A | WT | NM | PL2 | |
| F155A | Dark | 5.8×10−7 | 25.2 | PL2 |
| F155I | Dark | 8.1×10−4 | 35217 | PL2 |
| F155L | Dark | 4.1×10−6 | 178.3 | PL2 |
| G156A | WT | NM | PL2 | |
| P157A | WT | NM | PL2 | |
| F162A | Dark | 9.3×10−5 | 4043 | TM5 |
| F163A | Dark | 8.7×10−6 | 378.3 | TM5 |
| L166A | Dark | NA | TM5 | |
| L166R | Dark | 2.3×10−7 | 10.0 | TM5 |
| V170A | WT | NM | TM5 | |
| T173A | WT | NM | TM5 | |
| N176A | WT | NM | TM5 | |
| S184N | Sensitive | 1.1×10−8 | 0.5 | CL2 |
| K186A | WT | NM | CL2 | |
| L187A | WT | NM | CL2 | |
| A190T | WT | NM | CL2 | |
| K191A | WT | NM | CL2 | |
| Y194A | WT | NM | TM6 | |
| G198A | WT | NM | TM6 | |
| I199A | WT | NM | TM6 | |
| F202A | Dark | NA | TM6 | |
| F202Y | Dark | 7.1×10−7 | 30.9 | TM6 |
| S205A | WT | 3.2×10−8 | 1.4 | TM6 |
| S205P | Dark | NA | TM6 | |
| T206A | Dark | 3.1×10−7 | 13.5 | TM6 |
| I209F | WT | 3.9×10−8 | 1.7 | TM6 |
| G212A | WT | 3.6×10−8 | 1.6 | TM6 |
| T214A | Dark | 4.5×10−7 | 19.6 | TM6 |
| T214I | Dark | 7.0×10−5 | 1043 | TM6 |
| D219A | Dark | 1.3×10−7 | 5.7 | PL3 |
| F220A | Dark | 1.9×10−4 | 8261 | PL3 |
| F220I | Dark | 7.1×10−4 | 30870 | PL3 |
| S221A | Dark | 1.9×10−7 | 8.3 | PL3 |
| W224A | Dark | 1.9×10−7 | 8.3 | TM7 |
| L225A | WT | 5.5×10−8 | 2.4 | TM7 |
| P226A | Dark | 2.3×10−4 | 10000 | TM7 |
| P226T | Dark | NA | TM7 | |
| P227A | Dark | 4.0×10−6 | 173.9 | TM7 |
| P227L | Dark | 3.9×10−3 | 169565 | TM7 |
| L229A | WT | NM | TM7 | |
| S230A | WT | NM | TM7 | |
| S232A | Dark | 4.0×10−7 | 17.4 | TM7 |
| S232N | WT | 4.1×10−8 | 1.8 | TM7 |
| E233A | Dark | NA | TM7 | |
| M234I | WT | NM | TM7 | |
| M234A | WT | NM | TM7 | |
| G238A | WT | NM | TM7 | |
| Y239A | WT | NM | TM7 | |
| R245L | Sensitive | 4.8×10−9 | 0.21 | CL4 |
| V249I | WT | NM | CL4 | |
| G271D | Sensitive | 3.7×10−9 | 0.16 | TM8 |
| F163A/R245L | Dark | 3.7×10−6 | 160.9 | |
| F163A/R245L/S184N | Dark | 1.4×10−7 | 6 | |
| F163A/R245L/S184N/G271D | Sensitive | NA |
Fold change in EC50 value with respect to wild-type EC50 value.
Bold indicated 100% conserved amino acids. (See Figure S2)
TM (Trans-Membrane Domain)
CL (Cytoplasmic Loop)
PL (Periplasmic Loop)
NM (Not Measured)
NA (Not Applicable)
The LRS5 mutation, which confers a dark phenotype, is a single base-pair deletion at position 634 causing a premature stop codon at amino-acid residue 213 (Table S1). This mutation is perplexing because the kinase domain of LuxN should not be synthesized, making it unclear how this mutation could confer a dark phenotype. To investigate this, a FLAG-epitope tag was fused to the C-terminus of this mutant LuxN and the protein was probed by western blot analysis. A truncated version of LuxN lacking approximately the first 220 amino acids is synthesized (data not shown). We therefore suspect that an alternative ribosome binding site exists downstream of the LRS5 deletion, enabling translation of a truncated form of LuxN. Because this truncation eliminates almost the entire proposed AI-1 binding domain from LuxN, only the cytoplasmic kinase domain is produced which, because it is unable to bind to AI-1, constitutively acts as a kinase causing a dark phenotype.
LuxN mutant phenotypes
To characterize the signaling capabilities of the single-amino-acid-substituted LuxN mutants, we carried out a series of quantitative phenotypic analyses. First, we measured bioluminescence in stationary-phase cultures of strains carrying either wild-type luxN or each luxN allele conferring a dark phenotype. The bioluminescence produced by the strain with wild-type luxN was set at 100% (Figure S1A). As negative controls, two luxN mutants harboring wild-type phenotypes (LuxN L138A and LuxN E154Q) which were randomly isolated from the screen were also included in the analysis and they produced the wild-type level of bioluminescence (Figure S1A). By contrast, the LuxN mutants F151A, I153F, F155I, L166R, T214I, F220A, P226T, and S232N exhibited at least an 80% reduction in bioluminescence relative to wild type (Figure S1A). To confirm that the dark phenotypes did not stem from increased LuxN protein levels, FLAG-epitope tags were incorporated at the C-terminus of a representative subset of the LuxN mutants shown in Figure S1A as well as wild-type LuxN. Western blot showed that there were no differences in protein production (data not shown).
We reasoned that the LuxN mutants conferring dark phenotypes must be acting as kinases at high cell density, resulting in continued flow of phosphate through the quorum-sensing circuit. This in turn, should manifest itself in elevated qrr expression at high cell density (Figure 1A). To test this idea, we performed quantitative real-time PCR and measured Qrr4 transcript levels in each of the luxN mutant strains described above. As controls, we measured Qrr4 transcript levels in the wild type and the bright control strains, LuxN L138A and LuxN E154Q, and found that indeed, in these three strains, Qrr4 levels are low, consistent with these LuxN proteins acting as phosphatases at high cell density (Figure S1B). However, the luxN mutants exhibiting dark phenotypes (Figure S1A) all have significantly increased Qrr4 transcript levels (10 to 30-fold higher than wild type) (Figure S1B). This result confirms that the decrease in bioluminescence we observe in the dark LuxN mutants is the direct result of an alteration in signaling through the LuxN quorum-sensing pathway.
AI-1 dose-response curves
We considered two possible mechanisms underlying the dark LuxN phenotypes. First, a particular mutation could abolish AI-1 binding. If so, this type of mutation would cause LuxN to act as a kinase at high cell density in the presence of AI-1. Alternatively, a mutation could allow AI-1 binding, but disrupt the ability of LuxN to transduce the signal to the cytoplasm. We first determined which LuxN mutant proteins could bind AI-1 by measuring the AI-1 dose-response of each LuxN missense mutant. For this, V. harveyi strain HLS253 ΔluxMN, ΔluxPQ, ΔluxS was used. V. harveyi HLS253 is constitutively bright because the luxN and luxPQ genes, encoding the quorum-sensing receptors, have been deleted. Also, V. harveyi HLS253 does not produce AI-1 or AI-2, due to the luxM and luxS deletions, respectively. Introduction of a wild-type copy of luxN into V. harveyi HLS253 confers a dark phenotype because, in the absence of AI-1, LuxN acts as a constitutive kinase. However, addition of exogenous AI-1 to HLS253 harboring a wild-type copy of luxN induces bioluminescence. Introduction of a luxN mutant encoding a LuxN protein incapable of binding AI-1 or incapable of signaling the AI-1 binding event to the cytoplasm will confer a dark phenotype to HLS253. And, such defects will cause the LuxN proteins to remain as kinases even in the presence of AI-1. By contrast, if a particular LuxN mutant is introduced that is capable of binding AI-1, even with lower affinity than wild-type LuxN, these LuxN proteins will switch to phosphatase activity following the addition of sufficient AI-1, and bioluminescence will be induced.
To determine AI-1 EC50 values, wild-type LuxN and each LuxN mutant were assayed for response to AI-1 at concentrations ranging from 24 pM to 500 µM. A subset of the dose-response curves is shown in Figure 2A, and the remainder of the EC50 data is provided in Table 1. The EC50 for wild-type LuxN binding to AI-1 is 23 nM. The control mutants, LuxN L138A and LuxN E154Q, as expected, have EC50 values of 30 nM and 55 nM, respectively, similar to wild-type LuxN (Table 1). Many of the LuxN mutants have drastically increased EC50 values (Table 1). For example, LuxN I153F, F155A, F162A, T206A, and S232A have EC50 values of 130 nM, 580 nM, 93 µM, 310 nM, and 400 nM, respectively (Figure 2A). In five cases, LuxN L166R, F202A, S205P, P226T, and E233A, the mutants conferred a dark phenotype to V. harveyi even at 500 µM AI-1 (Figure 2B) and therefore we were unable to assign them EC50 values. Nonetheless, we successfully determined the AI-1 EC50 values for 25 of the 30 LuxN mutants that conferred a dark phenotype. We conclude that LuxN mutant proteins that produce measurable EC50 values, albeit higher than wild type, can bind AI-1 at least with some capacity.
