

Abstract
It has proven difficult to use systemic administration of small molecules to selectively modulate nociception. Over the past decade, we and others have developed non-replicating herpes simplex virus (HSV)-based vectors to treat chronic pain. Subcutaneous inoculation of an HSV vector effectively transduces sensory neurons in the dorsal root ganglion; release of transgene-coded inhibitory neurotransmitters or anti-inflammatory peptides reduces pain-related behaviors in rodent models of chronic inflammatory and neuro-pathic pain. A phase 1 trial of this therapy in patients is set to begin soon.
Keywords: gene therapy, herpes simplex virus, pain, enkephalin, GABA, cytokines
Acute pain is an unpleasant sensory experience with discriminative, affective, and cognitive components that helps alert and individual response to potentially harmful stimuli in the environment. Acute pain is short lasting and often manifests objective correlates that can be observed and described. It is usually related to discrete events and may occur intermittently or last up to several days (subacute pain). Chronic pain is more difficult to define precisely. It has been described as a painful sensation that persists beyond the duration of the inciting event, or, in the definition used by the U.S. Food and Drug Administration, a pain that persists for more than 3 months. Different classification schemes have been proposed to separate chronic pains according to their pathophysiology and clinical manifestations. For the purposes of the discussion in this review we will consider two major forms of chronic pain: inflammatory or nociceptive pain, a pain that results from tissue injury leading to the continued activation of peripheral nociceptors and neuropathic pain, a pain that results from injury to neural structures without evident damage of peripheral tissues.
The neuroanatomic pathways subserving nociception are well defined. Primary nociceptors, a subclass of neurons in the dorsal root (or trigeminal) ganglion (DRG), are specifically activated by painful chemical, physical or thermal stimuli. These neurons synapse in the dorsal horn of spinal cord (or the descending trigeminal nucleus) onto second order neurons that project primarily to the thalamus. Third order neurons in the pain pathway project from the thalamus to somatic sensory cortex to subserve the discriminative aspects of pain, and to a variety of limbic structures to subserve the affective components of the pain experience. In the setting of chronic persistent pain alteration of the components of the nociceptive pathway occur at every level of that pathway. Changes in transcription, translation, and post translational processing of proteins in the neurons of the DRG, spinal dorsal horn and central structures are well-described[1, 2]. In the brain, changes that can be mapped by measures of regional cerebral metabolism and by neuropsychological testing also occur[3, 4]. But these changes are not irreversible. For example patients with severe arthritis of a hip or knee may suffer chronic pain for a period of years, but successful replacement of the injured joint can result in the relief of pain in the disappearance of all of the manifestations of chronic pain.
For conditions in which the primary cause of the chronic pain is either not readily reversible or not identifiable the search for treatment has focused primarily on components of the ascending pain pathway. If one was able to selectively interfere with nociceptive neurotransmission one would have the ability to modulate the perception of pain despite the inability to correct the primary abnormality. Extensive efforts over the past several decades have been focused on the identification of small molecules that might act at neurotransmitters, ion channels or receptors that are specific for the nociceptive pathways. However, the redundant use of a relatively limited repertoire of neurotransmitters ion channels and receptors in many different functional pathways within the nervous system and in structures outside the nervous system has made it difficult to identify effective agents to modify nociceptive neurotransmission without causing “off target” adverse events mediated by neural or other structures outside of the nociceptive pathway.
One strategy that may be used to avoid “off target” adverse events of potent bioactive molecules is the use of gene transfer to express those molecules at specific sites within the nervous system. For example, selective expression of the inhibitory neurotransmitter in the amygdala, or the rostral a granular insular cortex can be used to selectively modify nociceptive neurotransmission. Other investigators have shown that transduction of the meninges by intrathecal injection of a gene transfer vector can be used to express antinociceptive peptides to provide an analgesic effect in models of neuropathic pain. Our own studies have focused on the synapse between the primary nociceptor and the second order neuron in the dorsal horn of spinal cord. It has been well-known for many years then nociceptive neurotransmission at this level is a subject of continuous and exquisite modulation effected by interneurons that release inhibitory or excitatory neurotransmitters under a descending neuromodulatory pain control system.
