

Abstract

Phosphine-catalyzed [4 + 2] annulations of α-alkylallenoates with activated olefins allow the efficient syntheses of cyclohexenes. Hexamethylphosphorous triamide (HMPT)-catalyzed [4 + 2] annulations of α-alkylallenoates with arylidenemalononitriles provided highly functionalized 5,5-dicyano-4,6-disubstituted cyclohex-1-enecarboxylates in excellent yields (77–98%) and moderate to high diastereoselectivities (1:2–12:1). Remarkably, the corresponding triarylphosphine-catalyzed [4 + 2] annulations of α-methylallenoate with arylidenemalononitriles manifested a polarity inversion of the 1,4-dipole synthon, providing 4,4-dicyano-5-substituted cyclohex-1-enecarboxylates in excellent yields (80–93%). The polarity inversion of α-alkylallenoates from one 1,4-dipole to another under phosphine catalysis presumably resulted from a change in the balance of the equilibrium between the phosphonium dienolate and the vinylogous phosphonium ylide intermediate.
The construction of suitably functionalized cyclohexene frameworks plays a central role in many natural product synthesis.1 Although the Diels–Alder reaction is among the most powerful tools for generating such carbocycles,2 it is often difficult to form systems that are highly congested or possess substituent arrays that are incompatible with the reaction.3 A number of alternative methods for synthesizing cyclohexenes have arisen from catalytic approaches, such as the phosphine-catalyzed Rauhut–Currier reaction,4 transition metal-catalyzed ring-closing metathesis (RCM),5 and cycloisomerization reactions.6 In contradistinction to the widespread use of these intramolecular processes, intermolecular counterparts for catalytic cyclohexene synthesis are less well developed.7
Nucleophilic phosphine catalysis has emerged recently as an efficient means of generating carbo- and heterocycles.8 In particular, Lu's [3+2] cycloaddition9 to form cyclopentenes from allenoates and alkenes under phosphine catalysis has been applied in the syntheses of several natural products.10 Nevertheless, phosphine catalysis has not been utilized previously for the formation of cyclohexenes. Building upon our phosphine-catalyzed [4 + 2] annulation for the synthesis of tetrahydropyridines,11 we reasoned that it might be possible to formulate an all-carbon variant of this strategy. Herein, we disclose the facile synthesis of cyclohexenes 3 and 4 via phosphine-catalyzed [4 + 2] annulations between allenoates 1 and activated olefins 2 (eq 1).
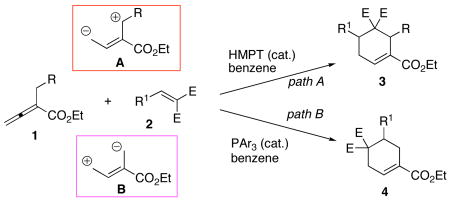 |
(1) |
We initiated our investigation by seeking a viable phosphine catalyst for the [4 + 2] annulation of the allenoate 1a and benzylidenemalononitrile 2a to provide the cyclohexene 3a (Table 1). The optimal conditions for tetrahydropyridine synthesis (20 mol% PBu3, CH2Cl2, rt) were ineffective for the formation of the cyclohexene (entry 1).12 Further examination revealed that hexamethylphosphorous triamide (HMPT) catalyzed the [4 + 2] reaction in benzene under reflux to provide 3a in 98% yield (entry 2). Interestingly, use of the less-nucleophilic13 PPh3 induced preferable formation of the regioisomeric cyclohexene 4a (entry 5). Among the triarylphosphines tested, we found that the more electron deficient the aryl groups, the greater the amount of 4a obtained (entries 3–7). Using tris(p-chlorophenyl)phosphine, we obtained isomer 4a exclusively (entry 7). Therefore, allenoate 1a serves as dipole A under the influence of HMPT and as inverted dipole B under triarylphosphine catalysis (eq 1).
Table 1.
