

Abstract
The flexibility of a promising protein target, human heat shock protein 90 (Hsp90), is investigated by molecular dynamics simulations. These simulations focus on: (i) the interactions between the protein and conserved water molecules; and (ii) the interactions of the ligand PU3, the conserved water molecules and the protein. This is followed by a virtual screening docking study of the PU3 family of compounds and Hsp90 incorporating several conserved water molecules.
Keywords: molecular dynamics, drug design, Hsp90
1. Introduction
The 90 kDa heat shock protein (Hsp90) family plays an important role in the folding and activation of a range of client proteins involved in cell cycle regulation, steroid hormone responsiveness and signal transduction (Stebbins et al. 1997; Roe et al. 1999; Piper 2001; Maloney & Workman 2002; Jez et al. 2003; Neckers 2003; Wright et al. 2004; Chiosis et al. 2006; Sharp 2007; Eccles et al. 2008; Taldone et al. 2008). Hsp90 is a molecular chaperone whose association is required for the stability and function of multiple mutated, chimeric and over-expressed signalling proteins that promote cancer cell growth and/or survival. Hsp90 client proteins include mutated p53, Bcr-Abl, Raf-1, Akt, HER2/Neu (ErbB2) and HIF-1α.
It has been proposed that Hsp90 inhibitors, by interacting specifically with a single molecular target, cause the destabilization and eventual degradation of Hsp90 client proteins. As such they have shown promising levels of anti-tumour activity in preclinical model systems, and one Hsp90 inhibitor, 17-AAG, is currently in Phase II clinical trials (Sharp 2007). In addition, several synthetic Hsp90 inhibitors are currently in Phase I evaluation, including a purine-scaffold agent CNF-2024 (Chiosis et al. 2006). Others, NVP-AUY922 (Eccles et al. 2008) and SNX-5422 (Taldone et al. 2008) developed by Serenex and Novartis, have also recently moved into clinical evaluation. Hsp90 inhibitors are unique in that, although they are directed towards a specific molecular target, they simultaneously inhibit multiple signalling pathways on which cancer cells depend for growth and survival. In addition, anti-cancer selectivity may derive from the simultaneous combinatorial effects of Hsp90 inhibitors on multiple cancer targets and pathways. Therefore, Hsp90 is an ideal protein target for anti-cancer research and this activity has been reviewed recently (Piper 2001; Maloney & Workman 2002; Neckers 2003).
Known Hsp90 inhibitors include: geldanamycin (GM; Stebbins et al. 1997), 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17-DMAG; Jez et al. 2003), 17-allylamino-17-demethoxygeldanamycin (17-AAG; NSC 330507; Jez et al. 2003), herbimycin A (NSC 305978; Piper 2001), radicicol (Roe et al. 1999) and PU3 (Wright et al. 2004). These represent a wide range of structures (electronic supplementary material).
Several crystal structures of Hsp90 are available (Prodromou et al. 1997a,b; Stebbins et al. 1997; Obermann et al. 1998; Roe et al. 1999; Jez et al. 2003; Soldano et al. 2003; Wright et al. 2004); in all cases the active site contains some bound water molecules. The structural, dynamical and functional importance of water molecules for biomacromolecular structure and recognition is well appreciated. Water is known to contribute significantly to the stability of biomacromolecules and to play a crucial role in molecular association (Westhof 1993). Complex structures have shown that waters can be very important in mediating the interaction between ligand and protein (Westhof 1993; Ladbury 1996; Minke et al. 1999; Ni et al. 2001; Glen & Allen 2003). That particular structural waters are crucial for the binding affinity or specificity of protein–ligand complexes is an important issue in the design of new ligands (Ladbury 1996).
To establish the flexibility of the protein structure and the stability of the conserved waters in the Hsp90 structures, molecular dynamics simulations on the unliganded protein were carried out. This also raised the question of whether ligand binding required water bridging via these conserved waters, which we have investigated by simulation of the complex with PU3. Thus we have shown which water molecules are persistent components of the binding site.
Conserved water molecules play an important role in the interactions between the Hsp90 and ligands. Having defined the binding site based on the protein structure with several conserved water molecules, virtual docking of a series of ligands of PU3 family molecules was performed, in order to show how conserved water molecules affect the interaction between the protein and the different ligands, which have the same scaffold and different substituents.
