

Abstract
Human immunodeficiency virus (HIV)-infected patients have a higher incidence of oxidative stress, endothelial dysfunction, and cardiovascular disease than uninfected individuals. Recent reports have demonstrated that viral proteins upregulate reactive oxygen species, which may contribute to elevated cardiovascular risk in HIV-1 patients. In this study we employed an HIV-1 transgenic rat model to investigate the physiological effects of viral protein expression on the vasculature. Markers of oxidative stress in wild-type and HIV-1 transgenic rats were measured using electron spin resonance, fluorescence microscopy, and various molecular techniques. Relaxation studies were completed on isolated aortic rings, and mRNA and protein were collected to measure changes in expression of nitric oxide (NO) and superoxide sources. HIV-1 transgenic rats displayed significantly less NO-hemoglobin, serum nitrite, serum S-nitrosothiols, aortic tissue NO, and impaired endothelium-dependent vasorelaxation than wild-type rats. NO reduction was not attributed to differences in endothelial NO synthase (eNOS) protein expression, eNOS-Ser1177 phosphorylation, or tetrahydrobiopterin availability. Aortas from HIV-1 transgenic rats had higher levels of superoxide and 3-nitrotyrosine but did not differ in expression of superoxide-generating sources NADPH oxidase or xanthine oxidase. However, transgenic aortas displayed decreased superoxide dismutase and glutathione. Administering the glutathione precursor procysteine decreased superoxide, restored aortic NO levels and NO-hemoglobin, and improved endothelium-dependent relaxation in HIV-1 transgenic rats. These results show that HIV-1 protein expression decreases NO and causes endothelial dysfunction. Diminished antioxidant capacity increases vascular superoxide levels, which reduce NO bioavailability and promote peroxynitrite generation. Restoring glutathione levels reverses HIV-1 protein-mediated effects on superoxide, NO, and vasorelaxation.
Keywords: acquired immunodeficiency syndrome, antioxidants, superoxide
Highly active antiretroviral therapy (HAART) has extended the life expectancy of patients infected with human immunodeficiency virus-1 (HIV-1) and has significantly reduced viral burden (43, 74). However, serious HIV-related complications including cardiac and skeletal myopathies, hypertension, hyperinsulinemia, hypertriglyceridemia, and insulin resistance have been shown to develop over time (16, 57). These complications increase the risk of patients developing diabetes and various cardiovascular diseases, which have become an important cause of morbidity in HIV-1 patients. Although much effort focuses on the contribution of HAART, there is increasing clinical evidence that HIV-1 itself increases the risk of developing cardiovascular disease. Studies preceding the era of HAART demonstrate that HIV-1-infected individuals exhibit vasculitis in small blood vessels and aneurysms in medium to large arteries (18, 61, 62), and HIV-1-infected children have been reported to display severe arteriopathy (47). Antiretroviral-naive patients display lower levels of high-density lipoprotein (HDL) cholesterol (39), as well as markers of endothelial dysfunction including von Willebrand factor, plasminogen activator inhibitor-1 (PAI-1) antigen, vascular cell adhesion molecule-1 (VCAM-1), and E-selectin (38, 51, 81). Recently, the Strategies for Management of Antiretroviral Therapy (SMART) Study Group concluded that cessation of antiretroviral therapy in HIV-1-positive patients increases their short-term risk of developing cardiovascular disease (29, 45, 90), implicating viral factors in causing this effect. Together, these clinical studies demonstrate increased cardiovascular risk in HIV-1-infected individuals, even in the absence of antiretroviral therapy.
Increased production of reactive oxygen species (ROS), such as superoxide, is closely associated with the progression of cardiovascular diseases. Under normal physiological conditions, vascular smooth muscle cells and the endothelium strictly regulate the production and detoxification of ROS (60). However, when stimuli chronically upregulate sources of superoxide production and/or downregulate endogenous antioxidant systems, cellular superoxide levels increase, leading to aberrant cell signaling, vascular smooth muscle cell hypertrophy and migration (95), endothelial dysfunction, and, potentially, apoptosis (83). High levels of superoxide also readily react with nitric oxide (NO) to produce the powerful oxidant peroxynitrite (93). These reactive oxygen and nitrogen species attack iron-sulfur centers of heme-containing molecules, as well as form damaging adducts on lipids, proteins, and DNA (17, 41). Moreover, NO reacting with superoxide reduces NO bioavailability (15, 40). This has dire consequences, because NO is the primary endogenous vasorelaxant, downregulates cell adhesion molecules, inhibits platelet and leukocyte adhesion to the vascular wall, and prevents vascular smooth muscle cell proliferation (32, 35). Superoxide plays a pivotal role in the development of cardiovascular diseases, not only through direct oxidative damage to the cell but also through the depletion of endogenous NO.