Survey of phosphine catalysts for [4 + 2] annulation of allenoate 1a and alkene 2aa
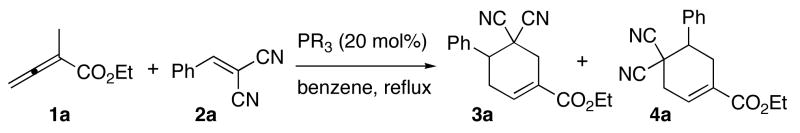 | |||
|---|---|---|---|
| entry | phosphine | 3a:4ab | % yieldc |
| 1d | PBu3 | NA | NR |
| 2 | P(NMe2)3 | 100:0 | 98 |
| 3 | P(4-MeOC6H4)3 | 33:67 | 96 |
| 4 | P(4-Me2NC6H4)Ph2 | 32:68 | 95 |
| 5 | PPh3 | 26:74 | 93 |
| 6 | P(4-FC6H4)3 | 8:92 | 98 |
| 7e | P(4-ClC6H4)3 | 0:100 | 93 |
Reaction conditions: 1a (1.2–1.4 mmol), 2a (1 mmol), and the phosphine (20 mol%) were heated under reflux in benzene (10 mL) for 14 h.
Determined through NMR spectroscopic analyses.
Isolated yields.
This transformation required a reaction time of 120 h.
Success at identifying phosphine catalysts for the efficient syntheses of both isomers of the cyclohexenes prompted us to probe the generality of the reaction using other activated olefins (Table 2). In the presence of HMPT, the allenoate 1a reacted with both electron-deficient and -rich arylidenes to provide the cyclohexenes 3a–3c in high yields (Table 2, entries 1–3). With tris(p-fluorophenyl)phosphine, the regioisomeric cyclohexenes 4a–c were obtained with high efficiency (entries 4–6). Notably, these conditions also worked well for activated heteroarylidenes, furnishing the cyclohexenes 4d–f (entries 7–9).
Table 2.
Survey of alkenes for [4 + 2] annulation with allenoate 1aa
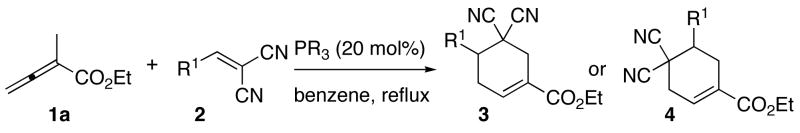 | ||||
|---|---|---|---|---|
| entry | R1 | phosphine | product | % yieldb |
| 1 | Ph (2a) | P(NMe2)3 | 3a | 98 |
| 2 | 4-MeOC6H4 (2b) | P(NMe2)3 | 3b | 94 |
| 3 | 4-BrC6H4 (2c) | P(NMe2)3 | 3c | 86 |
| 4 | Ph (2a) | P(4-FC6H4)3 | 4a | 93 |
| 5 | 4-MeOC6H4 (2b) | P(4-FC6H4)3 | 4b | 90 |
| 6 | 4-BrC6H4 (2c) | P(4-FC6H4)3 | 4c | 85 |
| 7 | 2-furyl (2d) | P(4-FC6H4)3 | 4d | 88 |
| 8 | 3-pyridyl (2e) | P(4-FC6H4)3 | 4e | 80 |
| 9 | N-Me-2-indolyl (2f) | P(4-FC6H4)3 | 4f c | 91 |
Reaction conditions: 1a (1.2 mmol), 2 (1 mmol), and the phosphine (20 mol%) were heated under reflux in benzene (10 mL) for 14 h.
Isolated yields.
Confirmed through single-crystal X-ray analysis.
The intriguing reversal of regioselectivity can be rationalized as indicated in Scheme 1. Under HMPT catalysis, the β-phosphonium dienolate intermediate 5, formed through conjugate addition of the phosphine to the allenoate 1a, adds to 2a at the γ carbon atom to give adduct 6. Zwitterion 6 converts into the allylic phosphonium intermediate 7 through proton transfer.11a Conjugate addition of the malononitrile anion in 7 and subsequent β-elimination of HMPT provide the cyclohexene 3a. On the other hand, the phosphonium dienolate-to-phosphorous ylide equilibrium (5 ⇆ 8) favors the ylide when a more-electron-withdrawing triarylphosphine is used.14 The vinylogous ylide 8 adds conjugatively to olefin 2a to give the adduct 9. Consecutive proton transfers provide the deconjugated enoate 10, which allows 6-endo cyclization to generate the cyclic ylide 11. Finally, 1,2-proton transfer and subsequent β-expulsion of the phosphine catalyst furnish the cyclohexene 4a.