The experimental paradigm is identical for all of the studies that we will describe. A gene transfer vector carrying the transgene of choice is injected subcutaneously in the skin; in all of the experiments reviewed, this injection is placed in the plantar surface of the hind foot of a rodent. From the site of inoculation the gene transfer vector is picked up by nerve terminals in the skin and carried by retrograde axonal transport to the neuronal perikaryon in the DRG. In the DRG gene transfer vector genome enters the nucleus to establish a persistent state from which the transgene is expressed, and the transgene product then transported in an anterograde direction in the bipolar DRG axon to the central terminals and the dorsal horn of spinal cord where it will modulate nociceptive neurotransmission (Fig. 1).
Fig. 1.
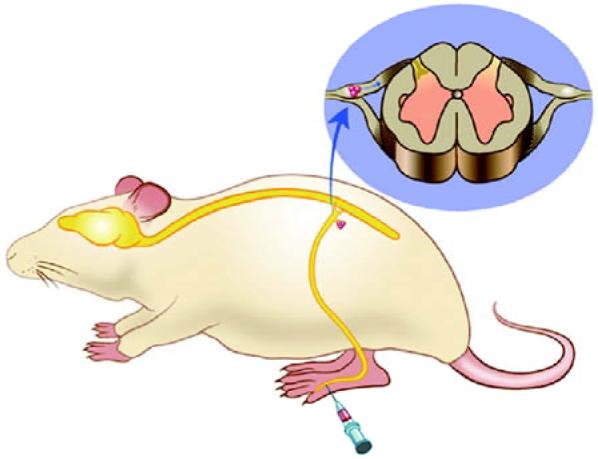
HSV-mediated gene transfer approach (schematic). The vector inoculated under the skin is taken up by sensory nerve terminals and carried to the neuronal cell body in the DRG by retrograde axonal transport. Latent vector genomes in the nucleus produce the transgene product that is then transported to the afferent terminals in the dorsal horn of spinal cord.
Many different gene transfer vectors have been developed, and the characteristics of some of the principal vectors are outlined in Table 1. Liposomes are cationic lipids complexed with DNA. Liposomes have been shown to be quite effective in transferring genes into cells of the meninges following intrathecal inoculation, but are not effective at transferring genes into neurons. Retroviruses are RNA viruses that can carry up to an 8 kb insert, but a corporation of the retroviral coded cDNA into the host cell genome requires cell division so that these vectors cannot be used for the nervous system. Lentiviral vectors have overcome this problem by the incorporation of a viral integrase into the viral genome that facilitates the integration of the cDNA into nondividing cells and lentiviral vectors are very effective for transferring genes into neurons. Similarly, the DNA virus based vectors constructed from adenovirus and adeno-associated virus (AAV) have been used extensively for gene transfer into neurons in brain. Gene transfer using these vectors requires injection of the vector genome in close proximity to the neuronal cell body.
Table 1.
Characteristics of vectors commonly used for gene transfer
| Vector | Size (nm) | Genome (kb) | Type | Insert (kb) |
|---|---|---|---|---|
| Lippspme | DNA | |||
| Retrovirus | 100 | 8 | ss-RNA | 8 |
| Adenovirus | 100 | 36 | ds-RNA | 8, 30 |
| AAV | 20 | 5 | ss-RNA | 5 |
| Lentivirus | 100 | 10 | ss-RNA | 8 |
| HSV | 200 | 152 | ss-RNA | 50 |
Herpes simplex virus (HSV) is uniquely suited for gene transfer to peripheral sensory neurons[5]. The wild type parental virus, which is responsible for cold sores, is naturally transmitted from host to host by contact of skin or mucous membrane. Following infection of the peripheral site the wild type virus is taken up by sensory nerve terminals and carried by retrograde axonal transport to the DRG neuron where the viral genome is capable of establishing a lifelong persistent state. This process of uptake and retrograde axonal transport is complex and highly evolved. The virus is an enveloped double stranded DNA virus with a structured protein capsid surrounded by an amorphous protein pigment within the lipid envelope. The initial interactions of the virus particle with the center nerve terminal are mediated by nonspecific interactions between the viral envelope glycoproteins gB and gC and heparan sulfate moieties on the sensory nerve terminal. These nonspecific interactions are followed by a selective high affinity interaction between the viral glycoprotein gD and the homophilic cell adhesion molecule nectin-1 which is also known as HveC. Finally interaction between the viral envelope glycoproteins gH and gL mediate fusion of the viral envelope with the cell surface membrane and entry of the viral particle into the sensory nerve terminal. Specific interactions between viral proteins and fee retrograde axonal motor dynein mediate the movement along microtubules long distances back to the neuronal cell body, where the viral DNA is injected through a nuclear pore into the nucleus. Latent HSV genomes exist as intranuclear extrachromosomal elements and may persist for the life of the host.
