

Abstract
Purpose:
This study evaluated epithelial cell death ELISAs that measure circulating Cytokeratin-18 (CK18) in mice bearing Small Cell Lung Cancer (SCLC) xenografts treated with a pro-apoptotic dose of the BH-3 mimetic ABT-737.
Experimental Design:
H146 tumour bearing and non bearing SCID/bg mice were treated with ABT-737 or vehicle control. Plasma collected before and 2-360h after treatment was analysed by M30 (caspase-cleaved CK18) and M65 (intact and cleaved CK18) ELISA. In parallel, tumours were interrogated for cleaved caspase-3 and cleaved CK18 as biomarkers of apoptosis.
Results:
ABT-737-treated tumours regressed by 48h (p<0.01) compared to controls, correlating with increased cleaved CK18 (p<0.01, 6 and 24 h) and increased intact CK18 (p<0.01, 24h). Cleaved CK18 levels decreased below baseline between 72-360h for ABT-737-treated and control mice whilst intact CK18 fell below the level of detection at 8 and 15 days in ABT-737 treated mice only. Apoptosis in tumours reflected changes in circulating CK18 (cleaved caspase-3, p<0.05 at 2h and p<0.001 at 6, 12 and 24h; caspase-cleaved CK18, p<0.05 at 15 days, for drug-treated versus controls).
Conclusions:
ABT-737 caused tumour regression by apoptosis in H146 xenografts that mapped to a drug-specific, early increase in circulating cleaved CK18 that subsequently declined. Circulating, intact CK18 levels correlated with tumour burden. Cleaved caspase-3 and caspase-cleaved CK18 in tumour correlated with treatment (p<0.05, 2 h; p<0.001, 6, 12, 24 h; cleaved caspase-3, p<0.05 15 days; caspase-cleaved CK18) indicating that events in plasma were tumour derived. These circulating biomarker data will be translated to clinical trials where serial tumour biopsies are rarely obtained.
Keywords: Cell Death Biomarkers, M30, M65, Bcl-2, ABT-737
Introduction
The arrival of molecularly targeted agents for the treatment of cancer brings added impetus to pharmacodynamic biomarker qualification and the need for biomarker-enhanced clinical trials where proof of mechanism (drug hits target) and proof of concept (appropriate tumour response is stimulated) are sought (1). Drugs that target components of apoptotic pathway(s) are currently in pre-clinical development and entering early clinical trials (2). Validated, proof of concept biomarkers that report apoptotic cell deaths in tumour, or, as is often the case when tumour is not available, in surrogates such as blood, are therefore, urgently required (3-5). With this in mind, this study evaluates circulating biomarkers of epithelial cell death in a responsive pre-clinical tumour model before and after treatment with ABT-737, a pro-apoptotic Bcl-2 family targeted novel agent (6).
The anti-apoptotic Bcl-2 family of proteins are attractive drug targets as they are frequently over-expressed in many human cancers and can mediate drug resistance (7). Targeting these molecules requires the specific interruption of protein-protein interactions between pro-and anti-apoptotic family members that involve their BH-3 domains (8). The highly potent BH-3 mimetic, ABT-737, is the leading member of a new class of small molecule drugs that is approaching or entering early clinical trials for cancer treatment (6). ABT-737 binds with nanomolar affinity to the BH-3 binding groove of anti-apoptotic proteins Bcl-2, Bcl-xL and Bcl-w, but not to those of Mcl-1 and A1 (6, 9-12). Upon binding, ABT-737 prevents these anti-apoptotic proteins from sequestering pro-apoptotic family members to trigger apoptosis via the intrinsic mitochondrial pathway (6). Pre-clinical studies have shown that ABT-737 sensitises many cancer cell types to conventional therapies in vitro (6, 11, 13-19) and it exhibited single agent activity in vivo in human tumour xenograft models of B-cell Lymphoma and Small Cell Lung Carcinoma (SCLC) (6). The impressive anti-tumour activity in vivo was demonstrated in mice bearing xenografts of a range of SCLC cell lines, including H146, where ABT-737 induced complete regression of 77% H146 tumours when dosed daily at 100 mg/kg/day for 21 days (6). Here, we examine the utility of circulating forms of cytokeratin 18 (CK18) as blood-borne biomarkers of ABT-737-driven tumour cell death by exploiting the well established, ABT-737 sensitive H146 SCLC tumour model.
The potential of CKs as circulating biomarkers of epithelial cell death resides in the knowledge they are not expressed in haematopoietic cells. CKs are expressed in most epithelial cells and in many carcinomas (20, 21) and fragmented/complexed CKs have been detected in the circulation of patients with epithelial malignancies where they have been evaluated as tumour biomarkers (20-23). The M65 and M30 ELISAs detect intact and caspase-cleaved forms of CK18 (Figure 1). The M65 assay detects both full-length and caspase-cleaved CK18 (24) and as such, is proposed as a biomarker of caspase dependent and independent cell death. The M30 assay detects only a CK18 neo-epitope generated following caspase cleavage at position 387-396 and is considered to be a specific assay for epithelial apoptosis (25-27). Several reports propose that levels of caspase-cleaved CK18 are predictive of tumour response to drug treatment (28) and may have prognostic significance (29).