Figure 2. LuxN AI-1 Dose-Response Curves.
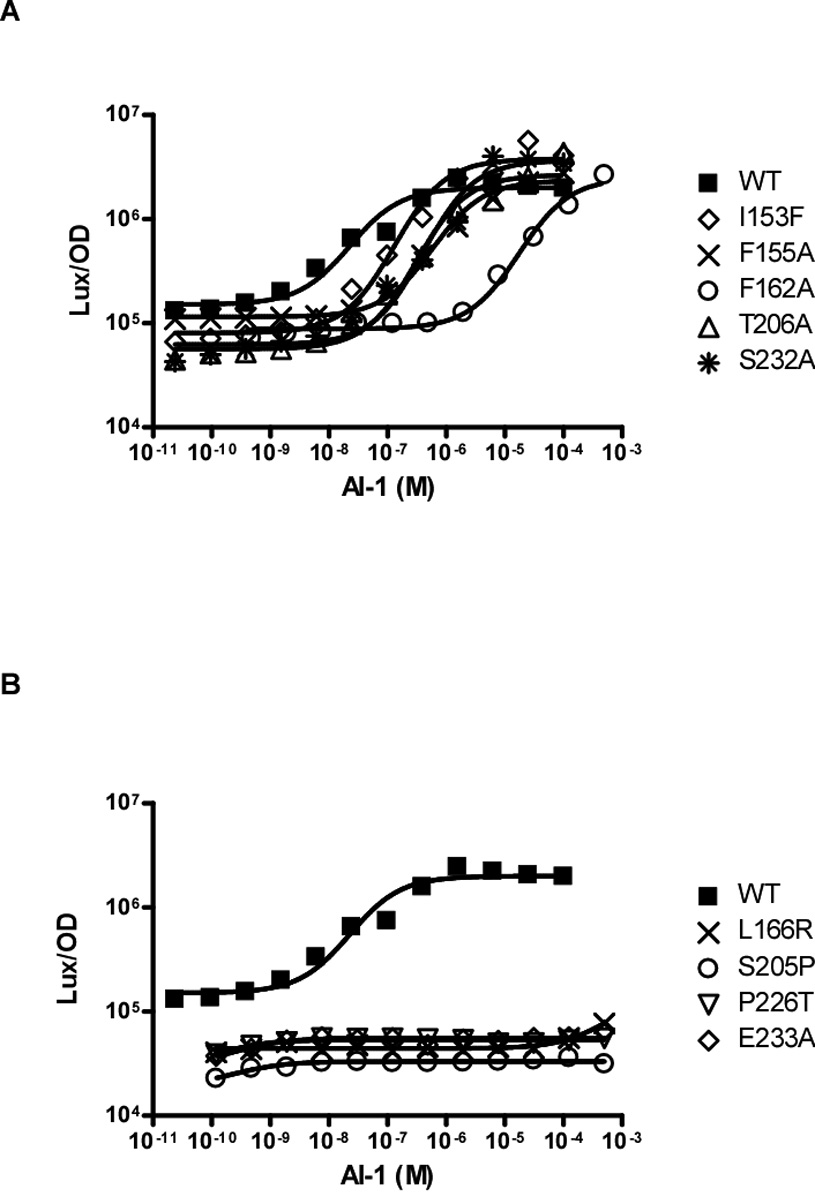
(A) Light production at various AI-1 concentrations is shown for wild-type LuxN and for representative LuxN mutants that have increased AI-1 EC-50 values. The data were fit with a variable-slope sigmoidal dose-response curve to determine the EC50 values. (B) Light production at various AI-1 concentrations is shown for wild-type LuxN and for representative LuxN mutations that cause constitutive dark phenotypes at all AI-1 concentrations. EC50 values were not determined for these mutants.
Identification of LuxN antagonists
To probe the LuxN/AI-1 interaction further, we identified small molecules that interfere with V. harveyi quorum sensing by disrupting the binding of AI-1 to LuxN. To do this, a high-throughput chemical screen (Broad Institute) was carried out, in which we identified small molecules that specifically antagonize LuxN signaling in V. harveyi. The V. harveyi strain, JMH624 ΔluxPQ, ΔluxM, which lacks the AI-2 receptor, LuxPQ, as well as the AI-1 synthase, LuxM, was used for the antagonist screen. V. harveyi JMH624 is dark because there is no AI-2 receptor and the lack of AI-1 causes LuxN to act as a kinase (Figure 1A). However, following exogenous addition of 20 nM AI-1, bioluminescence is induced because LuxN switches to phosphatase mode. Potential antagonist molecules were tested for the ability to reduce bioluminescence of V. harveyi JMH624 in the presence of 20 nM AI-1. To eliminate molecules causing general toxicity and those that interfere with luciferase or other downstream components of the quorum-sensing bioluminescence pathway, a second screen was carried out using a V. harveyi ΔluxN, ΔluxS control strain, JHM610. V. harveyi JMH610 lacks the AI-1 receptor LuxN and the AI-2 synthase, LuxS. In this case, because of the lack of AI-2, LuxQ acts as a kinase, and V. harveyi JMH610 is dark. However, following exogenous addition of AI-2, bioluminescence is induced because LuxQ switches to phosphatase mode (Figure 1A). Any molecule that reduced bioluminescence in both JMH610 in the presence AI-2 and JMH624 in the presence of AI-1 was eliminated from further analysis. Approximately 35,000 low-molecular-weight compounds were screened for specific inhibition of bioluminescence through the LuxN quorum-sensing pathway; 45 molecules were selected for further analysis, and a representative subset of these molecules with varying levels of antagonistic activity is shown in Figure 3A. For example, molecule C450-0730 has an IC50 value of 2.7 µM while a weaker antagonist, 3578–0898 has an IC50 of 62.3 µM. Interestingly, the molecular cores of two of the strongest LuxN antagonists, C450-0730 and C646-0078, are very similar (Figure 3A).
Figure 3. Molecules that Antagonize LuxN-AI-1 Binding or Signaling.

(A) Structures and designations of five molecules that inhibit LuxN signaling in response to AI-1. The IC50 value for each antagonist molecule is given below its structure. (B) Light production from wild-type LuxN and LuxN F163A was measured at the specified AI-1 concentrations in the presence of 0 µM, 1 µM, and 10 µM C450-0730. Data were fit as described above. (C) The light production values in panel B were collapsed as a function of f-ΔεWT as described in Experimental Procedures. f is the ligand-dependent free-energy difference between the kinase active (on) and kinase inactive (off) states of LuxN, and ΔεWT is the wild type value of f in the absence of ligand. The binding parameters used are as follows: The collapse was obtained by using Δε−ΔεWT = 3.2 for the LuxN F163A mutant.
We wondered whether the potent LuxN antagonist, C450-0730, was competing for the LuxN AI-1 binding site. To examine this, AI-1 EC50 values were determined in the presence of 0 µM, 1 µM, and 10 µM C450-0730. Our rational is that, if C450-0730 competes with AI-1 for binding, the AI-1 EC50 value should increase with increasing concentrations of C450-0730. Indeed, this is the case, as the AI-1 EC50 values are 23 nM, 76 nM, and 376 nM at 0 µM, 1 µM, and 10 µM C450-0730, respectively (Figure 3B). Indeed, the AI-1 dose-response curves at these three C450-0730 concentrations can be collapsed onto a single curve, consistent with competitive inhibition (Figure 3C and Experimental Procedures). The principal underlying the data collapse is that there is a fixed (albeit initially unknown) quantitative relation between measured bioluminescence and the free-energy difference between the active and inactive configurations of LuxN (Keymer et al., 2006). Therefore, all our dose-response curves should reproduce this same relation, i.e. the curves should “collapse” when bioluminescence is plotted versus free-energy difference. However, to plot the data this way we need to know how to relate ligand concentrations to free-energy differences, which means that we need to know the ligand dissociation constants KD for both the active and inactive configurations of LuxN. In practice, we iteratively improve our estimates for KD values by attempting to collapse the dose-response curves and infer the true values from the best data collapse. This is a reliable procedure in our case since the dose-response curves contain more data than the number of unknown KD values. A major benefit of collapsing the data in this way is that it allows us to deduce the state-dependent KD values for LuxN from the in vivo data: in the phosphatase (off) state , and in the kinase (on) state .