1.1 Hsp90 crystal structures
The Hsp90 family is composed of four members: Hsp90α, Hsp90β (cytoplasm), Grp94 (endoplasmic reticulum) and Trap-1 (mitochondria).
The N-terminal domain (hereafter Nt-Hsp90) has been studied by crystallography and contains an unusually shaped ATP binding cleft, known as the Bergerat fold, responsible for the ATPase activity important for function (Bergerat et al. 1997). The Nt-Hsp90 domain is shown in figure 1. In this work, we have focused on Apo-Nt-Human Hsp90α (code 1YES; Stebbins et al. 1997) and human Hsp90α in complex with the PU3 (1UY6; Wright et al. 2004).
Figure 1.

Crystal structure of human Hsp90α (code 1YES).
A number of crystal structures of Apo-Nt-Human Hsp90α have been determined showing two different conformations: the ‘open’ conformation (code 1YES) and the ‘closed’ conformation (1YER) as reported by Stebbins et al. (1997), and independently by Wright et al. (2004) (1UYL). It was observed that the structures of 1UYL and 1YER are essentially identical. The main variation between the open and closed structures is in the conformation of residues 104–111 (as shown in fig. 1A of Wright et al. 2004). These comparisons demonstrate that there is considerable plasticity in this region of the structure, which is at the entrance to the ATP binding site. Some of the residues seen as important for binding to ligands are in this loop, e.g. Leu 107.
Crystal structures are also available for a series of complexes between Nt-Hsp90α and the ligands PU3 (Wright et al. 2004), ADP (Prodromou et al. 1997a,b), geldanamycin (Stebbins et al. 1997) and radicicol (Roe et al. 1999). The key interaction with Asp93 is preserved in all complexes as part of a network of hydrogen bonds around the carboxylate of Asp93 involving Asn51, Ser52, Thr184, Gly97 and four water molecules consistently found in the same positions (as shown in fig. 3 of Wright et al. 2004). Three of these waters are hydrogen bonded to ADP, PU3 and geldanamycin. The important role played by the crystallographic waters in the recognition of the ligands by Hsp90 is further supported by the almost identical location of these water molecules across all the structures so far available.
2. Methods
2.1 Molecular dynamics simulation
The dynamic behaviour of the protein and ligand in an aqueous solution was investigated by molecular dynamics simulation at 300 K. This involved solvation of a single molecule of the ligand (or without ligand) and most of the protein in a water sphere of radius 25 Å containing TIP3P (Jorgensen et al. 1983) water molecules around the active site. The solvent sphere was centred at the point of the coordinates of the CG (gamma carbon) of TYR139 of the protein. This assembly was partitioned into a 21 Å/25 Å reaction region/buffer region for stochastic boundary molecular dynamics (Brooks & Karplus 1983). The solvent was first minimized and subjected to a 22.5 ps equilibration period at 300 K, during which the solute was restrained. A 10 ps unrestrained equilibration period was then performed followed by an unrestrained production simulation at 300 K. The duration of production simulation was 1.5 ns. The molecular dynamics simulations were performed using the CHARMM program (Brooks et al. 1983) (version 27b41) with the CHARMM22 force field (MacKerell 1998) and using the SHAKE (Ryckaert et al. 1977) algorithm to constrain bonds to hydrogen, allowing a time step of 1 fs. The simulations were performed using a deformable boundary potential with a Langevin friction coefficient of 62.0 ps−1 applied to the water oxygen atoms (Brunger et al. 1984).
The conformation of the crystal structure of the human Hsp90 protein of the unliganded form (code: 1YES) or the liganded form (code: 1UY6) was taken. All the water molecules observed in the crystal structure were retained. The protein (or the protein with the ligand) was then put into the solvated system and Langevin molecular dynamics simulation was performed.
2.1.1 Molecular dynamics simulation for Apo-Nt-Human Hsp90α
The open conformation of the crystal structure of the unliganded form of human Hsp90 protein (code: 1YES) was used, and all the 246 water molecules from its crystal structure were retained. The molecular dynamics simulation involved solvation of most of the protein in a water sphere of radius 25 Å containing 1208 TIP3P (Jorgensen et al. 1983) water molecules after removing those that overlap.
2.1.2 Molecular dynamics simulation for Nt-Human Hsp90α in complex with PU3
The crystal structure of the protein in complex with the ligand PU3 (code: 1UY6) was used with all 263 water molecules from its crystal structure. The molecular dynamics simulation involved solvation of a single molecule of the ligand and most part of protein in a water sphere of radius 25 Å containing 1207 TIP3P (Jorgensen et al. 1983) water molecules.