Figure 1. Schematic representation of cytokeratin 18 (CK18) caspase cleavage and the sites for M30 and M65 antibody recognition.
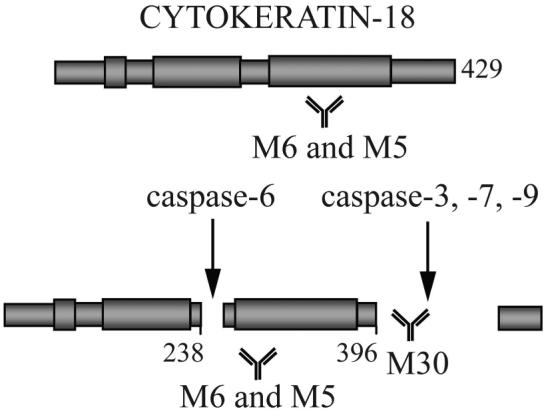
During apoptosis activated caspases-3, -6, -7 and -9 are able to cleave CK18 at specific peptide recognition sites. Caspase cleavage generates a neo-epitope which can be detected using the M30 and M65 assays, thus informing on the levels of apoptosis. In addition, the M65 antibody is also able to detect full length (intact) CK18, and thus provide information on the levels of necrotic cell death. The M65 ELISA uses the M6 antibody as the catcher antibody and M5 as the detection antibody. The M30 ELISA uses M5 as the capture antibody and M30 as the detection antibody.
M30 and M65 data presented here demonstrate that cleaved and intact CK18 are indeed useful blood borne biomarkers of ABT-737 induced tumour cell death and of tumour burden per se as significant correlations between the levels of these circulating biomarkers, tumour apoptosis and tumour regression were established. This study also showed that these circulating biomarkers confirmed absence of ABT-737-induced epithelial toxicity following analysis in non-tumour bearing animals treated with ABT-737. These promising pre-clinical data can now be translated directly to upcoming clinical trials of Bcl-2 family targeted drugs in epithelial tumours.
Materials and Methods
Cell culture
H146 cells were purchased from American Tissue Type collection and were cultured in RPMI supplemented with 10% FCS, 1% sodium pyruvate and 4.5g/L glucose in a 37°C humidified 5% CO2 incubator and routinely checked for mycoplasma infection.
H146 Xenograft studies
All in vivo studies were conducted as described previously (6) in accordance with guidelines established by the internal Institutional Animal Care and Use Committee. Female C.B.-17 SCID/bg (scid-bg) mice were inoculated s.c. with 5×106 NCI-H146 SCLC cells and tumour size was monitored by calliper measurements (V = LxW 2/2). All mice were survival bled via a retro-orbital method 24h before the first drug dose and the blood collected in heparin-coated tubes for determination of baseline antigen level for M65 and M30. Blood was processed to isolate plasma that was stored immediately at −70°C. When average tumour volumes reached approximately 400 mm3, mice were treated daily with i.p. administration of ABT-737 at 100mg/kg or vehicle control. ABT-737 was formulated in 30% propylene glycol, 5% Tween 80, 65% D5W (5% dextrose in water), pH 4–5. A final terminal bleed was conducted at the indicated time points with concomitant harvest of tumour tissue in 10% neutral buffered formalin. Plasma samples were placed in dry ice while in transit from Abbott laboratories to the Paterson Institute for Cancer Research, and upon receipt they were stored at −80°C before analysis within 1 week.
Detection of M65 and M30 antigens
The M65 and M30 (Apoptosense) ELISA kits (Peviva, Sweden) were used for the plasma biomarker analysis (3, 4, 30) incorporating a blocking agent (HBR) modification to permit maintenance of assay dynamic range in a mouse background. HBR functions by binding and removing the cross-reacting heterophillic antibodies which are thought to interfere with the sandwich ELISA by interacting with both the capture and the detection antibodies thus leading to false positive amplification of signal. In brief, 25μl of each sample (standard, QC or plasma sample) was added to each well of a 96 well plate coated with the mouse monoclonal ‘catcher’ antibody. Following this 75μl of HRP-conjugated ‘detection’ monoclonal antibody and 4 μl of the protein blocking agent HBR plus (Scantibodies Laboratory, Inc., Santee, CA) was added per well and samples were incubated at room temperature for 2 hours (M65) or 4 hours (M30) followed by removal of excess conjugate. After adding 200μl of TMB substrate and incubating 20min in the dark, 50μl of 1.0M sulphuric acid was added and the absorbance was read at 540nm. The concentration (U/L) of antigen was calculated based on a standard curve from known antigen concentrations. The dynamic ranges of the two ELISAs are 0-1000 U/L and 0-2000 U/L for the M30 and M65 assays, respectively. However, values below 20 U/L are considered at the limit of detection for both assays and data below this limit are excluded.