Scheme 1.

Mechanistic proposal for the formation of cyclohexenes 3a and 4a with polarity inversion of the 1,4-dipole synthon 1a
The reaction tolerated a wide range of allenylic β′ substituents on the allenoate 1, including aryl, ester, alkyl, and vinyl moieties, to provide the cyclohexenes 3 in excellent isolated yields (Table 3). For these β′-substituted allenoates 1, only 3 was formed, independent of the phosphine employed, presumably because of steric hindrance and the resulting diminished reactivity at the allenylic carbon atom. Good diastereoselectivities resulted when using α-benzylallenoates (entries 1–3), except when the benzyl group incorporated an ortho substituent (entry 4).15 The reactions of α-alkylallenoates occurred with improved diastereoselectivities upon increasing the size of the allenylic substituent (entries 5–7), although α-isobutylallenoate 1k manifested a unique preference for the trans isomer (entry 8). The diastereoselectivity was highest for the reactions of α-allylallenoates (entries 9 and 10).
Table 3.
Survey of allenoates 1 for [4 + 2] annulations with the alkene 2aa
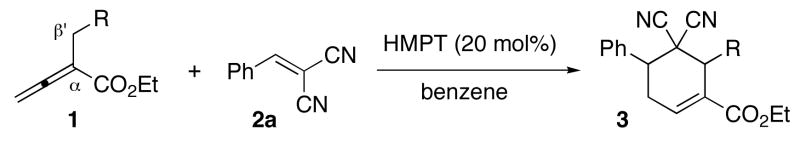 | |||||
|---|---|---|---|---|---|
| entry | R | product | T (°C) | cis:transb | % yieldc |
| 1 | Ph (1d) | 3dd | 45 | 82:18 | 93 |
| 2 | p-BrC6H4 (1e) | 3e | 45 | 84:16 | 96 |
| 3 | m-MeOC6H4 (1f) | 3f | 45 | 78:22 | 92 |
| 4 | o-MeC6H4 (1g) | 3g | 45 | 64:36 | 90 |
| 5 | CO2Et (1h) | 3h | rt | 66:33 | 96 |
| 6 | Me (1i) | 3i | 88 | 80:20 | 95 |
| 7 | Et (1j) | 3j | 88 | 92:8 | 98 |
| 8 | i-Pr (1k) | 3k | 88 | 34:66 | 77 |
| 9 | CH=CH2 (1l) | 3l | 45 | 91:9 | 94 |
| 10 | CH=CHPh (1m) | 3m | 45 | 91:9 | 93 |
Reaction conditions: 1 (1.2–1.4 mmol), 2a (1 mmol), and HMPT (20 mol%) were stirred in benzene (10 mL) for 14 h at the designated temperature.
Determined through NMR spectroscopic analysis and comparison with the spectra of cis-3d, for which a single-crystal X-ray structure was obtained.
Isolated yield.
Confirmed through single-crystal X-ray crystallographic analysis.
Scheme 2 demonstrates the potential utility of this [4 + 2] annulation for the synthesis of biologically active natural products: rapid entry into the tetracyclic framework 12 of nodulisporic acids16 via the Houben–Hoesch reaction of 4f.
Scheme 2.

One application of the allene–alkene [4 + 2] annulationa
a (a) Conc. HCl/EtOAc (10:1), cat. H2SO4. (b) EtOH, cat. H2SO4, 85% isolated yield over two steps.