We had reasoned that the dark phenotypes of our LuxN mutants could stem from (i) a defect in the ability to bind AI-1, (ii) a bias favoring the kinase state, (iii) a defect in signaling, or (iv) some combination of the above. The method of data collapse provides us with a powerful tool to distinguish among these possibilities. For example, we consider the case of the mutant LuxN F163A (Figure 3B) which has an AI-1 EC50 value 378-fold higher than that of wild-type LuxN and for which we obtained dose-response curves in the presence of 0 µM, 1 µM, and 10 µM of the antagonist C450-0730. First, we were able to collapse the three antagonist dose-response curves using the identical as we used to collapse the wild-type LuxN data, indicating that LuxN F163A is not defective in its ability to bind AI-1 (eliminating possibility (i)). Second, the LuxN F163A data could all be collapsed onto the wild-type LuxN antagonist curves simply by adjusting the free-energy bias between the kinase (on) and phosphatase (off) states (Figure 3C). This analysis allows us to conclude that LuxN F163A has an increased AI-1 EC50 value exclusively because it has an altered free-energy bias that favors the kinase (on) state, establishing that possibility (ii) accounts for the dark phenotype of this mutant. Similar analysis applied to our other dark mutants reveals examples of the different possibilities and allows us to deduce and quantify the origins of the dark phenotypes (see Discussion).
Antagonist Suppressor Analysis
To better understand the mechanism of C450-0730 interaction with LuxN, we performed a suppressor screen to identify LuxN mutants no longer antagonized by C450-0730. Using error-prone PCR, 2,000 mutants in the luxN N-terminal region were generated and conjugated into the V. harveyi ΔluxMN ΔluxPQ ΔluxS strain, HLS253, and arrayed in 96-well micro-titer plates. As mentioned, V. harveyi HLS253 is constitutively bright due to the absence of the quorum-sensing receptors, LuxN and LuxPQ, and both autoinducer synthases, LuxM and LuxS. To verify our strategy, a wild-type luxN control plasmid was also conjugated into V. harveyi HLS253, which conferred a dark phenotype because wild-type LuxN is a kinase in the absence of AI-1. Bioluminescence is restored to HLS253 containing wild-type luxN by the exogenous addition of 100 nM AI-1. We found that 800 nM C450-0730 was required to inhibit bioluminescence of HLS253 carrying wild-type luxN in the presence of 100 nM AI-1. The luxN mutant library was screened in the presence of 100 nM AI-1 and 800 nM C450-0730 for luxN alleles that enabled bioluminescence in V. harveyi HLS253. To eliminate luxN null mutants, the luxN mutant library was also screened in V. harveyi HLS253 in the absence of both AI-1 and C450-0730. The luxN alleles that conferred a bright phenotype in the absence of AI-1 were not examined further. Five LuxN mutant strains, LRS112, LRS311, LRS129, LRS147, and LRS1511 (Table S1) displayed dark phenotypes in the absence of AI-1 and C450-0730, but were bright in the simultaneous presence of AI-1 and C450-0730, suggesting that these LuxN proteins were no longer antagonized by C450-0730. The luxN mutations were sequenced to identify the alleles (Table S1). Interestingly, LuxN G271D was identified twice. From here forward this class of suppressor mutants is referred to as LuxN*.
Characterization of the LuxN* mutants
We speculated that the LuxN* mutants could have increased AI-1 sensitivity or decreased C450-0730 binding ability. To distinguish between these two possibilities, the LuxN* AI-1 EC50 values were determined (Figure 4A). As a reference, the dark mutant LuxN F163A is also included in Figure 4A. The EC50 value of wild-type LuxN is 23 nM, while LuxN* S184N is 11 nM, LuxN* I209F is 39 nM, LuxN* R245L is 4.8 nM, and LuxN* G271D is 3.7 nM (Table 1). Interestingly, three of the four LuxN* mutants, LuxN S184N, R245L, and G271D show increased sensitivity to AI-1, suggesting that these alleles circumvent C450-0730 antagonism through increased AI-1 binding or signaling or via a bias to the phosphatase state of LuxN (see Discussion). However, LuxN* I209F responded more like wild type to AI-1 as indicated by an AI-1 EC50 value of 39 nM (Table 1).
Figure 4. AI-1 Dose-response Curves of the LuxN* Suppressor Mutants.
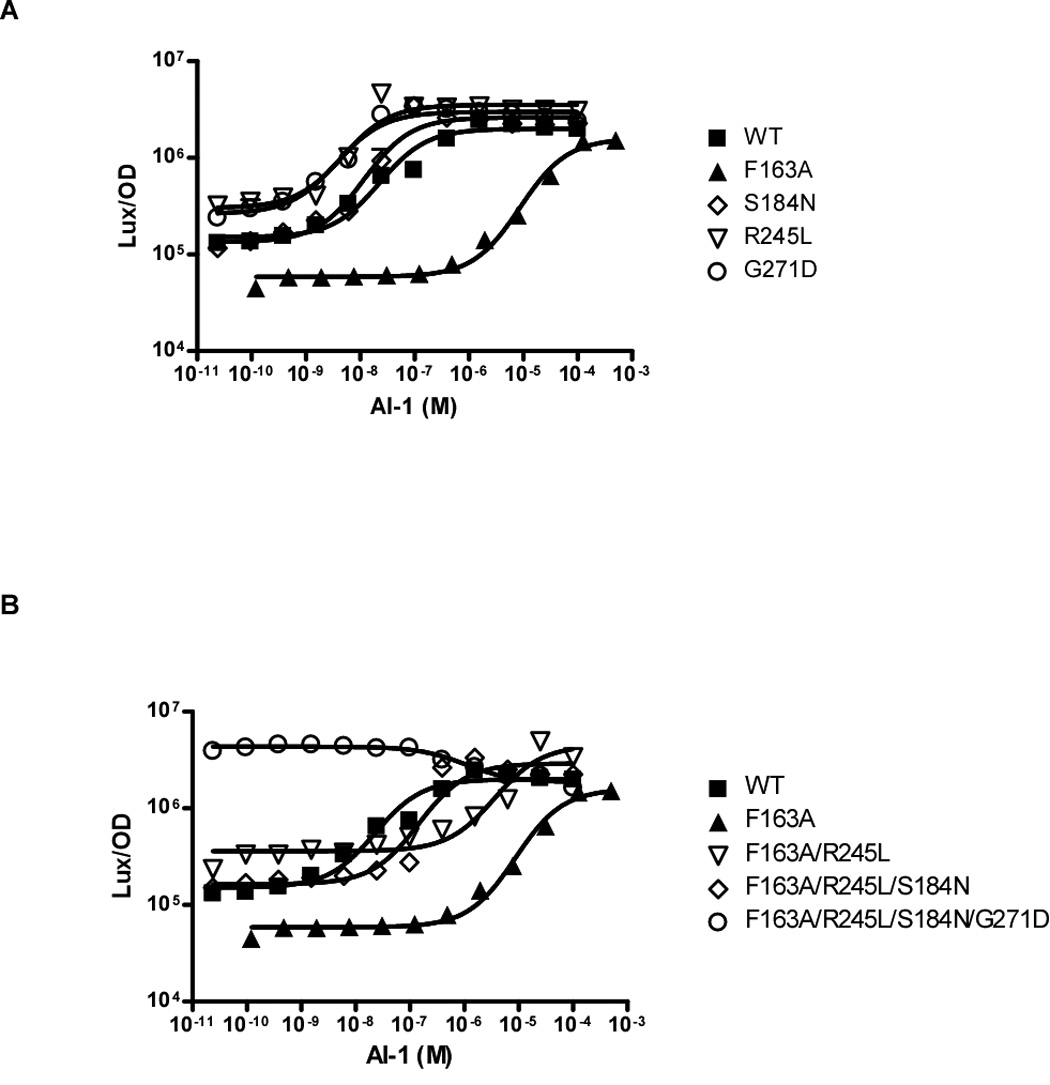
(A) Light production of the wild-type LuxN, the LuxN* mutants, and LuxN F163A at various AI-1 concentrations. The data were fit with a variable-slope sigmoidal dose-response curve to determine the EC50 value for each LuxN* mutant. (B) Light production of the dark LuxN F163A mutant harboring combinations of LuxN* mutations. Data were fit and AI-1 EC50 value was determined as above. An EC50 value could not be determined for the quadruple mutant because it is constitutively bright at all AI-1 concentrations.
In the reciprocal experiment, we determined the ability of C450-0730 to antagonize the LuxN* mutants. C450-0730 IC50 values were measured by titrating C450-0730 from 0.64 nM to 50 µM, while keeping the AI-1 concentration constant at 10 nM. The C450-0730 concentration required to inhibit LuxN* G271D, R245L, and S184N was similar to that required to inhibit wild-type LuxN, indicating that the observed “resistance” to C450-0730 was indeed due to increased sensitivity to AI-1 (data not shown). However, a 5-fold higher concentration of C450-0730 was required to antagonize LuxN* I209F. Therefore, the LuxN* I209F mutation appears to affect C450-0730 binding. Because I209 is located within our proposed AI-1 binding site (Figure 1B in green), and because it also affects C450-0730 antagonistic activity, we propose that C450-0730 could compete for the AI-1 binding site of LuxN. This hypothesis is strongly supported by the good data collapse in Figure 3C, which is based on competitive inhibition by C450-0730.