The herpesviral genome is complex containing more than 85 different genes. Conveniently, in the wild type virus these genes are expressed in a rigidly ordered temporal cascade. Only a limited subset of 5 “Immediate Early” (IE) genes can be transcribed in the absence of other viral protein synthesis; the transcription and translation of Early and Late viral genes depends on the prior production of IE gene products. Deletion of even a single essential IE gene from the viral genome results in the production of a recombinant that is incapable of replication even in the highly permissive environment of African green monkey kidney (Vero) cells that are used for viral propagation. Such recombinants can be produced to high titers in complementing cell lines constructed to express the missing IE gene product in trans from the cellular genome. Injected subcutaneously into animals, the replication incompetent recombinants target like wild type virus to the sensory neurons but are incapable of wild type infection or of reactivation. Because they cannot replicate these recombinants, spreads are limited to the first order neuron in the DRG after subcutaneous inoculation. The remainder of this review will summarize experiments using replication incompetent genomic vectors in models of inflammatory and neuropathic pain in rodent models.
We constructed a replication incompetent HSV vector containing the human proenkephalin coding sequence under the control of the human cytomegalovirus virus immediate early promoter (HCMV IEp) in the UL23 locus of an HSV vector deleted for both copies of the essential IE gene ICP4. Infection of primary DRG neurons in culture at a multiplicity of infection (MOI) of 1 resulted in the release of substantial amounts of enkephalin into the medium by the transduced cells. Subcutaneous inoculation into the plantar surface of the hind foot resulted in transduction of the L4-L6 DRG detected by PCR for HSV genomes and by RT-PCR for the human transgene sequences. Rats inoculated with the proenkephalin expressing vector 7 days prior to formalin test showed a substantial and statistically significant reduction in spontaneous pain behaviors in the delayed phase of the formalin test[6]. In a timecourse experiment we found that the analgesic effect was maximal in animals inoculated 7 days prior to formalin test and that by 28 days after inoculation proenkephalin vector inoculated animals were not statistically significantly different from control vector inoculated animals. Evidence that the analgesic effect was mediated by the opiate peptide gene product was provided by the observation that intrathecal injection of the opiate receptor antagonist naltrexone blocked the analgesic effect of the vector. Interestingly, reinoculation of the vector 28 days after the original inoculation resulted in a reestablishment of the analgesic effect, suggesting that the loss of effectiveness over the first month was due to a loss of expression rather than the development of tolerance to the transgene product[6]. This time-course of expression would be consistent with that which we have found in other experiments using HSV bass gene transfer vectors with the HCMV IEp driving transgene expression.