Evidence suggests that HIV-1 induces a state of chronic oxidative stress (6, 72). HIV-1 patients demonstrate higher plasma concentrations of hydroperoxides and malondialdehyde (31), indicative of polyunsaturated fatty acid peroxidation. The exact mechanism by which HIV-1 induces oxidative stress is unknown. Furthermore, the contribution of other factors in clinical studies (i.e., current antiretroviral therapy, disease stage, and smoking/drug abuse history, among others) complicates drawing definitive conclusions linking HIV-1 proteins to oxidative stress and cardiovascular disease. The NL4-3Δ gag/pol HIV-1 transgenic rat model provides a noninfectious and relevant way to study the physiological effects of HIV-1 proteins in vivo. This transgene expresses a nonreplicative HIV-1 provirus under the viral promoter and encodes for the viral genes env, tat, nef, rev, vif, vpr, and vpu (23, 49). Because the transgenic rat displays clinical manifestations that resemble those seen in HIV-1 patients, it is a useful model for studying HIV-1-associated neurological and cardiac pathologies (78). Unlike other models that can demonstrate atypical distribution of transgene expression, the HIV-1 transgenic rat expresses viral gene products in the blood and lymphoid tissue similar to those in humans (78). The endothelium is continually exposed to actively secreted viral proteins (19), viral particles released from lysed host cells (69), and infected monocytes due to its position between the blood and the vascular wall. Thus the transgenic HIV-1 rat model is a useful and appropriate tool for examining the physiological consequences of HIV-1 proteins on cells of the vasculature.
Because ROS play such a critical role in the development of cardiovascular diseases and HIV-1 proteins have been implicated in ROS production, we sought to explore a potential mechanistic link between HIV-1 protein-induced oxidative stress and vascular dysfunction. We hypothesize that HIV-1 protein expression in the transgenic rat model alters NO levels and that vascular oxidative stress plays a role in this effect. In this study, we investigated the effects of HIV-1 proteins on the vasculature, without the introduction of other complicating agents such as antiretroviral drugs, to determine the extent by which HIV-1 proteins by themselves lead to oxidative stress and promote cardiovascular complications.
MATERIALS AND METHODS
Wild-type and HIV-1 transgenic rats
Approximately 9-mo-old male Fisher 344/NHsd wild-type rats and NL4-3Δ gag/pol HIV-1 transgenic rats (Harlan, Indianapolis, IN) were used in experiments. Selected rats were given 0.35% 2-oxo-4-thiazolidinecarboxylic acid (procysteine; Sigma, St. Louis, MO) ad libitum for 3 wk. Experiments were performed with the consent of the Institutional Care and Use of Animals Committee at the Atlanta Veterans Affairs Medical Center. The investigation conforms with the “Guide for the Care and Use of Laboratory Animals,” published by the National Institutes of Health [DHEW Publication No. (NIH) 85-23, Revised 1996, Office of Science and Health Reports, DRR/NIH, Bethesda, MD 20205].
Aorta preparation
Rats were given a lethal intraperitoneal dose of pentobarbital. Aortas were excised, cleaned of loose fat and connective tissue, and maintained in physiological saline solution (Krebs-Henseleit buffer: 118 mM NaCl, 4.73 mM KCl, 1.2 mM MgSO4, 0.025 mM EDTA, 1.2 mM KH2PO4, 2.5 mM CaCl2, 11 mM glucose, and 25 mM NaH2CO3, pH 7.4, in 95%-5% CO2 at 37°C). Care was taken to ensure that aortas were free of blood. All chemicals were of highest purity and purchased from Sigma.
Systemic markers of NO production
Approximately 800 μl of whole blood were obtained through cardiac puncture using a 26-gauge needle, snap-frozen in liquid nitrogen, and analyzed by electron spin resonance (ESR) spectroscopy. The three-line hyperfine spectrum of the five-coordinate complex of NO-Hb was measured in a Bruker EMX ESR spectrometer (Bruker Instruments, Billerica, MA) with the following settings: microwave power, 10 mW; modulation frequency, 100 kHz; modulation amplitude, 3 G; field center, 3,320 G; sweep width, 320 G; microwave frequency, 9.39 GHz; conversion time, 655 ms; time constant, 5.24 s; number of scans, 2; and sweep time, 336 s (26). Amplitude of the NO-Hb signal was normalized to the weight of frozen whole blood.
Nitrite concentrations were determined using the nitrate/nitrite fluorometric assay kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer's instructions. Serum was immediately separated from whole blood after collection. Serum was then ultrafiltered through sterile 30-kDa molecular mass cut-off filters, 10 μl were incubated with 2,3-diaminonaphthalene and NaOH, and fluorescence was measured in a Wallac 1420 plate reader (Perkin Elmer, Boston, MA). Samples were run in triplicate from a total of four wild-type and four transgenic rats.