In order to account for natural biological variation a baseline measurement of M30 and M65 antigen was recorded for each individual mouse 24 hours prior to receiving either drug or vehicle. These values were then subtracted from subsequent readings for each individual mouse prior to grouping animals for statistical analysis. Mann Whitney U test in addition to one way ANOVA followed by a post hoc Bonferroni multiple range test (to determine where the differences were amongst multiple groups) were conducted on all pre-dose samples in order to test for significant differences in pre-dose data within, and between time-points for each treatment group.
Detection of cleaved caspase-3 and caspase-cleaved CK18 in Tumour
Cleaved Caspase-3
Paraffin-embedded tumour tissue was cut in 3μm sections which were deparaffinised and dehydrated. Sections were then microwaved for 25min in citric acid buffer (10mM, pH 6.0) followed by blocking of endogenous peroxidase by immersing in a 0.3% hydrogen peroxide solution for 30min. The slides were incubated with 10% casein solution for 1h to remove background staining. Slides were then incubated overnight with the primary antibody (anti-Cleaved Caspase-3 (asp175) Ab (Cell Signalling; 9661S) at 4°C in a humidified tray. After incubation, slides were washed in PBS and Goat Anti-Rabbit (Vectastain ABC kit from Vecta Lab. Inc; Ref: PK-4001) secondary antibody was added respectively for 30 minutes followed by PBS wash. ABC kit (Envision Kit, Vector Laboratories) to amplify signal was applied (according to manufacturer's instructions) and sections were washed in PBS before visualization of signal using DAB reagent (Dako K4011).
Caspase-cleaved CK18
The sections were cut and collected on Sugipath Xtra-adhesive slides and were then dried overnight at 37°C followed by 10min at 60°C. After dewaxing antigen retrieval was achieved by heating at 95°C for 12min followed by cooling in Dako (S3307) high pH retrieval solution. The following steps were performed using the i6000 automated IHC platform. 3% hydrogen peroxide solution was applied for 10min. Blocking was carried out by using the affinity purified goat anti-mouse Fab fragment (Jackson 115-007-003) for 15min at room temperature, washing and then incubating with 5% casein solution for 20min. The primary M30 Cytodeath™ antibody (Peviva, 10700) was applied for 120min at room temperature followed by a 30min incubation with the α mouse envision labelled polymer (Dako K4006) and eventually the signal was visualized using the DAB reagent. All sections were counterstained with haematoxylin so that negative cells can be identified followed by dehydration in increasing concentrations of ethanol solution (70%, 90%, 100%) for 1min and xylene for 5min before mounting.
Slides were analysed blinded by two independent scorers. Sections were chosen randomly for counting, but areas containing overt necrosis that gave non-specific staining (universally brown stained cells when using immunoglobulin isotype control) were unquantifiable and avoided. The number of positive cells was determined as the mean from 5 independent fields on each section and statistical significance was determined by using a 2-tailed Student's t test. Results were expressed as mean ± SEM.
Results
Pre-clinical studies were carried out using SCID/bg mice that were either non-tumour bearing, or carried an H146 human SCLC tumour xenograft. Tumour and non-tumour bearing mice were either treated with ABT-737 (100 mg/kg/day) or vehicle control. Blood was taken at various time-points during the study and processed to generate plasma samples. Samples were assayed for total CK18 (intact and caspase-cleaved) using the M65 ELISA, and the levels of caspase-cleaved CK18 were calculated using the M30 ELISA, both validated assays. Tumours were harvested and stained for biomarkers of apoptosis, cleaved caspase-3 and caspase-cleaved CK18 using validated IHC protocols.
Regression of H146 SCLC tumours after treatment with ABT-737 and growth of control tumours
Figure 2 confirmed that during this biomarker study and upon commencement of dosing with either ABT-737 or vehicle control, xenografts from mice receiving ABT-737 showed almost complete regression after 192h (8 days), an effect that was maintained for the duration of the study (15 days). In contrast, mice receiving vehicle control maintained relatively stable tumour sizes up to 8 days but by the end of the study these tumours were significantly larger than ABT-737 treated tumours and had reached over 800 mm3 (p<0.01 48h, p<0.001 72h, p<0.01 8 days and p<0.001 15 days, tumour volume ABT-737-treated versus vehicle-treated animals).
Figure 2. Effects of ABT-737 and vehicle on H146 human SCLC xenograft size.