We also examined the relative effectiveness of the proenkephalin expressing HSV vector in the selective L5 spinal nerve ligation model of neuropathic pain[7]. Subcutaneous inoculation of the vector 7 days after selective ligation of the L5 spinal nerve resulted in a substantial and statistically significant reduction in mechanical allodynia. Like the effect in the formalin model of inflammatory pain, the antiallodynic effect was reversed by intraperitoneal injection of the opiate receptor antagonist naloxone. The time-course of the vector mediate the effect in this bond obligation model of neuropathic pain was similar to that which we observed in the form of model of inflammatory pain; after 5 weeks there was no detectable effect of the vector, but reinoculation at that point reestablished the antiallodynic effect. The antiallodynic effect of the proenkephalin expressing vector was continuous over time. Animals tested in different time points several hours apart during a single day showed the same elevated threshold at all of the testing points. The effect of vector mediated proenkephalin expression was additive with morphine. In this model we found that the ED50 of morphine was 1.8 mg/kg in spinal nerve ligation control or control vector inoculated animals; animals inoculated with the proenkephalin expressing vector showed a leftward shift of the dose-response curve with an ED50 of 0.15 mg/kg[7]. Animals inoculated with the vector also showed a suppression in the activation of c-fos in neurons of the dorsal horn in response to non-noxious touch. The activation of c-fos by non-noxious touch is of unknown significance but serves as one histological marker that corresponds to the behavioral measures.
Enkephalin is the cognate ligand for the δ opiate receptor, but three quarters of the opiate receptors in the dorsal horns spinal cord are μ opiate receptors. The endogenous ligand for the μ opiate receptor has not been fully established. A pair of amidated tetrapeptides (endomorphin-1 and endomorphin-2) isolated from bovine spinal cord show high affinity binding to and activation of the μ opiate receptor, but a gene coding for endomorphin has not been identified. Nonetheless we constructed a synthetic endomorphin-2 gene by substituting the endomorphin coding sequence for the enkephalin coding sequences in the proenkephalin gene, with a glycine residue added at the end of each tetrapeptide to direct C-terminal amidation. The identity of the transgene product was confirmed by HPLC followed by radioimmunoassay, and by mass spectroscopy of the peptide released into the medium following transduction of DRG neurons in vitro (unpublished data). Subcutaneous inoculation of the endomorphin expressing vector into the punch surface of the hind foot of rats 7 days prior to injection of complete Freund’s adjuvant (CFA) resulted in a substantial and statistically significant reduction in mechanical allodynia that lasted for 2 weeks after CFA injection (3 weeks after vector inoculation) (Fig. 2). The antiallodynic effect of endomorphin expression was reversed by intrathecal inoculation of naloxone methiodide. In the spinal nerve ligation model of neuropathic pain inoculation of the endomorphin expressing HSV vector resulted in a production of mechanical allodynia and thermal hyperalgesia that was similar in magnitude to the effect produced by the enkephalin expressing vector. The antiallodynic effect of the endomorphin expressing vector was blocked by intrathecal injection of the selective μ opiate receptor antagonist CTOP (D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-amide).
Fig. 2.
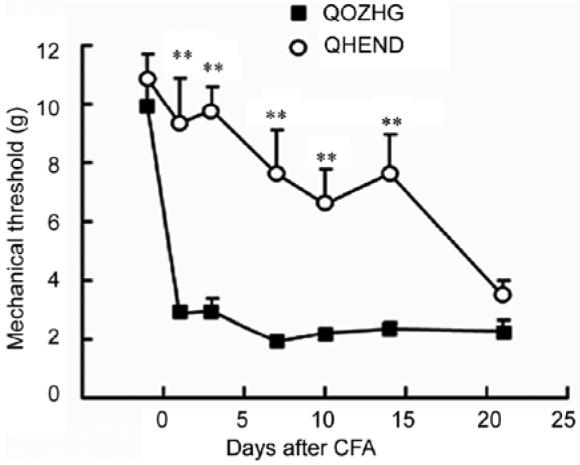
Subcutaneous inoculation of the endomorphin-2 expressing vector (QHEND) one week prior to complete Freund’s adjuvant (CFA) delays the onset of mechanical allodynia. The loss of effect by 4 weeks after vector inoculation (3 weeks after CFA) corresponds to the known timecourse of HSV mediated transgene expression driven by the HCMV IEp. QDZHG: the control vector.