Serum S-nitrosothiol (RSNO) concentration was determined colorimetrically using a standard curve from purified S-nitrosoglutathione (GSNO). Standard/sample (400 μl) was added to 200 μlof solution C (1 volume 5% HgCl2 in H2O and 10 volumes of 6.8% sulfanilamide in 0.4 N HCl). Next, 20 μl of solution D [4% N-(1-naphthyl)ethylenediamine dihydrochloride in 0.4 N HCl] were added and mixed. Samples were incubated for 10 min, 250 μl were transferred to a 96-well plate, and absorbance was measured at 550 nm. All reactions were kept on ice and run in duplicate.
Aortic NO measurements
Direct tissue NO levels were determined through ESR analysis of colloidal Fe(DETC)2-incubated aortas (26). Briefly, 7.2 mg of diethyldithiocarbamate (DETC) and 4.45 mg of FeSO4·7H2O were individually dissolved under nitrogen flow in two 10-ml volumes of ice-cold sterile/deoxygenated 0.9% NaCl and passed through sterile 0.22-μm filters. Aortic rings were placed in 12-well plates and incubated in 3.0 ml of cold Krebs-HEPES buffer (KHB: 99.0 mM NaCl, 4.69 mM KCl, 2.50 mM CaCl2·2H2O, 1.20 mM MgSO4·7H2O, 25.0 mM NaHCO3, 1.03 mM KH2PO4, 5.6 mM glucose, and 20.0 mM Na-HEPES, pH 7.4). DETC and FeSO4 were mixed 1:1, and 1.0 ml of the resulting colloidal Fe(DETC)2 was carefully added to each well at the same time. The final colloid concentration was 200 μM. As a positive control, some aortic rings were preincubated with the calcium ionophore A-23187 (5 μM) for 15 min to stimulate aortic NO production. As a negative control, other aortic segments were preincubated with the NO synthase inhibitor nitro-l-arginine methyl ester (l-NAME; 1 mM) for 60 min before A-23187 stimulation and incubation with Fe(DETC)2. The plates were incubated at 37°C for 60 min, whereupon the aortic rings were snap-frozen in the center of a 1-ml syringe containing ∼0.7 ml of KHB. The frozen samples were measured using a Bruker EMX ESR spectrometer. The ESR settings for Fe(DETC)2 were as follows: microwave power, 10 mW; modulation frequency, 100 kHz; modulation amplitude, 5 G; field center, 3,290 G; sweep width, 90 G; microwave frequency, 9.39 GHz; conversion time, 328 ms; time constant, 5.24 s; number of scans, 4; and sweep time, 168 s. Aortas were placed in an oven overnight to obtain the dry weights for normalization purposes.
Isometric force measurements
Segments (5 mm) of thoracic aorta were mounted between stainless steel wires in an organ chamber containing Krebs-Henseleit buffer and connected to a Harvard Apparatus differential capacitor force transducer. For each aorta, resting tension was adjusted to 50 mN over a 1-h period. This tension falls within the range for maximal active force generation. Data were recorded using PowerLab digital acquisition and analyzed using Chart software. Concentration-response curves were generated to the contractile agents KCl (0–50 mM) and phenylephrine (PE; 0.1 nM to 10 μM). After precontraction with 2–5 μM PE, a concentration that yields 80–90% maximum contraction, relaxation responses were examined in response to the endothelium-dependent vasorelaxant acetylcholine (0.1 nM to 100 μM) and the endothelium-independent NO-donor sodium nitroprusside (0.1 nM to 1 μM). To determine the effects of a superoxide dismutase (SOD) mimetic on vasorelaxation, we also preincubated some aortas with 1 mM tiron for 45 min before the addition of acetylcholine.
Western analysis of aortic tissue
Aortas were homogenized in lysis buffer (82), followed by sonication (10 × 2-s burst at low power). The lysate was spun at 28,000 g for 15 min, and protein concentrations were determined using a bicinchoninic acid assay (Pierce, Rockford, IL). Equal amounts of sample protein (50 μg/lane) were loaded onto a 4–12% bis-tris PAGE mini gel, separated by electrophoresis, and blotted to polyvinylidene difluoride. Membranes were incubated overnight at 4°C in a 1:1,000 dilution of primary antibodies for endothelial NO synthase (eNOS; BD-Transduction, Lexington, KY), eNOS-Ser1177 (Cell Signaling Technology, Danvers, MA), Cu/Zn-SOD (EMD Biosciences, San Diego, CA), Mn-SOD (Stressgen Biore-agents, Victoria, BC, Canada), actin (Santa Cruz Biotechnology, Santa Cruz, CA), GAPDH (Santa Cruz Biotechnology), Nox1 (Santa Cruz Biotechnology), Nox2 (BD-Transduction), p22phox (BD-Transduction), p47phox (Upstate Biotechnology, Lake Placid, NY), or xanthine oxidase (XO; Santa Cruz Biotechnology). The rabbit polyclonal antibody for Nox4 was kindly provided by Dr. David Lambeth (Emory University, Atlanta, GA). Proteins were visualized using LumiGlo (Millipore, Billerica, MA), and densitometry was accomplished using Bio-Rad Quantity One (version 4.5.0) software.