Sensitive LuxN* mutations are epistatic to the LuxN dark mutations
For chemotaxis receptors in E. coli, adaptive methylation of specific cytoplasmic residues is known to additively bias receptors toward a kinase-active state (Endres et al., 2007). By analogy, we wondered whether some of our single-residue mutations might bias LuxN toward kinase or phosphatase states in an additive manner. To determine whether the LuxN G271D, R245L, and S184N mutants which have lower than wild type AI-1 EC50 values are biased toward the phosphatase state, these mutations were engineered into the LuxN F163A mutant to test if they could shift the high EC50 of LuxN F163A back toward a low EC50. As a reminder, the F163A LuxN mutation has an increased AI-1 EC50 value of 8.7 µM as compared to 23 nM for wild-type LuxN; therefore, it requires approximately 378 times more AI-1 to switch LuxN F163A into the phosphatase mode than the amount of AI-1 required to switch wild-type LuxN. A double mutant (LuxN F163A/R245L), a triple mutant (LuxN F163A/R245L/S184N), and a quadruple mutant (LuxN F163A/R245L/S184N/G271D) of LuxN were tested for their ability to respond to AI-1 (Figure 4B). The incorporation of each LuxN* mutation into the context of the F163A mutation successively decreased the AI-1 EC50 value approximately 10-fold, while the quadruple mutant had a constitutively bright phenotype (Table 1). This analysis suggests that the LuxN* mutations are additive in their ability to bias LuxN toward the phosphatase mode.
Discussion
LuxN is the founding member of a growing family of recognized membrane-bound receptors that detect and respond to acylated homoserine lactone (AHL) quorum-sensing autoinducers. AHLs are used by a broad spectrum of Gram-negative bacteria to control quorum-sensing behaviors, including virulence (Fuqua et al., 1996; Waters and Bassler, 2005). Typically, cytoplasmic LuxR-type proteins detect and respond to the accumulation of an AHL signal (Fuqua et al., 1996; Fuqua et al., 1994) (Engebrecht and Silverman, 1984, 1987). Little is known about how membrane-bound kinase proteins, like LuxN, detect AHLs. Our mutagenesis strategy identified the AI-1 binding domain of LuxN from V. harveyi as TM helices 4, 5, 6, and 7 as well as the intervening periplasmic loops 2 and 3 (Figure 1B). All of the identified amino-acid mutations that affect AI-1 signaling cluster in TM helices near the periplasmic face, or are located within periplasmic loops, indicating that LuxN most likely binds AI-1 on the periplasmic side of the membrane (Figure 1B). This observation indicates that AI-1 is released from V. harveyi, accumulates in the extracellular space, and subsequently triggers the LuxN quorum-sensing cascade. This mechanism is distinct from the previously characterized LuxR-type AHL-signaling mechanism. Typically, LuxR-type AHL receptors require significant intracellular AHL concentrations for folding (Zhu and Winans, 2001). Thus, at low cell densities the LuxR proteins do not fold properly and are degraded, so quorum sensing does not occur (Zhu and Winans, 1999). Degradation of the LuxR-type proteins in the absence of the AHL signal is presumed to be a mechanism preventing premature activation of quorum sensing in canonical LuxR-AHL systems (Zhu and Winans, 2001). Apparently, V. harveyi has evolved a distinct mechanism to circumvent short circuiting its quorum-sensing pathway, namely by compartmentalizing the cytosolic production of AI-1 in a location inaccessible to the periplasmic sensing domain of LuxN. This spatial uncoupling of AI-1 production from AI-1 binding allows V. harveyi to exclusively monitor extracellular levels of AI-1. It must be noted that V. harveyi has three quorum-sensing circuits, all of which have similar architectures (Henke and Bassler, 2004b). Thus, all three systems have signal production spatially uncoupled from signal detection (Figure 1A).
The large number of mutations identified in this work that affect AI-1 binding suggest that LuxN makes multiple contacts with AI-1. Further supporting our hypothesis that TM4, TM5, TM6, and TM7 and periplasmic loops 2 and 3 encode the AI-1 binding domain of wild-type LuxN, a LuxN homolog was recently discovered that lacks the first 80 amino acids, which encode TM1, TM2, and periplasmic loop 1, indicating that this region of LuxN is dispensable for AI-1 binding and signaling (Figure S2 and NCBI database). This truncated LuxN homolog retains all of the critical regions identified in our proposed AI-1 binding domain, indicating that this LuxN variant can still respond to an autoinducer molecule (Figure S2). Interestingly, the most highly conserved domain in LuxN is centered at position P226, and contains a PPAL motif that is 100% conserved among all known LuxN homologs (Figure S2). Both proline residues of this motif were identified as critical for LuxN signaling by our random mutagenesis screen. Therefore, we hypothesize that the PPAL motif is essential for LuxN signal transduction.
In a high-throughput chemical screen, fifteen small-molecule antagonists were identified that specifically antagonize the LuxN/AI-1 quorum-sensing circuit in V. harveyi. These are the first antagonist molecules that target an AHL membrane-bound sensor kinase. Interestingly, the antagonists identified by this screen are not similar in structure to AI-1 (Figure 3A). Therefore, it is unlikely that rational-design experiments would have predicted these molecules as AHL antagonists. We wondered whether our antagonists competed with AI-1 for binding to LuxN. To explore this, we performed an antagonist-suppressor screen, and we identified LuxN* I209F, which is not antagonized by C450-0730. Importantly, this mutation lies on the periplasmic side of TM 6, in the center of our proposed AI-1 binding domain, consistent with the possibility that C450-0730 competes for the AI-1 binding site (though the LuxN* mutation I209F does not affect AI-1 signaling). We also obtained AI-1 dose-response curves in the presence of different concentrations of C450-0730 for both wild-type LuxN and LuxN F163A, and obtained a good data collapse indicative of competitive inhibition (Figure 3B and 3C). Combined, these results strongly suggest that the C450-0730 antagonist is competing for the AI-1 binding pocket of LuxN. Because the LuxN* I209F mutation only affects the antagonistic ability of C450-0730, but does not interfere with AI-1 signaling, we hypothesize that C450-0730 makes at least some contacts with LuxN that are distinct from those made by AI-1.
Our discovery of LuxN mutations with both increased and decreased sensitivity to AI-1 suggests an analogy to chemotaxis receptors in E. coli. Chemotaxis receptors are known to adjust their sensitivity to ligand via reversible methylation of specific cytoplasmic residues (Goy et al., 1977; Kehry and Dahlquist, 1982a, b; Springer and Koshland, 1977; Stock and Koshland, 1978; Terwilliger and Koshland, 1984). We wondered whether mutations in LuxN could play the same role as methylation/demethylation in chemotaxis, namely could they shift the free-energy bias between receptor kinase and phosphatase states? The resulting model is shown schematically in Figure 5A and 5B. We propose that each LuxN can exist in any of four states: kinase (on) or phosphatase (off), with ligand bound or unbound. Receptor activity is determined by the thermal equilibrium among these states, characterized by the free-energy difference f between the on and off states of LuxN (see Experimental Procedures). Within the model, the measured output, bioluminescence, is the same unknown function of f for all strains, reflecting the fact that bioluminescence depends only on receptor activity, which at equilibrium depends only on f. The model predicts that mutations can cause EC50 to increase or decrease depending on the sign of the shift in Δε,the free-energy bias between kinase and phosphatase states. Indeed, the model is nicely supported by the data collapse in Figure 3C, where the bioluminescence for the LuxN F163A mutant collapses well with wild type assuming only a shift in Δε More generally, we have found that the bioluminescence data for many of our LuxN mutants collapse well with the combined wild-type and LuxN F163A data, allowing us to deduce changed Δε values and in some cases also changed binding affinities (Figure 6A and 6B). This analysis supports a close functional analogy between LuxN and E. coli chemotaxis receptors, and suggests the general relevance of two-state, free-energy models for bacterial sensor kinases.
Figure 5. LuxN Signal Transduction Can Be Described by a Two-State Model.