The magnitude of the analgesic effects of both the enkephalin-expressing and the endomorphin-expressing vectors are substantial in the models of inflammatory pain, the effect on mechanical allodynia was less than a 50% amelioration of the mechanical threshold. An emerging literature has suggested that in the presence of peripheral nerve injury there is a specific reduction in GABAergic inhibitory tone that correlates with the reduction in neuropathic pain. We therefore constructed a vector to express glutamic acid decarboxylase (GAD), the enzyme that catalyzes the conversion of glutamate to GABA (gamma amino butyric acid) [8]. In preliminary studies we showed that the vector produces the GAD protein in DRG neurons in vivo, and that following subcutaneous inoculation of the vector GAD67 protein is transported to the afferent terminals of DRG neurons in the dorsal horn spinal cord. DRG neurons transduced in vitro release GABA in a constitutively active manner that appears to be mediated by the GABA transporter running in the reverse direction, since it can be substantially inhibited by the GABA-transporter blocker NO-711 [9].
Subcutaneous inoculation of vector into the hind feet of rats 7 days after selective spinal nerve ligation produced an antiallodynic effect that essentially returned the mechanical threshold to normal levels[10]. The timecourse of the effect of GAD-mediated GABA expression was similar to the timecourse that we observed with the vectors expressing enkephalin, which employed the same HCMV IEp to drive transgene expression. Vector-mediated expression of GABA also substantially reduced thermal hyperalgesia in the spinal nerve ligation model of peripheral neuropathic pain. We also examined the effect of the GAD-expressing vector in the model of below-level central neuropathic pain caused by lateral hemisection of the spinal cord. Inoculation of the vector subcutaneously into the plantar surface of the foot of animals 1 week after T9 lateral hemisection resulted in a statistically significant reduction in both mechanical allodynia and thermal hyperalgesia in the hind limbs[8] (Fig. 3). The analgesic effects of the vector were partially blocked by intrathecal administration of either bicuculline or phaclofen, suggesting that these effects are mediated by both GABAA and GABAB receptors in the spinal cord.
Fig. 3.
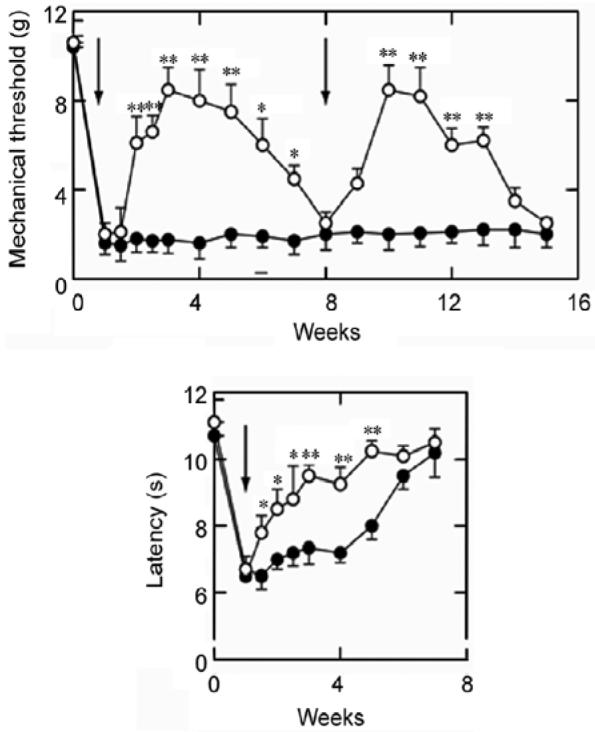
Subcutaneous inoculation of the GAD-expressing vector (QHGAD67) one week after selective L5 spinal nerve ligation (vertical arrow) provides a substantial antiallodynic and anti-hyperalgesia effect that can be reestablished by reinoculation.
All of the experiments that described above used the nonreplicating HSV vector to express inhibitory neurotransmitters to modulate nociceptive neurotransmission at the level of the dorsal horn of spinal cord. But several lines of recent evidences have come together to indicate that a “neuroinflammatory” reaction involving glia in the dorsal horn contributes to the phenotypic changes that are characteristic of chronic neuropathic pain. We therefore constructed a nonreplicating HSV vector to express the antiinflammatory cytokine interleukin-4 (IL-4)[11]. IL-4 is a prototypical anti-inflammatory cytokine that drives T cells towards a Th2 phenotype and reduces expression of pro-inflammatory cytokines. Like the inhibitory neurotransmitters, cytokines are highly potent bioactive peptides that have effects on a number of different systems and are therefore difficult to administer systemically without causing major “off target” adverse events.