Tetrahydrobiopterin analysis
Tetrahydrobiopterin (BH4) analysis was determined using HPLC as described previously (53).
In situ superoxide measurement
Dihydroethidium (DHE) staining for superoxide was described previously (27, 65). Cleaned aortas were embedded in optimal cutting temperature compound (Electron Microscopy Sciences, Hatfield, PA) for cryosectioning, and 30-μm sections were cut and placed on glass slides. DHE (10 μM) was added, coverslips were placed over the sections, slides were incubated at 37°C for 30 min, and images were taken using a laser scanning confocal microscope (Olympus, Melville, NY). DHE fluorescence intensity was analyzed using NIH ImageJ software as previously described (54).
ESR measurement of superoxide
Superoxide was determined by measuring the oxidation of the spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH; Alexis Biochemicals, San Diego, CA) (27, 28). Aortic rings were cleaned and stored in cold, sterile, deoxygenated KHB with chelating agents (5 μM DETC and 25 μM deferoxamine). Aortic rings were placed in 12-well plates and incubated at 37°C for 60 min with 0.5 mM CMH prepared in KHB with chelating agents. Selected aortas were preincubated with 50 U/ml of polyethylene glycol (PEG)-SOD for 4 h before addition of CMH. PEG-SOD was also included during CMH incubation of those samples. The rings were frozen in 1-ml syringes containing KHB and snap-frozen. ESR measurements were done on the Bruker EMX ESR spectrometer with the following settings: microwave power, 10 mW; modulation frequency, 100 kHz; modulation amplitude, 5 G; field center, 3,340 G; sweep width, 80 G; microwave frequency, 9.39 GHz; conversion time, 655 ms; time constant, 5.24 s; number of scans, 2; and sweep time, 60 s. Aortas were placed in an oven overnight to obtain the dry weights for normalization purposes.
Real-time RT-PCR
Cleaned aortas were homogenized in 1 ml of RNA-Bee (Tel-Tests, Friendswood, TX), and RNA was purified according to the manufacturer's instructions. Total RNA (5 μg) was reverse transcribed using random nanomer primers. Real-time PCR was then performed on the LightCycler (Roche Diagnostics, Indianapolis, IN) using primers for β-globin, XO, Nox1, Nox2, p47phox, Nox4, and p22phox (SuperArray Bioscience, Frederick, MD). mRNA expression was determined using the ΔΔCT (threshold cycle) method (58).
3-Nitrotyrosine analysis
Aortas were homogenized in sterile phosphate-buffered saline containing protease inhibitors and centrifuged at 12,000 relative centrifugal force (rcf) for 10 min at 4°C. Nitrotyrosine levels were quantified via ELISA (Northwest Life Science Specialties, Vancouver, WA) according to the manufacturer's protocol.
NADPH oxidase activity
Thoracic aortas were isolated, dissected free of adventitia, snap-frozen, and then used to prepare membrane fractions, as described previously (66) with minor modifications. In brief, the frozen aortic segments were homogenized in 350 μl of 50 mM phosphate buffer (treated for 2 h with 5 g/100 ml Chelex-100 and filtered) containing the protease inhibitors aprotinin (10 μg/ml), leupeptin (0.5 μg/ml), pepstatin (0.7 μg/ml), and phenylmethylsulfonyl fluoride (0.5 mM) (pH 7.4 at 4°C). The homogenate was centrifuged at 400 g for 10 min, and the supernatant was sonicated (at 4-W power) for 10 s on ice and centrifuged at 28,000 g for 15 min at 4°C. The membrane pellet was resuspended in 150 μl of the homogenization buffer, and 20-μg protein aliquots were used for ESR measurement of superoxide radicals induced by NADPH with 1 mM CMH, 200 μM NADPH, and 0.1 mM diethylenetriaminepentaacetic acid in a total volume of 100 μl of Chelex-treated PBS. The ESR spectra were recorded with an EMX ESR spectrometer (Bruker) and a super-high-Q microwave cavity exactly as described previously (66).