In summary, we have developed novel phosphine-catalyzed [4 + 2] annulation processes that enable regio-differentiating syntheses of cyclohexenes. In these all-carbon [4 + 2] annulations, simply switching the catalyst from HMPT to a triarylphosphine changes the reactivity of the α-alkylallenoate from that of a 1,4-dipole A to an inverted dipole B. These highly efficient and regioselective processes serve as rapid conduits toward the scaffolds of many natural products. Our future efforts will focus on developing asymmetric variants of these reactions and applying them to the construction of other biologically significant natural products.
Supplementary Material
Representative experimental procedures and spectral data for all new compounds (PDF). Crystallographic data for 3d, 4f, and 12 (CIF). This information is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This study was funded by the NIH (R01GM071779). We thank Dr. Saeed Khan for performing the crystallographic analyses. O.K. thanks Dr. Chulbom Lee for helpful discussions.
References
- 1.Ho TL. Carbocycle Construction in Terpene Synthesis. Wiley-VCH; Weinheim: 1988. [Google Scholar]
- 2.(a) Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis GE. Angew Chem, Int Ed Engl. 2002;41:1668. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]; (b) Takao KI, Munakata R, Tadano KI. Chem Rev. 2005;105:4779. doi: 10.1021/cr040632u. [DOI] [PubMed] [Google Scholar]
- 3.Jung ME, Ho DG. Org Lett. 2007;9:375. doi: 10.1021/ol062980j. and references therein. [DOI] [PubMed] [Google Scholar]
- 4.(a) Wang LC, Luis AL, Agapiou K, Jang HY, Krisch MJ. J Am Chem Soc. 2002;124:2402. doi: 10.1021/ja0121686. [DOI] [PubMed] [Google Scholar]; (b) Frank SA, Mergott DJ, Roush WR. J Am Chem Soc. 2002;124:2404. doi: 10.1021/ja017123j. [DOI] [PubMed] [Google Scholar]; (c) Krafft ME, Haxell TFN. J Am Chem Soc. 2005;127:10168. doi: 10.1021/ja052146+. [DOI] [PubMed] [Google Scholar]
- 5.Grubbs RH, Miller SJ, Fu GC. Acc Chem Res. 1995;28:446. [Google Scholar]
- 6.Kim H, Goble SD, Lee C. J Am Chem Soc. 2007;129:1030. doi: 10.1021/ja067336e. and references therein. [DOI] [PubMed] [Google Scholar]
- 7.Enders D, Huttl MRM, Grondal C, Raabe G. Nature. 2006;441:861. doi: 10.1038/nature04820. [DOI] [PubMed] [Google Scholar]
- 8.For reviews, see: Lu X, Zhang C, Xu Z. Acc Chem Res. 2001;34:535. doi: 10.1021/ar000253x.Methot JL, Roush WR. Adv Synth Catal. 2004;346:1035.Lu X, Du Y, Lu C. Pure Appl Chem. 2005;77:1985.Nair V, Menon RS, Sreekanth AR, Abhilash N, Biji AT. Acc Chem Res. 2006;39:520. doi: 10.1021/ar0502026.