H146 SCLC cells were implanted into SCID/bg mice and when tumours reached ∼400 mm3 were treated with either ABT-737 at 100 mg/kg/day for 7 days (black bars) or vehicle (white bars). Each column represents 3 mice plotted as mean ± SD. p<0.01 (**); p<0.001 (***)
Levels of biomarkers of epithelial cell death in tumour bearing mice treated with ABT-737
In order to conclude that significant changes in M30 and M65 antigen levels in tumour bearing mice treated with ABT-737, compared to those receiving vehicle were as a result of treatment comprehensive statistical evaluation was carried out. Pre-dose data were compared across all time-points using one-way ANOVA followed by a post hoc Bonferroni analysis to adjust for multiple sampling within groups and Mann Whitney U testing was used to identify differences between groups. Figure 3 shows that there were no statistically significant differences in the pre-dose M30 and M65 levels across both treatment groups, and thus any changes seen post dosing were indeed as a result of drug treatment, and not as a consequence of biological variation.
Figure 3. Variation in pre-dose (baseline) levels of M30 and M65.
One way ANOVA followed by a post hoc Bonferonni analysis was carried out to test for significant variation in 24 h pre-dose (baseline) levels of M30 (grey bars) and M65 (white bars) antigens in vehicle and ABT-737-treated animals.
In agreement with our collaborators at the Karolinska Institute we have found that the M30 and M65 assays were much less sensitive at detecting endogenous mouse CK18 when compared to human CK18 (manuscript in preparation). As such, baseline levels of circulating CK18 were up to 4 fold higher in tumour bearing compared to non-tumour bearing mice indicative of a tumour-derived biomarker signal. In the case of non-tumour bearing animals treated with ABT-737 or vehicle M30 and M65 signals were close to, or at the lower limit of detection for the assay (20 U/L, data not shown). Changes in M65 (Figure 4A) and M30 readings (Figure 4B) were calculated for each individual mouse by subtracting the reading at baseline in that mouse of the circulating antigen measured 24h prior to the first dose of ABT-737. M30 and M65 levels were recorded for individual mice prior to receipt of their first dose such that baseline, or background levels for these markers could be obtained. Baseline subtraction was carried out to account for effects on M30 and M65 levels due to natural biological variation making it then possible to more closely monitor drug, or tumour specific changes in CK18 (Figure 4).
Figure 4. Levels of circulating CK18 (M65) [A] and CK18-neo (M30) [B] in H146 SCLC human xenograft bearing mice treated with either ABT-737 or vehicle.

Female age and sex-matched SCID/bg mice were implanted with H146 SCLC cells until the tumours reached ∼400 mm3, at which point animals received either daily ABT-737 at 100 mg/kg/day via i.p. administration (black bars) or vehicle (white bars) and terminal bleed plasma samples were taken and assayed for A. intact CK18 and B. cleaved CK18 using M65 and M30 assays, respectively. Levels of CK18 or cleaved CK18 were calculated by subtraction of baseline levels taken 24 hours prior to dosing via retro-orbital survival bleed. Data are from 10 (6, 12, 24 and 192 h), 7 (2 h) or 3 (48, 72 and 360 h) ABT-737-treated mice per group and 10 (6, 12, 24 and 192 h), 7 (2 h) or 3 (48, 72 and 360 h) vehicle control treated mice plotted as mean ± SEM. p<0.05 (*); p<0.01 (**).
Figure 4A shows that in ABT-737 treated tumour-bearing mice M65 levels were raised 2-48 h after drug treatment compared to baseline levels, and compared to mice receiving vehicle. The M65 signal for ABT-737 treatment groups at 72, 192 and 360 hours was below the limit of detection for the assay (20 U/L) and thus no comparison could be made between ABT-737 and vehicle treatment groups at these time-points. The lack of M65 signal for these animals is perhaps reflective of the low tumour volumes observed at these time points and that tumours had completely regressed by day 15 (Figure 2).
Vehicle control treated tumour-bearing animals also exhibited increased levels of M65 antigen at 2-24h though the readings at 24h were significantly lower than for ABT-737 treated animals at this time-point (p<0.01). Notably, there was no difference in tumour volume for ABT-737 and vehicle control treated mice up to 24h (Figure 2). The M30 results showed a similar biomarker profile to those for M65 with two notable exceptions (Figure 4B). The overall difference between the increased levels of M30 antigen 2-48h after treatment with ABT-737 compared to vehicle control was greater, reaching significance at 6h (p<0.01, >3 fold increase) and at 24h (p<0.01, >3 fold). In contrast to the M65 data, the M30 antigen levels were not increased for vehicle control at 15 days.
Overall, the M30 data more sensitively reported a drug-induced effect at early time points prior to clear changes in tumour volume. In particular, Figure 4A shows that in tumour-bearing mice, those that have been treated with ABT-737 (black bars) the levels of CK18 measured by M65 2h after dosing were 2-3 fold higher than in those mice that received vehicle control (white bars) and these levels decreased throughout the course of the experiment as the tumours regressed. The data presented confirm that caspase-cleaved CK18 is a major contributor to the M65 signal in this study as anticipated for a pure apoptosis inducer. This comparison of M65 and M30 will be of relevance in clinical trials of this type of agent.