SOD activity
Aortic SOD activity was measured using the superoxide dismutase assay kit from Cell Technology (Mountain View, CA) according to the manufacturer's instructions. Mn-SOD activity was determined by measuring fluorescence in the presence of 3 mM NaCN.
GSH analysis
Approximately 50 mg of aortic tissue were homogenized and sonicated in 500 μl of a 10% (wt/vol) perchloric acid solution containing 0.2 M boric acid and 10 μM γ-Glu-Glu. Homogenates were centrifuged at 10,000 rcf for 10 min at 4°C and derivatized with iodoacetic acid and dansyl chloride for HPLC analysis according to the method of Jones (46).
Statistics
A Student's t-test analysis was done for simple comparison of two groups. A two-way ANOVA with Bonferroni post hoc test was used for comparison of multiple parameters. Statistical significance was defined as P ≤ 0.05, and all values are means ± SE.
RESULTS
HIV-1 transgenic rats display decreased circulating and aortic NO
Decreased NO bioavailability disrupts vascular homeostasis and enhances the risk of developing cardiovascular disease. To examine bioavailable NO in the wild-type and HIV-1 transgenic rats, we measured markers of endothelium-derived NO in the blood and serum, as well as NO levels in the aorta. HIV-1 transgenic rats demonstrate decreased markers of circulating NO in blood and serum. Levels of NO-Hb in whole blood were ∼47% higher in wild-type rats compared with HIV-1 transgenic rats (Fig. 1A), and wild-type rats also had ∼4.7-fold higher levels of the NO decomposition product nitrite in their serum (Fig. 1B). Levels of another circulating marker of bioactive NO (88), serum RSNO, were also significantly higher in the wild-type rats compared with their transgenic counterparts (Fig. 1C; 78.52 ± 6.31 vs. 51.15 ± 5.08 nmol/ml, respectively). To directly and quantifiably measure NO in the aortic tissue, we incubated isolated aortic rings with the colloid Fe(DETC)2 and measured the amplitude of the NO-Fe(DETC)2 signal using ESR spectroscopy. The HIV-1 transgenic rats had significantly less baseline aortic NO-Fe(DETC)2 than wild-type rats (Fig. 1D). Rings were treated with the calcium ionophore A-23187 or the NO synthase inhibitor l-NAME as positive or negative controls, respectively, for ESR measurement of NO. As expected, A-23187 stimulation significantly increased the signal for NO-Fe(DETC)2, which was attenuated with l-NAME treatment.
Fig. 1.

Serum and aortic levels of nitric oxide (NO) are decreased in human immunodeficiency virus-1 (HIV-1) transgenic rats. A: whole blood from wild-type (WT) and HIV-1 transgenic rats was frozen in liquid N2, and levels of nitrosyl-hemoglobin (NO-Hb) were analyzed by electron spin resonance (ESR) spectroscopy. Results are expressed as relative units/mg frozen blood (n = 6–12). B: serum nitrite levels, a marker of endothelium-derived NO decomposition, were determined by incubating rat serum with 2,3-diaminonaphthalene. Samples were run in triplicate (n = 4). C: S-nitrosothiol (RSNO), a marker of bioactive NO production, was measured in rat serum using a colorimetric nitrosylation assay. Samples were run in duplicate (n = 4). D: basal NO levels in aortic tissue are significantly decreased due to HIV-1 transgene expression, as determined by ESR measurement of Fe(DETC)2-treated aortic rings. Open bars, WT aortas; shaded bars, HIV-1 transgenic aortas. Some rings were treated with the calcium ionophore A-23187 to stimulate NO production as a positive control, and others were treated with A-23187 and the NO synthase inhibitor nitro-l-arginine methyl ester (l-NAME) as a negative control. NO is expressed as relative units/mg dry weight of aorta (n = 3–10). *P < 0.05; **P < 0.01; ***P < 0.001 vs. untreated and l-NAME-treated groups.
Endothelium-dependent relaxation is impaired in HIV-1 transgenic rats
NO deficiency in the HIV-1 transgenic rats suggests that they may be predisposed to developing endothelial dysfunction. To determine whether transgene status affects vascular smooth muscle contraction, we determined aortic isometric force measurements in the presence of the contractile agents KCl and PE. No differences in sensitivity to KCl or PE were observed (data not shown). Next, we measured aortic relaxation by precontracting aortas with PE to 80% maximum contraction and completed concentration-relaxation curves to the endothelium-dependent vasorelaxant acetylcholine (Fig. 2A). Endothelium-dependent relaxation in response to acetylcholine was significantly impaired in aortas from HIV-1 transgenic rats. This decrease in relaxation is not attributable to reduced smooth muscle sensitivity to NO because aortas from wild-type and HIV-1 transgenic rats showed comparable responses to the NO donor sodium nitroprusside (Fig. 2B).