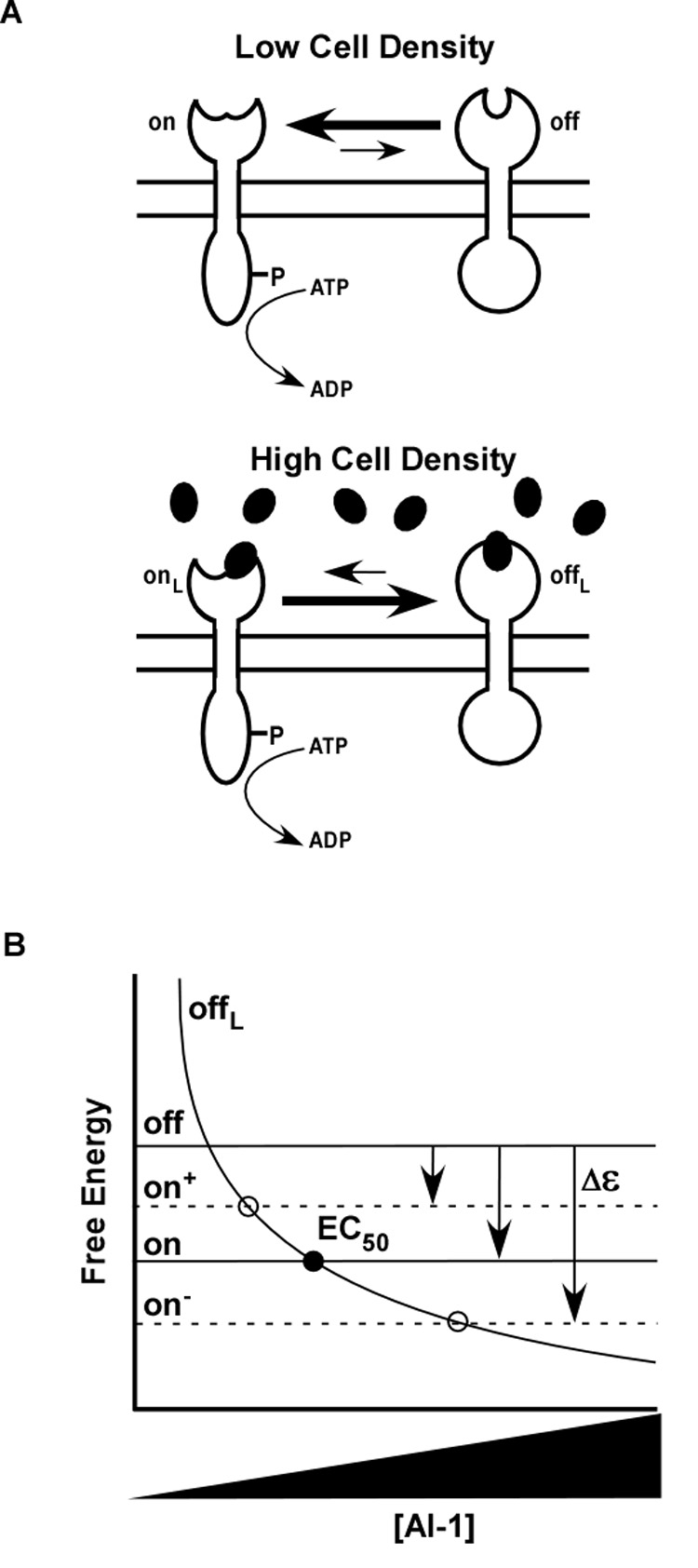
(A) Wild-type LuxN toggles between two conformations indicated by the open and closed periplasmic domains. At low cell density, when the AI-1 concentration is negligible, LuxN is strongly biased toward its kinase state represented by the open periplasmic structure. At high cell density, in the presence of AI-1 (dark ovals), LuxN is biased toward the phosphatase state represented by the closed periplasmic structure. (B) This two-state model is represented by a free-energy diagram that describes the two ligand-free forms of the protein as on (open periplasmic domain) or off (closed periplasmic domain). The free energies of these two states are independent of ligand concentration and are represented by horizontal black lines. The free energy of the on state is lower than the free energy of the off state, producing the bias toward the kinase mode at low cell densities (i.e. low autoinducer concentration). The free energy of LuxN in its phosphatase state and bound to ligand (offL) is represented by the descending solid curve. The point at which the free energy of the offL state equals the free energy of the on state (solid circle) corresponds to the EC50 value for AI-1. LuxN mutants identified in the genetic screen that possess increased AI-1 EC50 values are represented as on−. Compared to wild-type LuxN, they have lower on state free energies and therefore exhibit larger AI-1 EC50 values. By contrast, the three LuxN* mutants that exhibit a bias toward the phosphatase state are represented as on+. These mutants possess higher on state free energies than wild-type LuxN and therefore have decreased AI-1 EC50 values. The EC50 values of the on− and on+ mutants are represented by the open circles.
Figure 6. Data Collapse for LuxN*, LuxN Bias, and Combined LuxN*-Bias Mutants.
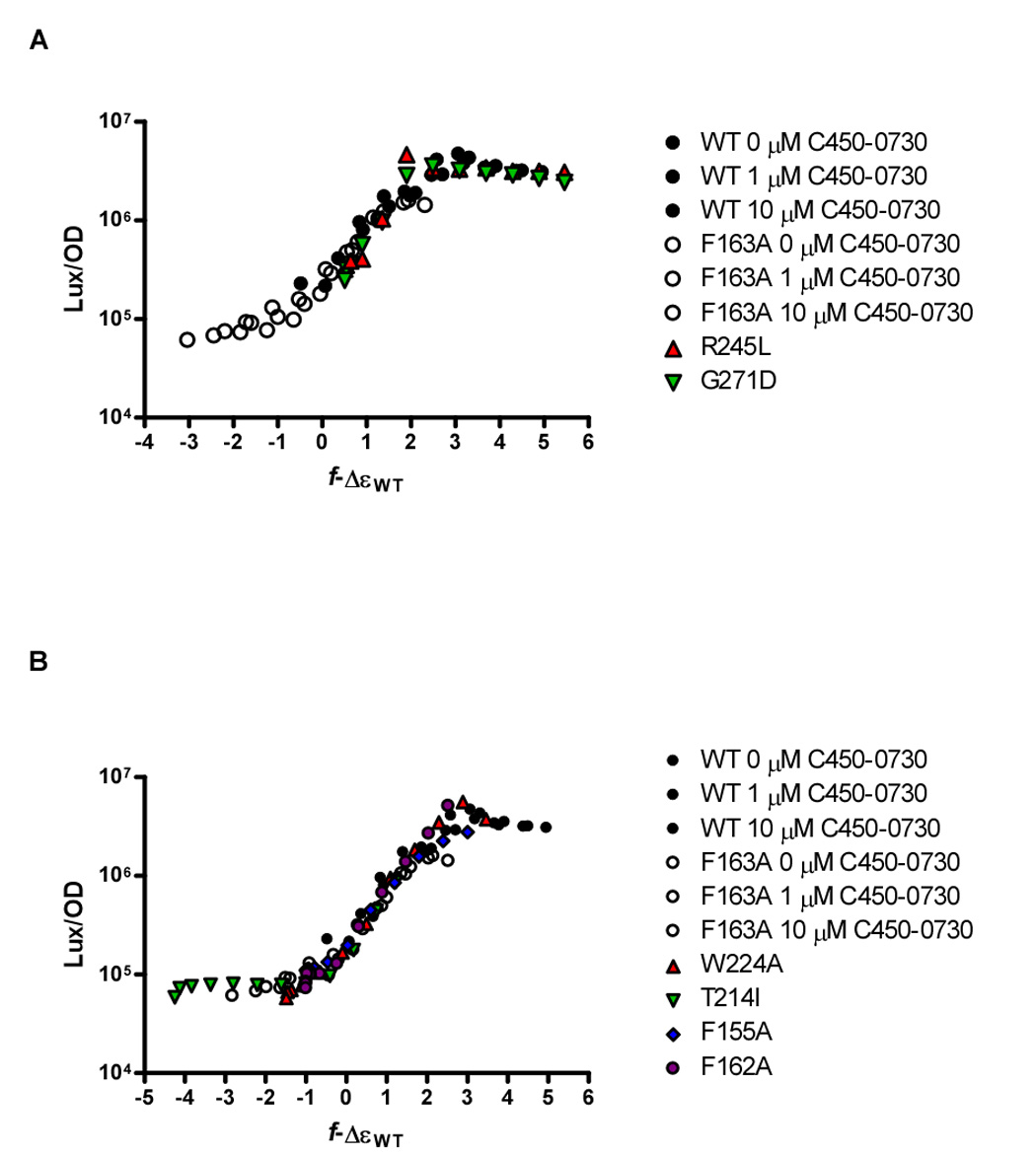
(A) Collapse of the dose-response data from LuxN* R245L and G271D mutants with the combined wild-type/LuxN F163A antagonist collapse from Figure 3C. These LuxN* curves were collapsed by adjusting only the bias Δε−ΔεWT to +0.5. (B) Collapse of dose-response curves from representative dark LuxN mutants with the combined wild-type/LuxN F163A antagonist collapse from Figure 3C. The LuxN W224A and LuxN T214I dose-response curves were collapsed by adjusting only the bias Δε−ΔεWT to −1.5 and −4.3, respectively. The LuxN F155A and LuxN F162A dose-response curves were collapsed by adjusting the bias Δε−ΔεWT parameter and increasing the for LuxN F155A, Δε−ΔεWT = −1.0 and , for LuxN F162A, Δε−ΔεWT = −1.0 and .
The genetic screen used here enabled us to identify three classes of LuxN mutants. The first class of LuxN mutants identified as LuxN* was unexpectedly revealed in the antagonist-suppressor screen. Among these suppressor mutants, LuxN G271D, S184N and R245L, have increased sensitivity to AI-1 as indicated by their reduced AI-1 EC50 values. These mutations are not located in the proposed AI-1 binding domain. Rather, two of these mutations, S184N and R245L are located within cytoplasmic loops 3 and 4, respectively, while G271D is located at the periplasmic interface of TM 8 (Figure 1B). We propose that these mutations bias LuxN toward the phosphatase state, such that lower AI-1 is needed to switch these mutant proteins into the kinase (off) state than is required by wild-type LuxN. Consistent with this idea, the AI-1 dose-response data for LuxN R245L and LuxN G271D collapsed onto the data for wild type LuxN and LuxN F163A from Figure 3C when only the Δε parameter was altered while keeping the binding parameters constant at the wild-type values (Figure 6A). Specifically, LuxN R245L and G271D required only an additional 0.5 be added to Δε to collapse the data, while we had to decrease Δε by 0.4 to collapse the data for the LuxN S184N mutant (not shown). Therefore, LuxN R245L and LuxN G271D behave as predicted if their mutations bias them toward the kinase (off) state. While we do not understand why LuxN S184N does not follow this pattern, we suspect that the LuxN S184N mutation must, in addition to affecting the free-energy bias, also affect the rate of phosphotransfer or the rate of phosphatase activity, two parameters not determined in either our experimental or theoretical analyses.