Biomarkers of Apoptosis in Tumour
In order to relate the circulating biomarkers of cell death (M65/M30) described above to cell fate within ABT-737-treated tumours, immunohistochemical evaluation of cleaved caspase-3, a classical biochemical measurement of apoptosis, was undertaken throughout the time-course of the study (Figure 5). Figure 5A shows that at 6-24h post treatment, the levels of cleaved caspase-3 increased 3-4 fold in sections of ABT-737-treated versus vehicle control-treated tumours (p<0.05, 2h; p<0.001, 6, 12 and 24h). After 8 days, cleaved caspase-3 staining levels returned to a similar level to that which was seen in the vehicle control and ABT-737-treated tumours at 2h. In addition to this, IHC analysis of caspase-cleaved CK18 could be directly assessed in the tumour using the M30 antibody (Figure 5B). Although the absolute numbers of M30 positive tumour cells were low, (perhaps reflecting the later stage in apoptosis prior to phagocytic removal of apoptotic cells), this is not an uncommon feature of M30 IHC (31) and showed that levels of caspase-cleaved CK18 were higher in xenografts from animals treated with ABT-737, compared to animals that had received vehicle, and were significantly higher after 15 days (p<0.05). The kinetics corroborate the theory that caspase-3 activation occurs at an earlier time-point during the onset of apoptosis, after which it is possible to monitor cleavage of downstream targets (in this case CK18) at later time-points. Figure 5 therefore corroborates results shown for circulating biomarkers of cell death in tumour bearing mice that had received ABT-737 (Figure 4) and demonstrates an ABT-737 tumour-specific apoptosis.
Figure 5. Analysis of biomarkers of apoptosis in H146 SCLC xenograft sections from mice treated with either ABT-737 or vehicle.

SCID/bg mice were implanted with H146 SCLC cells and when tumours reached ∼400 mm3 were treated with either ABT-737 at mg/kg/day for 7 days (black bars) or vehicle control (white bars) and immunohistochemical analysis of the number of cells that scored positive for A. cleaved caspase-3 or B caspase-cleaved CK18 (M30) was determined between 2 and 192 hours after dosing commenced. Data are from 7 ABT-737 and 7 vehicle treated mice per time-point and are shown as the average from 5 fields of view from each tumour section as determined by 2 independent analysts who were blinded to tumour group identity. p<0.05 (*); p<0.001 (***). Error bars represent mean ± SEM.
Levels of Circulating biomarkers of epithelial cell death in non tumour bearing mice treated with ABT-737
The contribution of host cell death to the biomarker signatures obtained above was explored in a study of ABT-737 non-tumour bearing mice using a protocol that included positive quality assurance controls. ABT-737 treatment resulted in no significant increase in the circulating levels of intact CK18 or cleaved CK18 in the plasma of non-tumour bearing mice when compared to baseline levels taken 24h prior to treatment (data not shown). These data suggest that either ABT-737 exhibits no epithelial host toxicity measurable with these assays, or that these assays do not detect mouse CK18. To examine this further we treated mice with a dose of cisplatin (10mg/kg i.p.) known to induce epithelial toxicity associated with observed animal weight loss and again saw no significant change in M30 or M65 in plasma (data not shown). Overall these data suggest that the M30 and M65 assays do not detect mouse CK18 and thus the biomarker data obtained for tumour bearing mice is derived from the human tumour xenograft.
Discussion
Avoidance of apoptosis is a hallmark of cancer (31) and novel agents that target components of the apoptotic pathway are currently in pre-clinical and early clinical development. Selective tumour cell kill by these pro-apoptotic drugs is anticipated since unlike normal cells, cancer cells exist in hostile micro-environments that prime them for apoptosis. The premise is that such cancer cells survive because their adaptive up-regulation of anti-apoptotic proteins such as members of the Bcl-2 and IAP families, maintain inability to couple stress inducing stimuli to the activation of apoptosis. Bcl-2 family targeted agents such as ABT-737, a highly potent and specific inducer of apoptosis in vitro and in preclinical models in vivo (6), are entering Phase I/II Trials and therefore qualified pharmacodynamic biomarkers of drug-induced apoptosis are required as the hypothesis underlying selective tumour cell killing begins to be tested in patients with cancer.