Fig. 2.
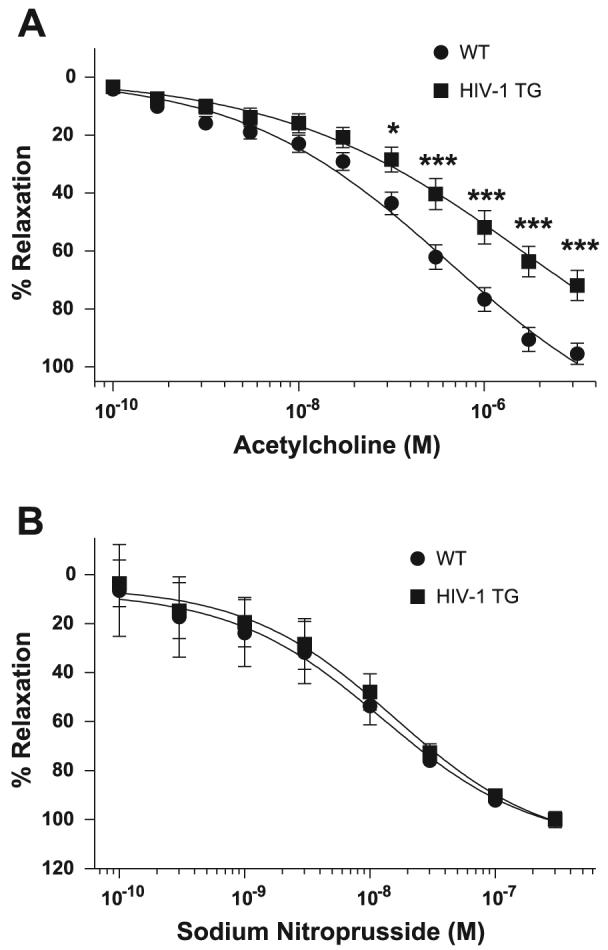
HIV-1 transgenic rats demonstrate impaired endothelium-dependent relaxation. Isometric force measurements were determined from WT and HIV-1 transgenic rat aortas. Aortas were incubated with phenylephrine to 80% maximum contraction and treated with increasing concentrations of the endothelium-dependent vasorelaxant acetylcholine (A; n = 8) or the endothelium-independent vasorelaxant sodium nitroprusside (B; n = 4). Statistical analysis was performed using two-way ANOVA with Bonferroni's posttest repeated measures. *P < 0.05; ***P < 0.001 compared with WT. TG, transgenic.
NO depletion is not due to changes in eNOS
The principal source of NO in the vasculature is the constitutively active endothelial isoform of nitric oxide synthase (eNOS). To determine whether alterations in eNOS protein expression are responsible for the observed decrease in NO, we performed Western blot analysis on aortic tissue. Overall, there were no differences in total eNOS protein expression between wild-type and HIV-1 transgenic rat aortas (Fig. 3A). We next investigated whether phosphorylation of eNOS-Ser1177 was altered, since phosphorylation of this residue is one of the enzyme's main stimulatory posttranslational modifications (67). Similar to total eNOS protein, eNOS-Ser1177 phosphorylation levels were comparable between the wild-type and HIV-1 transgenic rats (Fig. 3B). Aortic levels of the eNOS cofactor BH4, although similar to previously published values (52), also were not affected by transgene expression (Fig. 3C), and the ratio of oxidized to reduced BH4 was unaffected as well (Fig. 3D).
Fig. 3.

Transgenic HIV-1 protein expression does not alter total endothelial NO synthase (eNOS) protein levels, eNOS-Ser1177 phosphorylation (pSer1177-eNOS), or tetrahydrobiopterin (BH4) levels. Total eNOS (A; n = 3) and pSer1177-eNOS (B; n = 6–8) levels were determined by Western blotting and normalized to actin or total eNOS, respectively. Insets are representative images of blots. C:BH4 levels were measured by HPLC and normalized to total protein (n = 3). D: the ratio of oxidized BH2 to reduced BH4 levels was determined (n = 3).