The second class of mutants includes LuxN T139I, V143A, I153F, F155I, F163A, T206A, T214I, F220A, W224A, P226A, and P227A bind and respond to AI-1, but are less sensitive to AI-1 than is wild-type LuxN. Thus, this second class of mutants requires increased AI-1 concentration to convert LuxN from a kinase to a phosphatase. We favor the idea that these mutations diminish LuxN's ability to respond to AI-1 by biasing LuxN toward kinase mode, and indeed, the dose-response curves of many of these mutants (e.g. LuxN T214I and LuxN W224A) collapse well onto the combined wild-type and LuxN F163A data collapse from Figure 3C using the wild-type binding-affinity parameters, , while changing only the free-energy-bias parameter Δε (Figure 6B). For these mutants, we note that doing the reverse, i.e., changing the binding-affinity parameters while maintaining the free-energy bias Δε at the wild-type level, does not allow us to collapse the data. The second class also contains mutants such as LuxN F155A and LuxN F162A that have greatly increased AI-1 EC50 values compared to wild-type LuxN yet, when we adjust only Δε, their dose-response curves do not collapse onto the combined wild-type LuxN and LuxN F163A data in Figure 3C. We could successfully collapse their data by simultaneously adjusting Δε and increasing to 10 nM and 100 nM, respectively. This finding suggests that these two LuxN mutations impair AI-1 binding in addition to shifting the LuxN free-energy bias Δε toward the kinase (on) state. Data from several other LuxN mutants (LuxN N133A, F151A, I153A, F155L, L166A, F202Y, T214A, D219A, F220I, S221A, P227L, S232A) could not be collapsed even by simultaneously adjusting the values, suggesting that these mutations have additional effects on receptor function.
The third class of mutants includes LuxN L166R, F202A, S205P, P226T, and E233A. These LuxN mutant proteins do not respond to AI-1 even at 500 µM. We interpret this to mean that this particular class of LuxN mutants either cannot bind AI-1 or is impaired in the ability to transduce the AI-1 signal to the kinase domain of LuxN. These mutations all lie within our proposed AI-1 binding domain, suggesting that this region of LuxN is required for both signal detection and signal transduction across the bacterial membrane. As additional evidence supporting the idea that this class of mutants is incapable of signaling, rather than having AI-1 EC50 values shifted beyond our test range, we combined one of these mutations (LuxN S205P) with one of our heightened-sensitivity LuxN* mutations (LuxN R245L). The double mutant remained dark at all AI-1 concentrations tested, suggesting that the LuxN S205P mutation irreversibly cripples signaling.
Our data-collapse method for deducing signaling parameters did not require prior knowledge of the input-output function relating receptor activity to bioluminescence. The approach can therefore be applied very generally, even to poorly characterized pathways, as long as the final output is a function of receptor activity. Indeed, one product of our analysis is the curve in Figure 3C expressing bioluminescence as a function of f -ΔεWT for LuxN. One consideration in using data collapse is the possibility of overfitting. For example, each of our AI-1 dose-response curves can be described by a relatively simple function, and requires at most four parameters to describe (e.g., max, min, EC50, and Hill coefficient). Therefore, a data collapse that allowed adjustment of four parameters for each curve would occur trivially, and would not necessarily reflect the underlying biology. In our case, we were able to obtain a good collapse with one or at most two parameter adjustments per curve. The collapse of the antagonist data for LuxN F163A onto the wild-type curves in Figure 3C is even more parsimonious as it requires adjustment of only one parameter, Δε-ΔεWT, to collapse all three curves.
One oversimplification in Figure 5B is that the EC50 value is indicated to occur at the crossing of the free energies of the kinase (on) state and the phosphatase (off) state with bound AI-1, i.e. where LuxN is active as a kinase approximately 50% of the time. However, our measured EC50 values pertain to bioluminescence, which is an output at the end of the quorum-sensing pathway. Since the relation between bioluminescence and LuxN activity may be nonlinear, the EC50 value measured for bioluminescence may be shifted from the point of half-maximal LuxN kinase activity. Nevertheless, as long as this shift is not large, we can estimate the wild-type bias Δε from the wild-type EC50 value. Taking the observed EC50 value of 23 nM to be the concentration of AI-1 at which wild-type LuxN is active as a kinase 50% of the time, implies that f = 0 in Eq. 2 at this AI-1 concentration (Experimental Procedures). Using the values of from our data collapse in Eq. 2 then allows us to estimate the wild-type bias as ΔεWT = −3.2 kBT. The corresponding probability for LuxN to be active as a kinase in the absence of AI-1 is 1/[1 + e−3.2] = 96%. This strong preference for the kinase state in the absence of AI-1 explains the large ratio between the observed wild-type EC50 value ~20 nM and the underlying binding constant of ~1 nM in the phosphatase (off) state. As shown schematically in Figure 5A and 5B, for a large (negative) bias Δε, the ligand concentration required to switch from kinase to phosphatase grows exponentially In this expression, the exponential factor e|Δε| must be large to achieve a high kinase probability in the absence of AI-1, so the EC50 value (~ 20 nM) must be much larger than the binding constant (~ 1nM). Interestingly, this large ratio (~ 20) of the EC50 value to the underlying binding constant is opposite to that observed for signaling by the chemotaxis network at low ligand concentrations, where receptor clustering and approximately equal kinase and phosphatase probabilities lead to a small ratio (~ 0.1) of the EC50 to the Koff of the chemotaxis receptors (Keymer et al., 2006).
We suggest that the distinct design properties inherent in the quorum sensing and chemotaxis signaling systems have evolved to optimally solve very different biological problems. In chemotaxis, bacterial cells must respond rapidly to small, differential changes in ligand concentration. Consistent with this, chemotaxis receptors are poised to change signaling strength, spending nearly half their time in the on state. Moreover, the chemotaxis receptors cluster which promotes signal amplification (Ames et al., 2002; Kim et al., 1999; Maddock and Shapiro, 1993; Sourjik and Berg, 2002) That is, when a particular receptor-dimer binds a ligand, that event is transduced to nearby, un-bound receptors, amplifying the cell’s response to small changes in ligand concentration (Bray et al., 1998). Thus, the chemotaxis system seems particularly well suited to facilitate biased swimming in a chemical gradient. By contrast, the quorum-sensing receptors (e.g., LuxN) have dramatically different signaling properties. First, in the absence of ligand the quorum-sensing receptors are nearly always in the on state, and thus require a significant threshold ligand concentration to switch off. Second, the quaternary arrangement of receptors (at least in the case of LuxPQ) precludes clustering, and thus excludes chemotaxis-style signal amplification (Neiditch et al., 2005; Neiditch et al., 2006). Therefore, the quorum-sensing apparatus appears designed to respond slowly to the accumulation of ligand. Critically, no alterations in gene expression occur following chemotaxis signaling – the output of the system is exclusively a change in swimming direction. In contrast, a global alteration in gene expression is induced by quorum sensing. Thus, in chemotaxis, the fate of the cell likely does not hinge on a single erroneous signal-transduction event, whereas quorum sensing establishes a committed program, possibly making signaling mistakes fatal. We suggest that this dramatic difference in the output of the two systems selects for high-sensitivity differential signaling accompanied by amplification for chemotaxis, while selecting against exactly those features in quorum-sensing signaling. The striking difference between the resting states of the E. coli chemotaxis system and the V. harveyi quorum-sensing system suggests a broad utility in classifying other two-component regulatory systems into their proper kinase-bias regimes. Such categorization would help define whether a particular two-component system promotes a fast switch in behavior or a slow quasi-developmental process.
Experimental Procedures
Bacterial strains and media
All V. harveyi strains were derived from V. harveyi BB120 and grown aerobically at 30° C in either Luria-Marine (LM) broth or Autoinducer Bioassay (AB) broth. Plasmids were maintained in E. coli strain XL10Gold (Stratagene) at 37° C in LB broth. Tri-parental conjugations were performed with the helper plasmid pRK2013 as described (Ditta et al., 1980). When needed, chloramphenicol (Cm) was added to a final concentration of 10 µg/ml and IPTG to a final concentration of 500 µM. A list of strains and plasmids used in this study is provided in the supplemental material (Table S2).