2.2 Docking study
To understand the interactions between the protein and the small molecules, to find the general principles of the interactions and to give some hints for drug design, docking studies were performed using the program LigandFit in the Cerius2 package (2003) (Venkatachalam et al. 2003).
The interaction between PU3 and human Hsp90α was simulated by a docking study starting from the crystal structure PU3·Hsp90α (code 1UY6). In this work, the docking study was carried out with the protein and the several conserved water molecules, to test whether the correct docking result can be obtained by including the conserved water molecules. The protein was prepared by including several important water molecules, and removing all the other water molecules and the ligand. Then a set of PU3 family molecules (Chiosis et al. 2002; Wright et al. 2004) containing a purine scaffold, which have shown good experiment binding affinity with the protein Hsp90, were docked into that same protein.
In LigandFit there are two ways of searching for binding sites (Venkatachalam et al. 2003; Cerius2 2003): (i) when the protein and the ligand are known, the sole binding site can be found using the ‘docked ligand’ mode; and (ii) protein shape only, which employs a cavity finding algorithm for detecting invaginations in the protein as possible candidate active site regions. In this case, several sites can be found. By comparison with the crystal structure, the active binding site was selected.
The binding site search was carried out using a protein-shape based cavity search method with an eraser size of 7.0 Å. It was found that the active binding site corresponds to the largest cavity site. This was also confirmed by the crystal structure. The found binding site was then enlarged several times and adjusted until it covered the whole ligand.
In LigandFit, different ligand conformations were generated for docking and then filtered by Monte Carlo conformational search. Selection of a pose was based on the comparison of the shape of the ligand conformation with that of the site. The quality of docking was evaluated using a grid-based energy calculation for estimating the interaction energy of the ligand with the protein.
The following parameters were adopted: CFF force field (Peng et al. 1997; Ewig 2001) (cff1.02) and partial charges assigned using the Gasteiger method (Gasteiger & Marsili 1978) as implemented in Cerius2. The binding site was partitioned into four parts corresponding to a total of 10 binding sites (modes). The number of trials was fixed to 25 000; the maximum number of conformers saved for each ligand after docking was 20. Rigid body minimization of the ligand based on dock energy after docking using shape alignment was adopted. All docked conformations obtained for each molecule were clustered using a leader algorithm in Cerius2 with an RMS threshold of 1.5 Å.
3. Results and discussion
3.1 Molecular dynamics simulation of Apo-Nt-Human Hsp90α
In the protein crystal structure the B-factors (Brooks et al. 1988), which are directly related to the mean-square atomic fluctuations Δr2, are calculated by the following equation:
| (3.1) |
To reveal the most flexible regions of the protein structure, B-factors (Brooks et al. 1988) were calculated for the Cα atoms (averaged over 1500 ps time frames) of 1yes. As shown in figure 2, the B-factors display major peaks in wide regions of residues (21–40, 98–146, 165–168, 190–195) with the highest B-factors in the region of 99–102, which is the position of α-helix 4 of the protein. The very low B-factors between residues 50 and 97 correspond to regions of the protein close to or beyond the boundary of the simulation. In figure 2, the experimental B-factor values for the Cα atoms from the protein crystal structure are also shown.
Figure 2.

Calculated (solid curve) and the experimental (dotted curve) B-factors for the Cα atoms of the protein (code: 1YES). The calculated value was averaged over 1500 ps time frames of the molecular dynamics simulation.
Examination of the φ and ψ traces within the flexible region shows that a significant conformational change takes place at approximately 0.75 ns. These traces are shown for some of the more important residues in the electronic supplementary material. It is observed that the important conformational changes take place at residues Met98, Leu107, Val136, Phe138 and Tyr139.
Figure 3 shows the mean-square fluctuation of the water oxygens averaged over the entire simulation. Waters 1–246 originated from the crystal structure; the remainder were added in the solvation step of the simulation setup. Examination of these values shows two distinct classes of behaviour: a cluster of water molecules with Δr2 less than 3 Å2, and the remainder showing fluctuations between 5 and 20 Å2. Strikingly, this appears to be independent of the origin of the water molecules, suggesting that most of the water molecules originating from the crystal have exchanged and equilibrated with the bulk solvent.