Vector S4IL4 is deleted in both copies of the HSV IE gene ICP4 and contains a human IL-4 coding sequence in the HSV UL41 locus under the control of the HSV ICP4 promoter. DRG neurons in vitro transduced with S4IL4 release substantial amounts of IL-4 into the medium, and subcutaneous inoculation of the vector into the hind foot results in the expression of IL-4 in DRG neurons in vivo. In the selective L5 spinal nerve ligation model of neuropathic pain, subcutaneous inoculation of the IL-4 expressing vector one week after spinal nerve ligation resulted in a substantial and statistically significant reduction in both mechanical allodynia and thermal hyperalgesia[11]. Like the effects of the HSV vectors expressing inhibitory neurotransmitters, the IL-4 expressing vector blocked the induction of c-fos in dorsal horn neurons by non-noxious touch. Expression of IL-4 from the vector also blocked the phosphorylation of the MAP kinase p38 in microglia of the dorsal horn, and prevented the increases in IL-1β and PGE2 that occur in the dorsal horn following spinal nerve ligation.
In order to determine whether expression of the anti-inflammatory cytokine could prevent the development of neuropathic pain, we inoculated the vector 7 days prior to spinal nerve ligation. In this case, the onset of mechanical allodynia and thermal hyperalgesia was delayed for a period of weeks[11], corresponding to the timecourse of expression that we observed in the experiments, in which the vector was inoculated after the spinal nerve ligation. These results suggest that while the neuroinflammatory glial reaction participates in the development of chronic pain behaviors, it does not simply establish that phenotype, but rather continued neuroinflammatory processes are involved in the perpetuation of the pain.
We constructed a second anti-inflammatory HSV vector to express the soluble truncated p55 tumor necrosis factor (TNF) α receptor (sTNFR) that serves to naturally terminate the action of TNFα[12]. Expression of the gene product was confirmed in vitro and in vivo. Subcutaneous inoculation of the TNFα expressing vector seven days after spinal nerve ligation resulted in a reduction in mechanical allodynia and thermal hyperalgesia (unpublished data) that was similar in magnitude to the response that we observed following inoculation of the IL-4 expressing vector. Expression of the p55 TNFR resulted in a reduction in phosphorylation of p38 and a reduction in the expression of TNFα and IL-1β in the dorsal horn of spinal cord. Like the effects of vector mediated IL-4, inoculation of the sTNFR-expressing vector 1 week prior to the ligation delayed the onset of mechanical allodynia and thermal hyperalgesia but did not prevent the ultimate development of these phenomena. The sTNFR-expressing vector also provided an analgesic effect against below-level central neuropathic pain in the lateral hemisection model of central neuropathic pain[12].
HSV-mediated gene transfer has been shown to be safe following direct intracranial inoculation into glioblastoma multiformae in several completed phase 1 and phase 2 human trials, but there have been no previous studies of HSV-mediated gene transfer for the treatment of pain in human patients. We therefore examined the effect of vector mediated veggie expression in the osteogenic sarcoma model of pain caused by cancer in bone. Inoculation of each of the vectors resulted in a substantial reduction in spontaneous pain related behaviors in these mice with a check sarcoma in the distal femur (Fig. 4). In this model, proenkephalin proved to be the most effective vector[13], while the GAD-expressing vector was far less effective. We are therefore in the process of proposing a phase 1 safety and dose finding study in patients with cancer metastatic to a vertebral body causing pain that is unresponsive to maximal conventional management. The vector will be inoculated intradermally in the doorman told that corresponds to the radicular distribution of the pain. The results of this phase 1 study will set the stage for later phase 2 efficacy studies of the opioid peptide expressing vectors in cancer or inflammatory pain, and the GAD-expressing vector in neuropathic pain.
Fig. 4.

The effect of vector inoculation on spontaneous and dilatory pain in mice with osteogenic sarcoma in the head of the femur (inset).