The CK18-based M30 and M65 assays have been used in the clinic to monitor cell death induced by a variety of different cancer chemotherapeutic agents in a range of malignancies (3). Although the M30 and M65 ELISAs have been validated in vitro (3, 4, 30), the levels of circulating forms of CK18 as biomarkers of prognosis and treatment response in patients have yet to be qualified. The studies have so far largely supported the use of circulating levels of cleaved CK18 to inform on tumour cell apoptosis and thus determine treatment response (29, 32, 33) and several reports recently suggested that levels of circulating caspase-cleaved CK18 correlate with poor survival rates in some cancers (6, 28, 29). Apoptotic tumour cells have been demonstrated in the blood using the M30 antibody (34) yet it remains unclear as to whether the majority of caspase-cleaved CK18 present in the blood of drug-treated cancer patients is tumour derived, or derives from other epithelial sites as a result of drug toxicity or secondary effects of the malignancy (5).
This study represents the first assessment of the M30 and M65 assays in response to an apoptosis-targeted drug in a pre-clinical in vivo setting. Because only animals whose tumours were responding to ABT-737 treatment were included in the study biomarker data were not affected by non-responders. The rationale behind this pre-clinical study was therefore to inform on the validity of M30 and M65 levels as circulating biomarkers of tumour cell death in a tumour model with known sensitivity to the Bcl-2 antagonist, ABT-737 where a parallel assessment of tumour cell death with the circulating biomarkers could be performed. In addition, it was envisaged that this study could provide information on optimum time-points for blood sampling from clinical trial patients receiving BH-3 mimetics such as ABT-737 to allow kinetics of tumour cell death to be inferred from circulating biomarker profiles with some confidence when serial tumour biopsies were unavailable for analysis.
This type of study was previously hampered by technical issues presented by the high background signal in mouse blood due to cross-reactivity between mouse immunoglobulins and the mouse antibodies used in these assays. Here this is overcome through use of a heterophillic blocking reagent that minimises the high background signal often seen in mouse plasma samples.
In this study, cleaved CK18 levels in plasma were higher in animals receiving drug, compared to those receiving vehicle control, with a drug specific effect apparent as early as 6h after receiving ABT-737 (Figure 4B). Figure 4B shows a statistically significant difference between M30 levels in ABT-737 treated and control mice at 6 and 24 h (p<0.01) post-dosing. That these levels decline over the course of the experiment and that this correlates with tumour regression (Figure 2) strongly suggests that ABT-737 is indeed causing cell death by apoptosis, and the levels of cleaved CK18 in the blood is indicative of a drug response.
In contrast, M65 levels in animals with SCLC xenografts receiving ABT-737 were not significantly different compared to those that received vehicle at very early time-points (6 h). However, after 24h the levels of both M30 and M65 were significantly higher in animals receiving ABT-737 compared to vehicle control treated mice (p<0.01). This suggests that the rapid kinetics of ABT-737 induced tumour regression by apoptosis may progress to secondary necrosis at later times (24h). This observation is consistent with the proposed mechanism of action of the drug and is corroborated by an agreement of drug-induced changes in M65 and M30 which suggests that cell death occurs predominantly via an apoptotic mechanism.
Of particular note, both the M30 and M65 assays were able to detect changes in circulating biomarkers in mice with SCLC xenografts up to 24h after drug treatment, whereas changes in tumour volume were not significantly different until 48h after receiving ABT-737 (p<0.01). This is an important consideration when assessing the utility of a biomarker in the clinic. Indeed, these preclinical data suggest that the M30 and M65 blood borne assays may provide a valuable tool for detecting early tumour response to apoptosis-inducing therapy that we speculate could occur prior to discernable changes in tumour volume measured by imaging and thus may ultimately have use in predicting an appropriate time point for imaging patients.
While it is known that ABT-737 treatment induces an apoptosis-like response in platelets resulting in enhanced platelet clearance by the reticuloendothelial system (35), the M30 and M65 assays would not detect this haematopoietic cytotoxicity. The finding that levels of M30 and M65 antigen are raised in tumour bearing mice compared to control and non-tumour bearing mice and that significant changes in the levels of circulating CK18 occurs only in ABT-737-treated animals bearing xenografts provides further compelling evidence that CK18 measured via this method is a reliable measure of events occurring within the tumour. This is consistent with data in Figure 5 that showed both caspase-3 and CK18 cleavage to be elevated in tumours receiving ABT-737 treatment. The lack of biomarker signals in non-tumour bearing mice treated with a dose of cisplatin known to provoke epithelial toxicity and weight loss further supports that the circulating CK18 biomarker signatures seen in ABT-737-treated mice are derived from the human tumour xenograft. Complementing this study, we have recently published a corresponding induction of drug induced apoptosis in human xenograft tumour measured by M30 and reported in plasma with M30 in a nude rat model (31).
The kinetics of apoptosis caused by ABT-737 as detected by the M30 and M65 assays in this study is consistent with other studies of ABT-737 responses in vitro whereby maximal caspase-3 activity was seen 6-24 h after treatment (15, 18, 19), followed by degradation of CK18 to generate the caspase-cleaved neo-epitope recognised by M30 after 192 h. Similarly, the kinetics of ABT-737 induced release of CK18 from dying cells is in agreement with other clinical studies, where elevated levels of circulating intact and cleaved CK18 were seen in plasma following conventional chemotherapeutic treatment of lung (29), hormone refractory prostate (24) and breast cancers (28, 32) between 24-48h post treatment. These observations are all consistent with the primary mechanism of action of the drug being induction of apoptosis.