Figure 3.

Calculated mean-square fluctuation of the oxygen atoms of all the solvent water molecules in the molecular dynamics simulation for protein (with water) averaged over 1500 ps time frames. (In the graph the value of the line that is vertical to the x-axis is 246, the points on the left-hand side of the line represent the fluctuation of the crystal water molecules, the points on the right-hand side of it represent the fluctuation of the added solvent water molecules; the value of the line that is vertical to the y-axis is 3, the points under it mean that the fluctuation is very small.)
Of the 37 waters that show small fluctuations, all but six appear to be close to the boundary of the simulation sphere and their apparent lack of motion must be related directly to the boundary potential. The remaining six water molecules are of considerable interest. Three of these correspond to the conserved water molecule positions seen in all Hsp90α crystal structures, and in the complex structures these form a bridge between the purine ring and the protein. A fourth water molecule is also found to be strongly constrained within a network of hydrogen bonds, but comparison with crystal structure data shows that it would be displaced by a bound ligand. During the simulation the molecule corresponding to water 527 in the pdb structure 1yes moves into the bulk; however its position is taken by another water molecule. The remaining ‘immobile’ waters form several hydrogen bonds to Tyr139, Asn106 and Ile110, Gly98, Asn105. (see figure 4a). The distances between these water molecules and their main contacts on the protein chain are shown in figure 4b.
Figure 4.

(a) The structure of the protein Hsp90α (code 1YES, in yellow) and the position of the six fixed water molecules during the molecular dynamics simulation (red: water 515, 302 and 365, which correspond to the conserved water molecules positions seen in all Hsp90 crystal structures; green: water 376, 371 and 525). (b) Distances between the fixed waters and some residues on the proteins. Distance between the water 365 and OG of Ser52 (shown in red), between the water 515 and OD1 of Asp93 (shown in green), between the water 302 and OD2 of Asp93 (shown in purple), and between water 376 and OD2 of Asp93 (shown in black).
3.2 Molecular dynamics simulation for Nt-Human Hsp90α in complex with PU3
B-factors were calculated for the Cα atoms (averaged over the entire simulation of 1500 ps) of the protein in complex with the ligand PU3 (code: 1UY6), revealing the most flexible regions of the protein structure. As shown in figure 5, the calculated B-factors display major peaks across a wide range of residues (101–118, 119–128, 129–136) with the highest B-factors in the region of 121–126, which corresponds to the position of α-helix 5, α-helix 6 and especially the loop between them. The very low B-factors between residues 50 and 100 correspond to regions of the protein close to or beyond the boundary of the simulation. The calculated values for helix 5 and 6 are very close to the experimental ones.
Figure 5.

Calculated (solid curve) and the experimental (dotted curve) B-factors for the Cα atoms of the protein (code: 1UY6). The calculated value was averaged over 1500 ps time frames of the molecular dynamics simulation.
Examination of the φ and ψ traces within the flexible region shows that a significant conformational change takes place at approximately 1.25 ns. These traces are also shown in the electronic supplementary material for some of the more important residues. The initial and the final structures are superimposed (electronic supplementary material). It was found that during the simulation, the conformation of the butyl group of the ligand PU3 changed, and a dramatic conformational change also takes place on Val136, which participates in hydrophobic interactions with the butyl group of the ligand PU3, via both in its backbone and side chain (see figure 6).
Figure 6.

(a,b)The changes of the dihedral angle of CG1 : CB : CA : N of Val136, and the dihedral angle between the N9 and the three continuous connecting carbon atoms of the butyl group of PU3.
Figure 7 shows the mean-square fluctuation of the water oxygens averaged over the entire simulation. Waters 1–263 originated from the crystal structure; the remainder were added in the solvation step of the simulation setup. Examination of these values shows two distinct classes of behaviour: a cluster of water molecules with Δr2 less than 3 Å2, and the remainder showing fluctuations between 5 and 20 Å2. Again this appears to be independent of the origin of the water molecules, suggesting that most of the water molecules from the crystal structure have exchanged and equilibrated with the bulk solvent.
Figure 7.