Increasing understanding of the anatomic pathways of nociception and pathogenic mechanisms underlying the development of chronic pain have led to the identification of products that may be used to modulate chronic pain. We have shown that recombinant replication defective HSV based vectors can be used to deliver genes to DRG by subcutaneous inoculation in the skin. HSV vectors expressing opioid peptides provide an analgesic effect in models of inflammatory and cancer related pain. An HSV based vector expressing GAD to produce GABA is particularly effective in models of central and peripheral neuropathic pain. Vectors expressing anti-inflammatory peptides can also be used in these models. The path to a human trial has been defined; the experience over the next few years will determine whether this approach will have utility in the treatment of chronic pain in human patients.
REFERENCES
- 1.Ji RR, Woolf CJ. Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis. 2001;8(1):1–10. doi: 10.1006/nbdi.2000.0360. [DOI] [PubMed] [Google Scholar]
- 2.Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413(6852):203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- 3.Borsook D, Ploghaus A, Becerra L. Utilizing brain imaging for analgesic drug development. Curr Opin Investig Drugs. 2002;3(9):1342–1347. [PubMed] [Google Scholar]
- 4.Casey KL, Lorenz J, Minoshima S. Insights into the pathophysiology of neuropathic pain through functional brain imaging. Exp Neurol. 2003;184(Suppl 1):S80–88. doi: 10.1016/j.expneurol.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 5.Glorioso JC, Fink DJ. Herpes vector-mediated gene transfer in treatment of diseases of the nervous system. Annu Rev Microbiol. 2004;58:253–271. doi: 10.1146/annurev.micro.58.030603.123709. [DOI] [PubMed] [Google Scholar]
- 6.Goss JR, Mata M, Goins WF, Wu HH, Glorioso JC, Fink DJ. Antinociceptive effect of a genomic herpes simplex virus-based vector expressing human proenkephalin in rat dorsal root ganglion. Gene Ther. 2001;8(7):551–556. doi: 10.1038/sj.gt.3301430. [DOI] [PubMed] [Google Scholar]
- 7.Hao S, Mata M, Goins W, Glorioso JC, Fink DJ. Transgene-mediated enkephalin release enhances the effect of morphine and evades tolerance to produce a sustained antiallodynic effect. Pain. 2003;102:135–142. doi: 10.1016/s0304-3959(02)00346-9. [DOI] [PubMed] [Google Scholar]
- 8.Liu J, Wolfe D, Hao S, Huang S, Glorioso JC, Mata M, Fink DJ. Peripherally delivered glutamic acid decarboxylase gene therapy for spinal cord injury pain. Mol Ther. 2004;10(1):57–66. doi: 10.1016/j.ymthe.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 9.Liu J, Tai C, de Groat WC, Peng XM, Mata M, Fink DJ. Release of GABA from sensory neurons transduced with a GAD67-expressing vector occurs by non-vesicular mechanisms. Brain Res. 2006;1073-1074:297–304. doi: 10.1016/j.brainres.2005.12.091. [DOI] [PubMed] [Google Scholar]
- 10.Hao S, Mata M, Wolfe D, Glorioso JC, Fink DJ. Gene transfer of glutamic acid decarboxylase reduces neuropathic pain. Ann Neurol. 2005;57(6):914–918. doi: 10.1002/ana.20483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hao S, Mata M, Glorioso JC, Fink DJ. HSV-mediated expression of interleukin-4 in dorsal root ganglion neurons reduces neuropathic pain. Mol Pain. 2006;2:6. doi: 10.1186/1744-8069-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng X, Zhou Z, Glorioso JC, Fink DJ, Mata M. Tumor necrosis factor alpha contributes to below-level neuropathic pain after spinal cord injury. Ann Neurol. 2006;59:843–851. doi: 10.1002/ana.20855. [DOI] [PubMed] [Google Scholar]
- 13.Goss JR, Harley CF, Mata M, O’Malley ME, Goins WF, Hu X, Glorioso JC, Fink DJ. Herpes vector-mediated expression of proenkephalin reduces pain-related behavior in a model of bone cancer pain. Ann Neurol. 2002;52:662–665. doi: 10.1002/ana.10343. [DOI] [PubMed] [Google Scholar]