HIV-1 transgenic rats display enhanced superoxide-mediated NO degradation
Increased superoxide levels may react with NO, thereby reducing NO availability and producing peroxynitrite. To qualitatively assess superoxide levels in wild-type and HIV-1 transgenic rats, we incubated aortic rings with DHE (Fig. 4A), which oxidizes to ethidium in the presence of superoxide and fluoresces upon DNA intercalation (8, 27). With the use of this technique, the HIV-1 transgenic aortas demonstrated ∼1.7-fold greater fluorescence intensity than wild-type aortas, suggesting elevated superoxide levels in the transgenic rats (Fig. 4B). Because DHE may be oxidized by other means (86) (14), aortic superoxide was also measured using ESR spectroscopy. ESR analysis using the superoxide spin probe CMH provides specific and quantitative measurements of superoxide. Aortas from HIV-1 transgenic rats had 2.4-fold higher superoxide levels than those from wild-type rats with this technique (Fig. 4C). Preincubation with the superoxide scavenger PEG-SOD decreased the amplitude of the superoxide signal, confirming the specificity of the CMH spin probe for superoxide.
Fig. 4.
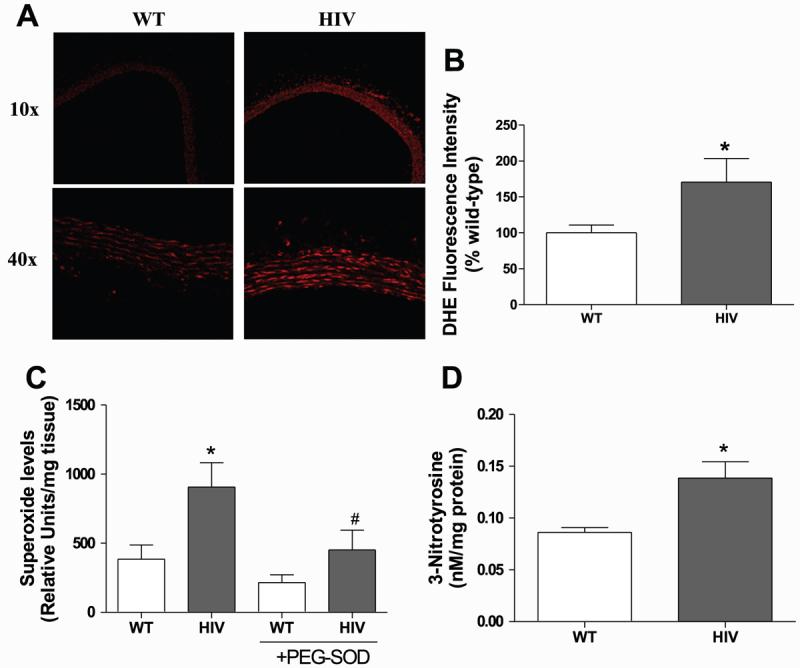
Superoxide and 3-nitrotyrosine are increased in HIV-1 transgenic rat aortas. A: sections were stained with dihydroethidium (DHE) and analyzed using laser scanning confocal microscopy to qualitatively examine superoxide levels. Images are representative of 4 experiments. B: DHE fluorescence intensity in aortic slices was measured and expressed as a percentage of WT fluorescence (n = 6–8). C: rings were incubated with the superoxide spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH), and the CM· product was measured with ESR. Selected aortas were incubated with 50 U/ml of polyethylene glycol-superoxide dismutase (PEG-SOD) for 4 h. Superoxide is expressed as relative units of CM· per mg dry weight of aorta (n = 5). D: aortic 3-nitrotyrosine was measured by ELISA and normalized to total protein (n = 4). *P < 0.05 compared with WT. #P < 0.05 compared with HIV.
Uncontrolled regulation of superoxide favors its reaction with NO to form the powerful and damaging oxidant peroxynitrite. This ROS nitrates protein tyrosine residues to create nitrotyrosines, which can be used as a footprint for the production of peroxynitrite and, thus, superoxide-mediated NO degradation. As determined by ELISA, levels of aortic 3-nitrotyrosine were significantly increased in the HIV-1 transgenic rats compared with wild-type rats (Fig. 4D).
XO and NADPH oxidase expression are not increased in HIV-1 transgenic rats
To determine whether upregulation of superoxide-generating systems accounts for the elevated superoxide levels, we analyzed mRNA and protein expression of XO and NADPH oxidase components. No differences in mRNA levels of XO (Fig. 5A), Nox1 (Fig. 5C), Nox2 (Fig. 5E), p47phox (Fig. 5G), Nox4 (Fig. 5I), or p22phox (Fig. 5K) were detected between wild-type and HIV-1 transgenic aortas. Similarly, protein expression of XO (Fig. 5B), Nox1 (Fig. 5D), Nox2 (Fig. 5F), p47phox (Fig. 5H), Nox4 (Fig. 5J), and p22phox (Fig. 5L) were comparable between the two groups. Because NADPH oxidase components may potentially be regulated by means other than differences in expression, we directly measured NADPH oxidase activity in isolated aortic membrane preparations using ESR (Fig. 6). In accordance with the lack of a difference in NADPH oxidase subunit expression between wild-type and HIV-1 transgenic rat aortas, no significant difference in NADPH activity was observed between our two experimental groups.