Calculated mean-square fluctuation of the oxygen atoms of all the solvent water molecules in the molecular dynamics simulation for protein (with water and ligand) averaged over 1500 ps time frames. (In the graph the value of the line that is vertical to the x-axis is 263, the points on the left-hand side of the line represent the fluctuation of the crystal water molecules, the points on the right-hand side of it represent the fluctuation of the added solvent water molecules; the value of the line that is vertical to the y-axis is 3, the points under it mean that the fluctuation is very small.)
Of the 46 waters that show small fluctuations, all but eight appear to be close to the boundary of the simulation. These remaining eight water molecules are of considerable interest as four correspond to the four conserved water molecules positions seen in all Hsp90 crystal structures. The remaining four immobile waters are found at the position near to the methoxy groups of the PU3, which make some hydrogen bond interactions with the methoxy group of the PU3 and form several hydrogen bonds to Gln23, Leu103 and Tyr139 (see figure 8a). The distances between these water molecules, PU3 and their main contacts on the protein chain are shown in figure 8b. These four conserved water molecules are more stable at their positions than the others. Such information should be taken into account in future inhibitor design.
Figure 8.

(a) The structure of the protein Hsp90 (code: 1UY6, in yellow), PU3 (in purple) and the position of the eight fixed waters during the molecular dynamics simulation (red: water 56, 133, 137 and 262; green: water 10, 148, 149 and 166). (b) Distance between the water 56 and OG of Ser52 (shown in red), between the water 133 and OD2 of Asp93 (shown in green), between the water 262 and ND2 of Asn51 (shown in purple), and between water 137 and the N1 of the purine of the ligand PU3 (shown in black).
3.3 Docking results for PU3 and PU3 family compounds
For each of the 12 PU3 family molecules (see table 1), the crystal structure in the complex with Hsp90α was selected (Wright 2004). The three-dimensional structure was then minimized in the CFF force field (Brunger et al. 1984; Venkatachalam et al. 2003) (cff1.02) and the structure was assigned partial charges using the Gasteiger method (Cerius2 2003) as implemented in Cerius2. Then, they were used to dock to a protein, which was obtained from the crystal structure PU3·Hsp90α (code 1UY6). After docking, the best conformation for each molecule was selected manually. The corresponding RMS error between the same scaffold (as shown in table 1, excluding all hydrogen atoms) of the ligand and that of the PU3 (obtained from the crystal structure PU3·Hsp90α (code 1UY6)) was calculated and shown in table 1. The docking results for the ligands and the protein Hsp90α with four or eight conserved water molecules are represented in table 1. For PU3 and most of the other molecules, the docking results to the protein Hsp90α with four or eight water molecules are good. In table 1, RMS>1 Å means that the ligand binding is not very stable due to some substitution. It was also found that, typically, the four conserved water molecules are more important in reproducing the experimental structures than other water molecules.
Table 1.
The docking results for the 12 PU3 family molecules.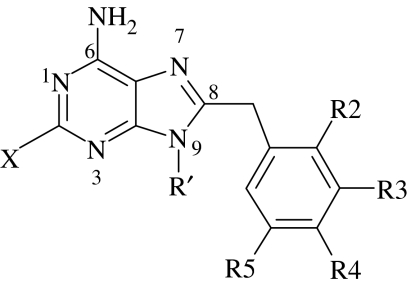
| Compounds | X | R2 | R3 | R4 | R5 | R′ | RMSa (Å) | RMSb (Å) |
|---|---|---|---|---|---|---|---|---|
| 1 PU3 | H | H | OMe | OMe | OMe | n-butyl | 0.34 | 0.83 |
| 2 | H | H | H | OMe | H | n-butyl | 0.43 | 0.80 |
| 3 | H | H | OMe | H | H | n-butyl | 3.17 | 0.88 |
| 4 | H | H | OCH2O | bridge | H | n-butyl | 0.46 | 0.78 |
| 5 | H | OMe | H | H | OMe | n-butyl | 0.47 | 0.68 |
| 6 | H | Cl | OMe | OMe | OMe | n-butyl | 0.59 | 0.89 |
| 7 | H | Cl | OMe | OMe | OMe | 1-pentynyl | 3.66 | 3.90 |
| 8 | F | Cl | OMe | OMe | OMe | 1-pentynyl | 3.01 | 1.07 |
| 9 | F | OMe | H | H | OMe | H | 0.73 | 2.87 |
| 10 | F | OMe | H | H | OMe | n-butyl | 0.60 | 1.21 |
| 11 | F | OMe | H | H | OMe | 1-pentynyl | 0.53 | 1.25 |
| 12 | F | H | OCH2O | bridge | H | n-butyl | 0.45 | 0.53 |
RMS: docking result when four conserved water molecules are included in the protein.