A recent 2007 review by Linder (5) highlighted the issue that measurement of circulating caspase-cleaved CK18 (M30) in patient samples is a promising method to determine the efficiency of cytotoxic drug treatment and could also be used to compare these with novel therapies. However, the author also stated that the use of M30 for monitoring treatment response in individual patients is yet to be unequivocally demonstrated. Further clinical studies are required to assign the use of M30 as a biomarker in the clinic. However, this is the first example of a pre-clinical study which should provide strength to the validity of the clinical use of the M30 and M65 assays, as changes in tumour volume directly correlate with changes in circulating biomarkers of cell death.
In light of this, the data shown here for a ‘pure’ apoptosis inducer, ABT-737, a drug targeted directly at apoptosis regulatory machinery, provide further evidence for the use of M30 as an indicator of drug-induced apoptosis that in this model occurred predominantly in tumour cells and correlated with tumour response. This study has proved highly informative both in terms of the validation of the M30 and M65 ELISAs and as a reliable means to provide pharmacodynamic information regarding tumour cell death following treatment in vivo, and to provide substantiating evidence for the mode of action in vivo for the novel anti-apoptotic agent, ABT-737. Furthermore, we have obtained valuable information regarding the kinetics for measurement of cell death products in the blood, which should prove valuable for the detailed and optimal assessment of this promising novel anticancer agent in the clinic.
Acknowledgements
C. Hodgkinson and A. Hogg for technical support. Grant sponsor: Cancer Research UK (to C.Dive); Grant number: C147. D. Micha is funded by a Cancer Research UK studentship.
References
- 1.Workman P. Cancer genome targets: RAF-ing up tumor cells to overcome oncogene addiction. Expert Rev Anticancer Ther. 2002;2:611–4. doi: 10.1586/14737140.2.6.611. [DOI] [PubMed] [Google Scholar]
- 2.Taylor K, Micha D, Ranson M, Dive C. Recent advances in targeting regulators of apoptosis in cancer cells for therapeutic gain. Expert Opin Investig Drugs. 2006;15:669–90. doi: 10.1517/13543784.15.6.669. [DOI] [PubMed] [Google Scholar]
- 3.Cummings J, Ranson M, Butt F, Moore D, Dive C. Qualification of M30 and M65 ELISAs as surrogate biomarkers of cell death: long term antigen stability in cancer patient plasma. Cancer Chemother Pharmacol. 2007;60:921–4. doi: 10.1007/s00280-007-0437-4. [DOI] [PubMed] [Google Scholar]
- 4.Cummings J, Ward TH, Greystoke A, Ranson M, Dive C. Biomarker method validation in anticancer drug development. Br J Pharmacol. 2007 doi: 10.1038/sj.bjp.0707441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linder S. Cytokeratin markers come of age. Tumour Biol. 2007;28:189–95. doi: 10.1159/000107582. [DOI] [PubMed] [Google Scholar]
- 6.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 7.Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 8.Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004;1644:83–94. doi: 10.1016/j.bbamcr.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 9.Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–91. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 10.Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Tahir SK, Yang X, Anderson MG, et al. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 2007;67:1176–83. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- 12.van Delft MF, Wei AH, Mason KD, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cragg MS, Kuroda J, Puthalakath H, Huang DC, Strasser A. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med. 2007;4:1681–89. doi: 10.1371/journal.pmed.0040316. discussion 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gong Y, Somwar R, Politi K, et al. Induction of BIM Is Essential for Apoptosis Triggered by EGFR Kinase Inhibitors in Mutant EGFR-Dependent Lung Adenocarcinomas. PLoS Med. 2007;4:e294. doi: 10.1371/journal.pmed.0040294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuroda J, Kimura S, Strasser A, et al. Apoptosis-based dual molecular targeting by INNO-406, a second-generation Bcr-Abl inhibitor, and ABT-737, an inhibitor of antiapoptotic Bcl-2 proteins, against Bcr-Abl-positive leukemia. Cell Death Differ. 2007;14:1667–77. doi: 10.1038/sj.cdd.4402168. [DOI] [PubMed] [Google Scholar]