DNA manipulations
DNA manipulations were performed as described in Sambrook et al (Sambrook et al., 1989). PCR reactions were performed using Herculase Enhanced DNA polymerase (Stratagene). Restriction endonucleases, dNTPs, and T4 ligase were purchased from New England Biolabs. Site-directed mutagenesis was performed using the Quickchange II Site-Directed mutagenesis kit (Stratagene). QIAGEN methods were used for plasmid preparations and PCR cleanups. Sequences of primers are available by request.
luxN mutant library construction
The luxN gene was amplified from wild-type V. harveyi BB120 by PCR and cloned into vector pFED343 at the EcoRI and NcoI sites, making pLS1001. Mutagenesis of the first 950 bases of luxN was performed using the error-prone PCR kit Genemorph II EZclone (Stratagene). Resulting mutations were cloned into vector pFED343. The luxN mutant library was conjugated into V. harveyi ΔluxN ΔluxPQ strain, JMH625. Ex-conjugates were selected on LM medium agar supplemented with Cm. Approximately 30,000 mutants were screened for reduced bioluminescence. Plasmids from dark mutants were isolated and backcrossed into V. harveyi JMH625 to confirm phenotypes. The luxN genes were sequenced and all mutations were engineered independently using Quikchange site-directed mutagenesis (Stratagene). All single luxN mutant constructs were conjugated into V. harveyi JMH625 to verify the phenotypes.
V. harveyi strain construction
To construct the V. harveyi ΔluxMN, ΔluxPQ, ΔluxS mutant strain, HLS253, the luxMN operon was deleted from strain FED119 (Neiditch et al., 2006). Specifically, cosmid pBB1754, carrying luxMN was modified by deleting DNA specifying the entire luxMN open reading frame. The resulting plasmid, p1754:: ΔluxMN, was introduced into V. harveyi FED119, and the deletion transferred to the chromosome to generate V. harveyi strain, HLS253.
Bioluminescence Assays
AI-1 dose-response curves were generated in V. harveyi strain HLS253 containing a vector with wild-type luxN or one of the luxN mutants. V. harveyi strains were grown overnight in LM medium containing Cm and diluted 1:10000 in AB medium plus Cm and 0.5 mM IPTG in triplicate in 96-well microtiter plates. AI-1 was added at either 100 µM or 500 µM and serial 4-fold dilutions were made to final AI-1 concentrations of 24 pM and 119 pM, respectively. The cultures were allowed to grow to stationary phase, at which time bioluminescence and optical density were measured using a Perkin Elmer Envision plate reader.
Quantitative real-time PCR analysis
Wild type and luxN mutant V. harveyi strains were grown in LM medium in triplicate to an OD600 of 1.0 after which cell pellets were isolated and flash-frozen using liquid nitrogen. Pellets were stored at −80° C prior to RNA isolation. RNA was isolated and treated with DNase using the Ribo-Pure-Bacteria kit (Applied Biosystems). RNA was quantified and 1 µg of RNA was converted to cDNA using Superscript II reverse transcriptase (Invitrogen). Quantitative real-time PCR analysis was performed with primers for qrr4 and hfq, where hfq served as an internal control (Tu and Bassler, 2007).
Screen for LuxN antagonists
The V. harveyi strains, JMH624 and JMH610 were grown overnight in AB medium and diluted 1:100 prior to the exogenous addition of either 20 nM AI-1 or 20 nM AI-2, respectively. The diluted cultures were dispensed into 384 well micro-titer plates and the potential antagonist molecules were added to each well. Each micro-titer plate was duplicated to eliminate variance. The 35,000 molecule library was supplied by the Broad Institute and the Initiative for Chemical Genetics (Cambridge, Mass). Antagonist activity was measured as a function of bioluminescence on a PerkinElmer Envision plate reader.
LuxN suppressor screen
The luxN mutant library was conjugated into V. harveyi strain HLS253 and selected on LM containing Cm. Colonies were inoculated into 96-well micro-titer plates containing LM broth and Cm and grown at 30° C with aeration to stationary phase. Glycerol was added to a final concentration of 20%, and the library was stored at −80° C. Frozen stocks were partially thawed and used to inoculate duplicate 96-well micro-titer plates containing AB medium with Cm and IPTG. To one plate, 100 nM AI-1 and 800 nM antagonist C450-0730 was added, while the duplicate control plate had neither AI-1 nor C450-0730 added. The plates were incubated at 30° C with aeration until the cultures reached stationary phase, at which time bioluminescence was measured and the two plates compared. Strains from wells that produced light in the AI-1/C450-0730 plates but did not produce light in the control plates (no AI-1/no C450-0730) were analyzed further. The luxN mutant plasmids were sequenced to determine the mutations responsible for the observed phenotypes and the mutations were reengineered using Quikchange site-directed mutagenesis (Stratagene).
LuxN free energies, competitive binding, and data collapse
In equilibrium, the probability for a LuxN to be active as a kinase is determined by the free-energy difference, f = fon – foff, between its kinase (on) and phosphatase (off) states according to
| (Eq. #1) |
(We measure all energies in units of the thermal energy kBT.) Assuming competitive binding of AI-1 and C450-0730, one obtains
| (Eq. #2} |
where is the dissociation constant for the given state and ligand, and the “bias” Δε is the value of f at zero ligand concentration (Keymer et al., 2006).
To test for competitive binding of C450-0730 to LuxN, we assume that bioluminescence is some (unknown) function of the fraction of LuxN proteins that are active as kinases, i.e. bioluminescence is a function of f. We therefore plot bioluminescence as a function of f – ΔεWT, as given in Eq. #2, and search for the values of that collapse all of our data onto a single curve. The results are shown in Figure 3C.
To quantitatively test whether LuxN mutations that shift AI-1 EC50 values can be attributed to changes in the bias Δε and/or the AI-1 binding affinities, we attempted to collapse the AI-1 dose-response curves for each mutant onto the wild-type curve (Figure 6B) using Δεmutant -ΔεWT and in some cases as fitting parameters. The collapse was satisfactory for many but not all cases, as discussed in the text.
Supplementary Material
Acknowledgements
This work was supported by HHMI, NIH grant 5R01GM065859, NIH grant 5R01 AI 054442, and NSF grant MCB-0343821 to B.L.B., and an NIH postdoctoral fellowship GM787552 to L.R.S. The antagonist screen was partly funded with federal funds supplied to the National Cancer Institute's Initiative for Chemical Genetics, National Institutes of Health, under Contract No. N01-CO-12400 and has been performed with the assistance of the Chemical Biology Platform of the Broad Institute of Harvard and MIT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ames P, Studdert CA, Reiser RH, Parkinson JS. Collaborative signaling by mixed chemoreceptor teams in Escherichia coli. Proc Natl Acad Sci U S A. 2002;99:7060–7065. doi: 10.1073/pnas.092071899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler BL, Greenberg EP, Stevens AM. Cross-species induction of luminescence in the quorum-sensing bacterium Vibrio harveyi. J Bacteriol. 1997;179:4043–4045. doi: 10.1128/jb.179.12.4043-4045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler BL, Wright M, Showalter RE, Silverman MR. Intercellular signalling in Vibrio harveyi: sequence and function of genes regulating expression of luminescence. Mol Microbiol. 1993;9:773–786. doi: 10.1111/j.1365-2958.1993.tb01737.x. [DOI] [PubMed] [Google Scholar]
- Bassler BL, Wright M, Silverman MR. Sequence and function of LuxO, a negative regulator of luminescence in Vibrio harveyi. Mol Microbiol. 1994;12:403–412. doi: 10.1111/j.1365-2958.1994.tb01029.x. [DOI] [PubMed] [Google Scholar]
- Beringer JE, Beynon JL, Buchanan-Wollaston AV, Johnston AWB. Transfer of the drug resistance transposon Tn5 to Rhizobium. Nature. 1978;276:633–634. [Google Scholar]
- Bray D, Levin MD, Morton-Firth CJ. Receptor clustering as a cellular mechanism to control sensitivity. Nature. 1998;393:85–88. doi: 10.1038/30018. [DOI] [PubMed] [Google Scholar]
- Cao JG, Meighen EA. Purification and structural identification of an autoinducer for the luminescence system of Vibrio harveyi. J Biol Chem. 1989;264:21670–21676. [PubMed] [Google Scholar]
- Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassler BL, Hughson FM. Structural identification of a bacterial quorum-sensing signal containing boron. Nature. 2002;415:545–549. doi: 10.1038/415545a. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditta G, Stanfield S, Corbin D, Helinski DR. Broad host range DNA cloning system for gram-negative bacteria: construction of a gene bank of Rhizobium meliloti. Proc Natl Acad Sci U S A. 1980;77:7347–7351. doi: 10.1073/pnas.77.12.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres RG, Falke JJ, Wingreen NS. Chemotaxis receptor complexes: from signaling to assembly. PLoS computational biology. 2007;3:e150. doi: 10.1371/journal.pcbi.0030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engebrecht J, Silverman M. Identification of genes and gene products necessary for bacterial bioluminescence. Proc Natl Acad Sci U S A. 1984;81:4154–4158. doi: 10.1073/pnas.81.13.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engebrecht J, Silverman M. Nucleotide sequence of the regulatory locus controlling expression of bacterial genes for bioluminescence. Nucleic Acids Res. 1987;15:10455–10467. doi: 10.1093/nar/15.24.10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JA, Bassler BL. A genetic analysis of the function of LuxO, a two-component response regulator involved in quorum sensing in Vibrio harveyi. Mol Microbiol. 1999a;31:665–677. doi: 10.1046/j.1365-2958.1999.01208.x. [DOI] [PubMed] [Google Scholar]
- Freeman JA, Bassler BL. Sequence and function of LuxU: a two-component phosphorelay protein that regulates quorum sensing in Vibrio harveyi. Mol Microbiol. 1999b;31:665–677. doi: 10.1128/jb.181.3.899-906.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JA, Lilley BN, Bassler BL. A genetic analysis of the functions of LuxN: a two-component hybrid sensor kinase that regulates quorum sensing in Vibrio harveyi. Mol Microbiol. 2000;35:139–149. doi: 10.1046/j.1365-2958.2000.01684.x. [DOI] [PubMed] [Google Scholar]
- Fuqua C, Parsek MR, Greenberg EP. Regulation of gene expression by cell-to-cell communication: acyl-homoserine lactone quorum sensing. Annu Rev Genet. 2001;35:439–468. doi: 10.1146/annurev.genet.35.102401.090913. [DOI] [PubMed] [Google Scholar]
- Fuqua C, Winans SC, Greenberg EP. Census and consensus in bacterial ecosystems: the LuxR-LuxI family of quorum-sensing transcriptional regulators. Annu Rev Microbiol. 1996;50:727–751. doi: 10.1146/annurev.micro.50.1.727. [DOI] [PubMed] [Google Scholar]
- Fuqua WC, Winans SC, Greenberg EP. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol. 1994;176:269–275. doi: 10.1128/jb.176.2.269-275.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goy MF, Springer MS, Adler J. Sensory transduction in Escherichia coli: role of a protein methylation reaction in sensory adaptation. Proc Natl Acad Sci U S A. 1977;74:4964–4968. doi: 10.1073/pnas.74.11.4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer BK, Bassler BL. Quorum sensing controls biofilm formation in Vibrio cholerae. Mol Microbiol. 2003;50:101–104. doi: 10.1046/j.1365-2958.2003.03688.x. [DOI] [PubMed] [Google Scholar]
- Henke JM, Bassler BL. Quorum sensing regulates type III secretion in Vibrio harveyi and Vibrio parahaemolyticus. J Bacteriol. 2004a;186:3794–3805. doi: 10.1128/JB.186.12.3794-3805.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke JM, Bassler BL. Three Parallel Quorum-Sensing Systems Regulate Gene Expression in Vibrio harveyi. J Bacteriol. 2004b;186:6902–6914. doi: 10.1128/JB.186.20.6902-6914.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins DA, Pomianek ME, Kraml CM, Taylor RK, Semmelhack MF, Bassler BL. The major Vibrio cholerae autoinducer and its role in virulence factor production. Nature. 2007;450:883–886. doi: 10.1038/nature06284. [DOI] [PubMed] [Google Scholar]
- Jung K, Odenbach T, Timmen M. The quorum-sensing hybrid histidine kinase LuxN of Vibrio harveyi contains a periplasmically located N terminus. J Bacteriol. 2007;189:2945–2948. doi: 10.1128/JB.01723-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehry MR, Dahlquist FW. Adaptation in bacterial chemotaxis: CheB-dependent modification permits additional methylations of sensory transducer proteins. Cell. 1982a;29:761–772. doi: 10.1016/0092-8674(82)90438-x. [DOI] [PubMed] [Google Scholar]
- Kehry MR, Dahlquist FW. The methyl-accepting chemotaxis proteins of Escherichia coli. Identification of the multiple methylation sites on methyl-accepting chemotaxis protein I. J Biol Chem. 1982b;257:10378–10386. [PubMed] [Google Scholar]
- Keymer JE, Endres RG, Skoge M, Meir Y, Wingreen NS. Chemosensing in Escherichia coli: two regimes of two-state receptors. Proc Natl Acad Sci U S A. 2006;103:1786–1791. doi: 10.1073/pnas.0507438103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KK, Yokota H, Kim SH. Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature. 1999;400:787–792. doi: 10.1038/23512. [DOI] [PubMed] [Google Scholar]
- Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell. 2004;118:69–82. doi: 10.1016/j.cell.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Maddock JR, Shapiro L. Polar location of the chemoreceptor complex in the Escherichia coli cell. Science. 1993;259:1717–1723. doi: 10.1126/science.8456299. [DOI] [PubMed] [Google Scholar]
- Martin M, Showalter R, Silverman M. Identification of a locus controlling expression of luminescence genes in Vibrio harveyi. J Bacteriol. 1989;171:2406–2414. doi: 10.1128/jb.171.5.2406-2414.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFall-Ngai MJ, Ruby EG. Developmental biology in marine invertebrate symbioses. Curr Opin Microbiol. 2000;3:603–607. doi: 10.1016/s1369-5274(00)00147-8. [DOI] [PubMed] [Google Scholar]
- Miller MB, Bassler BL. Quorum sensing in bacteria. Annu Rev Microbiol. 2001;55:165–199. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- Neiditch MB, Federle MJ, Miller ST, Bassler BL, Hughson FM. Regulation of LuxPQ receptor activity by the quorum-sensing signal autoinducer-2. Mol Cell. 2005;18:507–518. doi: 10.1016/j.molcel.2005.04.020. [DOI] [PubMed] [Google Scholar]
- Neiditch MB, Federle MJ, Pompeani AJ, Kelly RC, Swem DL, Jeffrey PD, Bassler BL, Hughson FM. Ligand-induced asymmetry in histidine sensor kinase complex regulates quorum sensing. Cell. 2006;126:1095–1108. doi: 10.1016/j.cell.2006.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Saha SK, Inouye M. Two-domain reconstitution of a functional protein histidine kinase. Proc Natl Acad Sci U S A. 1998;95:6728–6732. doi: 10.1073/pnas.95.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Showalter RE, Martin MO, Silverman MR. Cloning and nucleotide sequence of luxR, a regulatory gene controlling bioluminescence in Vibrio harveyi. J Bacteriol. 1990;172:2946–2954. doi: 10.1128/jb.172.6.2946-2954.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourjik V. Receptor clustering and signal processing in E. coli chemotaxis. Trends Microbiol. 2004;12:569–576. doi: 10.1016/j.tim.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Sourjik V, Berg HC. Receptor sensitivity in bacterial chemotaxis. Proc Natl Acad Sci U S A. 2002;99:123–127. doi: 10.1073/pnas.011589998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourjik V, Berg HC. Functional interactions between receptors in bacterial chemotaxis. Nature. 2004;428:437–441. doi: 10.1038/nature02406. [DOI] [PubMed] [Google Scholar]
- Springer WR, Koshland DE., Jr Identification of a protein methyltransferase as the cheR gene product in the bacterial sensing system. Proc Natl Acad Sci U S A. 1977;74:533–537. doi: 10.1073/pnas.74.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock JB, Koshland DE., Jr A protein methylesterase involved in bacterial sensing. Proc Natl Acad Sci U S A. 1978;75:3659–3663. doi: 10.1073/pnas.75.8.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surette MG, Miller MB, Bassler BL. Quorum sensing in Escherichia coli, Salmonella typhimurium, and Vibrio harveyi: a new family of genes responsible for autoinducer production. Proc Natl Acad Sci U S A. 1999;96:1639–1644. doi: 10.1073/pnas.96.4.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsen SL, Waters CM, Bassler BL. A negative feedback loop involving small RNAs accelerates Vibrio cholerae’s transition out of quorum-sensing mode. Genes Dev. 2008;22:226–238. doi: 10.1101/gad.1629908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Koshland DE., Jr Sites of methyl esterification and deamination on the aspartate receptor involved in chemotaxis. J Biol Chem. 1984;259:7719–7725. [PubMed] [Google Scholar]
- Timmen M, Bassler BL, Jung K. AI-1 influences the kinase activity but not the phosphatase activity of LuxN of Vibrio harveyi. J Biol Chem. 2006;281:24398–24404. doi: 10.1074/jbc.M604108200. [DOI] [PubMed] [Google Scholar]
- Tu KC, Bassler BL. Multiple small RNAs act additively to integrate sensory information and control quorum sensing in Vibrio harveyi. Genes Dev. 2007;21:221–233. doi: 10.1101/gad.1502407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters CM, Bassler BL. QUORUM SENSING: Cell-to-Cell Communication in Bacteria. Annu Rev Cell Dev Biol. 2005;21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- Zhu J, Winans SC. Autoinducer binding by the quorum-sensing regulator TraR increases affinity for target promoters in vitro and decreases TraR turnover rates in whole cells. Proc Natl Acad Sci U S A. 1999;96:4832–4837. doi: 10.1073/pnas.96.9.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Winans SC. The quorum-sensing transcriptional regulator TraR requires its cognate signaling ligand for protein folding, protease resistance, and dimerization. Proc Natl Acad Sci U S A. 2001;98:1507–1512. doi: 10.1073/pnas.98.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.