RMS: docking result when eight conserved water molecules are included in the protein.
4. Conclusions
Several conserved water molecules exist at the binding site of protein Hsp90α which participate in the interactions between the protein Hsp90α and its inhibitor PU3. Simulation suggests they are very difficult to replace. As such the conserved water molecules determine the shape of the protein active binding site, and become a major factor in rational drug design.
The simulations also highlight the fact that Hsp90 is a difficult protein target because of the flexibility of the helix (residues 101–136 for 1UY6) and because there are several conserved waters participating in the interaction between it and its inhibitors. The simulations reveal that only four conserved water molecules (water molecules labelled as 56, 133, 137 and 262) are important for new inhibitor design, because their positions are stabilized when the conformation of the protein is changed.
Acknowledgements
This work has been supported by a grant from the National Foundation for Cancer Research.
Footnotes
One contribution of 9 to a Theme Supplement ‘Biomolecular simulation’.
Supplementary Material
additional figures
References
- Bergerat A., deMassy B., Gadelle D., Varoutas P.C., Nicolas A., Forterre P. An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature. 1997;386:414–417. doi: 10.1038/386414a0. [DOI] [PubMed] [Google Scholar]
- Brooks C.L., Karplus M. Deformable stochastic boundaries in molecular dynamics. J. Chem. Phys. 1983;79:6312–6325. doi: 10.1063/1.445724. [DOI] [Google Scholar]
- Brooks C.L., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S., Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 1983;4:187–217. doi: 10.1002/jcc.540040211. [DOI] [Google Scholar]
- Brooks C.L., III, Karplus M., Pettitt B.M. Wiley; New York, NY: 1988. Proteins: a theoretical perspective of dynamics, structure and thermodynamics. [Google Scholar]
- Brunger A., Brooks C.L., Karplus M. Stochastic boundary conditions for molecular dynamics simulations of ST2 water. Chem. Phys. Lett. 1984;105:495–500. doi: 10.1016/0009-2614(84)80098-6. [DOI] [Google Scholar]
- Cerius2 2003 Cerius2, v. 4.8. San Diego, CA: Accelrys, Inc. (http://www.accelrys.com/)
- Chiosis G., Lucas B., Shtil A., Huezo H., Rosen N. Development of a purine-scaffold novel class of Hsp90 binders that inhibit the proliferation of cancer cells and induce the degradation of Her2 tyrosine kinase. Bioorgan. Med. Chem. 2002;10:3555–3564. doi: 10.1016/S0968-0896(02)00253-5. [DOI] [PubMed] [Google Scholar]
- Chiosis G., Caldas L.E., Solit D. Heat shock protein-90 inhibitors: a chronicle from geldanamycin to today's agents. Curr. Opin. Investig. Drugs. 2006;7:534–541. [PubMed] [Google Scholar]
- Eccles S.A., et al. NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008;68:2850–2860. doi: 10.1158/0008-5472.CAN-07-5256. [DOI] [PubMed] [Google Scholar]
- Ewig C.S., et al. Derivation of class II force fields. VIII. Derivation of a general quantum mechanical force field for organic compounds. J. Comp. Chem. 2001;22:1782–1800. doi: 10.1002/jcc.1131. [DOI] [PubMed] [Google Scholar]
- Gasteiger J., Marsili M. A new model for calculating atomic charges in molecules. Tetrahedron Lett. 1978;34:3181–3184. doi: 10.1016/S0040-4039(01)94977-9. [DOI] [Google Scholar]
- Glen R.C., Allen S.C. Ligand-protein docking: cancer research at the interface between biology and chemistry. Curr. Med. Chem. 2003;10:763–777. doi: 10.2174/0929867033457809. [DOI] [PubMed] [Google Scholar]
- Jez J.M., Chen J.C., Rastelli G., Stroud R.M., Santi D.V. Crystal structure and molecular modeling of 17-Dmag in complex with human Hsp90. Chem. Biol. 2003;10:361–368. doi: 10.1016/S1074-5521(03)00075-9. [DOI] [PubMed] [Google Scholar]
- Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- Ladbury J.E. Just add water! The effect of water on the specificity of protein–ligand binding sites and its potential application to drug design. Chem. Biol. 1996;3:973–980. doi: 10.1016/S1074-5521(96)90164-7. [DOI] [PubMed] [Google Scholar]