- 9.(a) Zhang C, Lu X. J Org Chem. 1995;60:2906. [Google Scholar]; (b) Zhu G, Chen Z, Jiang Q, Xiao D, Cao P, Zhang X. J Am Chem Soc. 1997;119:3836. [Google Scholar]; (c) Shu LH, Sun WQ, Zhang DW, Wu SH, Wu HM, Xu JF, Lao XF. Chem Commun. 1997:79. [Google Scholar]; (d) O'Donovan BF, Hitchcock PB, Meidine MF, Kroto HW, Taylor R, Walton DRM. Chem Commun. 1997:81. [Google Scholar]; (e) Xu Z, Lu X. Tetrahedron Lett. 1999;40:549. [Google Scholar]; (f) Ung AT, Schafer K, Lindsay KB, Pyne SG, Amornraksa K, Wouters R, Van der Linden I, Biesmans I, Lesage ASJ, Skelton BW, White AH. J Org Chem. 2002;67:227. doi: 10.1021/jo010864i. [DOI] [PubMed] [Google Scholar]; (g) Du Y, Lu X, Yu Y. J Org Chem. 2002;67:8901. doi: 10.1021/jo026111t. [DOI] [PubMed] [Google Scholar]; (h) Lu X, Lu Z, Zhang X. Tetrahedron. 2006;62:457. [Google Scholar]; (i) Wilson JE, Fu GC. Angew Chem, Int Ed Engl. 2006;45:1426. doi: 10.1002/anie.200503312. [DOI] [PubMed] [Google Scholar]; (j) Xia Y, Liang Y, Chen Y, Wang M, Jiao L, Huang F, Liu S, Li Y, Yu ZX. J Am Chem Soc. 2007;129:3470. doi: 10.1021/ja068215h. [DOI] [PubMed] [Google Scholar]; (k) Mercier E, Fonovic B, Henry CE, Kwon O, Dudding T. Tetrahedron Lett. 2007;48:3617. [Google Scholar]; (l) Wallace DJ, Sidda RL, Reamer RA. J Org Chem. 2007;72:1051. doi: 10.1021/jo062170l. [DOI] [PubMed] [Google Scholar]
- 10.(a) Du Y, Lu X. J Org Chem. 2003;68:6463. doi: 10.1021/jo034281f. [DOI] [PubMed] [Google Scholar]; (b) Wang JC, Krische MJ. Angew Chem, Int Ed Engl. 2003;42:5855. doi: 10.1002/anie.200352218. [DOI] [PubMed] [Google Scholar]; (c) Pham TQ, Pyne SG, Skelton BW, White AH. J Org Chem. 2005;70:6369. doi: 10.1021/jo050827h. [DOI] [PubMed] [Google Scholar]
- 11.(a) Zhu XF, Lan J, Kwon O. J Am Chem Soc. 2003;125:4716. doi: 10.1021/ja0344009. [DOI] [PubMed] [Google Scholar]; (b) Tran YS, Kwon O. Org Lett. 2005;7:4289. doi: 10.1021/ol051799s. [DOI] [PubMed] [Google Scholar]; (c) Wurz RP, Fu GC. J Am Chem Soc. 2005;127:12234. doi: 10.1021/ja053277d. [DOI] [PubMed] [Google Scholar]; (d) Castellano S, Fiji HDG, Kinderman SS, Watanabe M, de Leon P, Tamanoi F, Kwon O. J Am Chem Soc. 2007;129:5843. doi: 10.1021/ja070274n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phosphines that resulted only in the oligomerization of the allenoate 1a without incorporation of benzylidenemalononitrile 2a (DCM, rt; or benzene, reflux) included Bu3P, Bn3P, Me3P, Me2PhP, MePh2P, P(OEt)3, (4-CF3C6H4)3P, (3,5-FC6H3)3P, and BINAP. DABCO, Et3N, and DBU were not effective catalysts for the desired transformation.
- 13.(a) Farrar DH, Poe AJ, Zheng Y. J Am Chem Soc. 1994;116:6252. [Google Scholar]; (b) Denmark SE, Chung W. J Org Chem. 2006;71:4002. doi: 10.1021/jo060153q. [DOI] [PubMed] [Google Scholar]
- 14.The exclusive formation of isomer 3 when using HMPT hints at the diminished acidity of the β′ proton in 5, presumably because of back bonding of the nitrogen atom's lone pair of electrons to the phosphonium center (P=N+Me2); see: Mark V. J Am Chem Soc. 1963;85:1884.
- 15.This result is consistent with our previous findings for allenoate/arylimine [4 + 2] annulations. See ref. 10a.
- 16.(a) Singh SB, Ondeyka JG, Jayasuriya H, Zink DL, Ha SN, Dahl-Roshak A, Greene J, Kim JA, Smith MM, Shoop W, Tkacz JS. J Nat Prod. 2004;67:1496. doi: 10.1021/np0498455. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Davulcu AH, Kurti L. Org Lett. 2006;8:1665. doi: 10.1021/ol060290+. and references therein. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative experimental procedures and spectral data for all new compounds (PDF). Crystallographic data for 3d, 4f, and 12 (CIF). This information is available free of charge via the Internet at http://pubs.acs.org.