- 16.Shoemaker AR, Oleksijew A, Bauch J, et al. A small-molecule inhibitor of Bcl-XL potentiates the activity of cytotoxic drugs in vitro and in vivo. Cancer Res. 2006;66:8731–9. doi: 10.1158/0008-5472.CAN-06-0367. [DOI] [PubMed] [Google Scholar]
- 17.Trudel S, Stewart AK, Li Z, et al. The Bcl-2 family protein inhibitor, ABT-737, has substantial antimyeloma activity and shows synergistic effect with dexamethasone and melphalan. Clin Cancer Res. 2007;13:621–9. doi: 10.1158/1078-0432.CCR-06-1526. [DOI] [PubMed] [Google Scholar]
- 18.Kline MP, Rajkumar SV, Timm MM, et al. ABT-737, an inhibitor of Bcl-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia. 2007;21:1549–60. doi: 10.1038/sj.leu.2404719. [DOI] [PubMed] [Google Scholar]
- 19.Kohl TM, Hellinger C, Ahmed F, et al. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia. 2007;21:1763–72. doi: 10.1038/sj.leu.2404776. [DOI] [PubMed] [Google Scholar]
- 20.Chu PG, Weiss LM. Keratin expression in human tissues and neoplasms. Histopathology. 2002;40:403–39. doi: 10.1046/j.1365-2559.2002.01387.x. [DOI] [PubMed] [Google Scholar]
- 21.Lane EB, Alexander CM. Use of keratin antibodies in tumor diagnosis. Semin Cancer Biol. 1990;1:165–79. [PubMed] [Google Scholar]
- 22.Hatzfeld M, Franke WW. Pair formation and promiscuity of cytokeratins: formation in vitro of heterotypic complexes and intermediate-sized filaments by homologous and heterologous recombinations of purified polypeptides. J Cell Biol. 1985;101:1826–41. doi: 10.1083/jcb.101.5.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinert PM, Roop DR. Molecular and cellular biology of intermediate filaments. Annu Rev Biochem. 1988;57:593–625. doi: 10.1146/annurev.bi.57.070188.003113. [DOI] [PubMed] [Google Scholar]
- 24.Kramer G, Erdal H, Mertens HJ, et al. Differentiation between cell death modes using measurements of different soluble forms of extracellular cytokeratin 18. Cancer Res. 2004;64:1751–6. doi: 10.1158/0008-5472.can-03-2455. [DOI] [PubMed] [Google Scholar]
- 25.Biven K, Erdal H, Hagg M, et al. A novel assay for discovery and characterization of proapoptotic drugs and for monitoring apoptosis in patient sera. Apoptosis. 2003;8:263–8. doi: 10.1023/a:1023672805949. [DOI] [PubMed] [Google Scholar]
- 26.Leers MP, Kolgen W, Bjorklund V, et al. Immunocytochemical detection and mapping of a cytokeratin 18 neo-epitope exposed during early apoptosis. J Pathol. 1999;187:567–72. doi: 10.1002/(SICI)1096-9896(199904)187:5<567::AID-PATH288>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 27.Schutte B, Henfling M, Kolgen W, et al. Keratin 8/18 breakdown and reorganization during apoptosis. Exp Cell Res. 2004;297:11–26. doi: 10.1016/j.yexcr.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 28.Demiray M, Ulukaya EE, Arslan M, et al. Response to neoadjuvant chemotherapy in breast cancer could be predictable by measuring a novel serum apoptosis product, caspase-cleaved cytokeratin 18: a prospective pilot study. Cancer Invest. 2006;24:669–76. doi: 10.1080/07357900600981307. [DOI] [PubMed] [Google Scholar]
- 29.Ulukaya E, Yilmaztepe A, Akgoz S, Linder S, Karadag M. The levels of caspase-cleaved cytokeratin 18 are elevated in serum from patients with lung cancer and helpful to predict the survival. Lung Cancer. 2007;56:399–404. doi: 10.1016/j.lungcan.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 30.Cummings J, Ward TH, LaCasse E, et al. Validation of pharmacodynamic assays to evaluate the clinical efficacy of an antisense compound (AEG 35156) targeted to the X-linked inhibitor of apoptosis protein XIAP. Br J Cancer. 2005;92:532–8. doi: 10.1038/sj.bjc.6602363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 32.Olofsson MH, Ueno T, Pan Y, et al. Cytokeratin-18 is a useful serum biomarker for early determination of response of breast carcinomas to chemotherapy. Clin Cancer Res. 2007;13:3198–206. doi: 10.1158/1078-0432.CCR-07-0009. [DOI] [PubMed] [Google Scholar]
- 33.Ueno T, Toi M, Biven K, Bando H, Ogawa T, Linder S. Measurement of an apoptotic product in the sera of breast cancer patients. Eur J Cancer. 2003;39:769–74. doi: 10.1016/s0959-8049(02)00865-1. [DOI] [PubMed] [Google Scholar]
- 34.Larson CJ, Moreno JG, Pienta KJ, et al. Apoptosis of circulating tumor cells in prostate cancer patients. Cytometry A. 2004;62:46–53. doi: 10.1002/cyto.a.20073. [DOI] [PubMed] [Google Scholar]
- 35.Zhang H, Nimmer PM, Tahir SK, et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14:943–51. doi: 10.1038/sj.cdd.4402081. [DOI] [PubMed] [Google Scholar]