- MacKerell A.D., Jr, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Maloney A., Workman P. HSP90 as new therapeutic target for cancer therapy: the story unfolds. Expert. Opin. Biol. Theory. 2002;2:3–24. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- Minke W.E., Diller D.J., Hol W.G.J., Verlinde C.L.M.J. The role of waters in docking strategies with incremental flexibility for carbohydrate derivatives: heat-labile enterotoxin, a multivalent test case. J. Med. Chem. 1999;42:1778–1788. doi: 10.1021/jm980472c. [DOI] [PubMed] [Google Scholar]
- Neckers L. Development of small molecule Hsp90 inhibitors: utilizing both forward and reverse chemical genomics for drug identification. Curr. Med. Chem. 2003;10:733–739. doi: 10.2174/0929867033457818. [DOI] [PubMed] [Google Scholar]
- Ni H.H., Sotriffer C.A., McCammon J.A. Ordered water and ligand mobility in the HIV-1 integrase-5CITEP complex: a molecular dynamics study. J. Med. Chem. 2001;44:3043–3047. doi: 10.1021/jm010205y. [DOI] [PubMed] [Google Scholar]
- Obermann W.M., Sondermann H., Russo A.A., Pavletich N.P., Hartl F.U. In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J. Cell. Biol. 1998;143:901–910. doi: 10.1083/jcb.143.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z., Ewig C.S., Hwang M.-J., Waldman M., Hagler A.T. Derivation of class II force fields. 4. Van der Waals parameters of alkali metal cations and halide anions. J. Phys. Chem. A. 1997;101:7243–7252. doi: 10.1021/jp964080y. [DOI] [Google Scholar]
- Piper P.W. The Hsp90 chaperone as a promising drug target. Curr. Opin. Investig. Drugs. 2001;2:1606–1610. [PubMed] [Google Scholar]
- Prodromou C., Roe S.M., Piper P.W., Pearl L.H. A molecular clamp in the crystal structure of the N-terminal domain of the yeast Hsp90 chaperone. Nat. Struct. Biol. 1997a;4:477–482. doi: 10.1038/nsb0697-477. [DOI] [PubMed] [Google Scholar]
- Prodromou C., Roe S.M., O'Brien R., Ladbury J.E., Piper P.W., Pearl L.H. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997b;90:65–75. doi: 10.1016/S0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- Roe S.M., Prodromou C., O'Brien R., Ladbury J.E., Piper P.W., Pearl L.H. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comp. Phys. 1977;23:327–341. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Sharp S.Y. Inhibition of the heat shock protein 90 molecular chaperone in vitro and in vivo by novel, synthetic, potent resorcinylic pyrazole/isoxazole amide analogues. Mol. Cancer Ther. 2007;6:1198–1211. doi: 10.1158/1535-7163.MCT-07-0149. [DOI] [PubMed] [Google Scholar]
- Soldano K.L., Jivan A., Nicchitta C.V., Gewirth D.T. Structure of the N-terminal domain of Grp94: basis for ligand specificity and regulation. J. Biol. Chem. 2003;278:48 330–48 338. doi: 10.1074/jbc.M308661200. [DOI] [PubMed] [Google Scholar]
- Stebbins C.E., Russo A.A., Schneider C., Rosen N., Hartl F.U., Pavletich N.P. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997;89:239–250. doi: 10.1016/S0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- Taldone, T., Gozman, A., Maharaj, R. & Chiosis, G. 2008 Targeting Hsp90: small-molecule inhibitors and their clinical development, Curr. Opin. Pharmacol. 8, 370–374. ( 10.1016/j.coph.2008.06.015) [DOI] [PMC free article] [PubMed]
- Venkatachalam C.M., Jiang X., Oldfield T., Waldman M. LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003;21:289–307. doi: 10.1016/S1093-3263(02)00164-X. [DOI] [PubMed] [Google Scholar]
- Westhof E. CRC Press; Boca Raton, FL: 1993. Water and biological macromolecules. [Google Scholar]
- Wright L., et al. Structure-activity relationships in purine-based inhibitor binding to HSP90 isoforms. Chem. Biol. 2004;11:775–785. doi: 10.1016/j.chembiol.2004.03.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
additional figures