Fig. 5.

HIV-1 transgene expression does not increase xanthine oxidase (XO) or NADPH oxidase expression. Real-time RT-PCR was performed on aortic homogenates for XO (A) and the NADPH oxidase subunits Nox1 (C), Nox2 (E), p47phox (G), Nox4 (I), and p22phox (K). Target mRNA expression was normalized to β-globin mRNA using the ΔΔCT method, and values are expressed as percentages of WT (n = 3). Western blots were performed on aortic homogenates for XO (B), Nox1 (D), Nox2 (F), p47phox (H), Nox4 (J), and p22phox (L). Target protein expression was analyzed by densitometry and normalized to GAPDH levels. Values are expressed as percentages of WT (n = 3–4). Insets are representative Western blots.
Fig. 6.
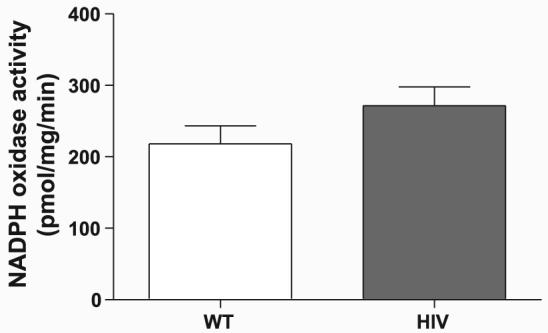
HIV-1 transgene expression does not affect aortic NADPH oxidase activity. Membrane preparations from WT and HIV-1 transgenic rat aortas were analyzed for NADPH-dependent superoxide production by ESR. Values represent PEG-SOD-inhibited superoxide production and are expressed as pmol of oxidized CMH per mg of total aortic protein per minute. Samples were run in duplicate (n = 3).
HIV-1 transgenic aortas have decreased antioxidant capacity
A decreased ability to scavenge ROS may contribute to increased superoxide levels. Therefore, we examined the expression of important antioxidant systems responsible for detoxifying superoxide as well as other toxic free radicals. HIV-1 transgene expression lowered aortic protein levels of the endogenous cytosolic isoform of SOD by 31% (Cu/Zn-SOD; Fig. 7A) but not the mitochondrial isoform Mn-SOD (Fig. 7B). Activity of the two isoforms correlated with their protein expression because Cu/Zn-SOD activity in aortas from HIV-1 transgenic rats was significantly decreased (Fig. 7C), whereas that of Mn-SOD was not (Fig. 7D). In addition, HIV-1 transgene expression significantly reduced levels of total aortic GSH (Fig. 7E) by 47%, reflecting a decreased ability to detoxify other damaging vascular free radicals.
Fig. 7.

Antioxidant levels are decreased in HIV-1 transgenic rat aortas. Aortic homogenates were analyzed by Western blot for Cu/Zn-SOD (A) and Mn-SOD protein levels (B) and normalized to actin expression. Insets show representative blots (n = 6–7). Activities of Cu/Zn-SOD (C; n = 6) and Mn-SOD (D; n = 4–6) were measured fluorometrically from aortic homogenates. E: total aortic tissue glutathione (GSH) levels were quantified by HPLC (n = 6). *P < 0.05; **P < 0.01 compared with WT.
GSH restoration decreases aortic superoxide and restores NO levels in HIV-1 transgenic rats
To further examine the importance of antioxidant depletion in HIV-1 protein-mediated vascular dysfunction, we gave rats the GSH precursor procysteine in drinking water (0.35%) for 3 wk and the measured superoxide and NO bioavailability. Our group (12) previously showed that this concentration of procysteine restores rat GSH levels and reduces pulmonary oxidative stress. Procysteine administration in HIV-1 transgenic rats significantly decreased aortic superoxide (Fig. 8A) and concomitantly restored aortic NO (Fig. 8B) and NO-Hb bioavailability (Fig. 8C) to wild-type levels. Aortic force measurement experiments also demonstrate that procysteine treatment of HIV-1 transgenic rats improved acetylcholine-mediated aortic relaxation (Fig. 9A) compared with vehicle-treated rats (Fig. 2A). Because changes in cellular GSH may directly modify signaling proteins independently of its effects on ROS levels, aortic endothelium-dependent relaxation was also measured in aortas preincubated with the SOD mimetic tiron (Fig. 9B). Similar to the data from procysteine-treated rats, our experiments show that tiron effectively restored endothelium-dependent relaxation in the HIV-1 transgenic rats, emphasizing the role of oxidative stress in endothelial dysfunction in HIV-1 transgenic rats.
Fig. 8.
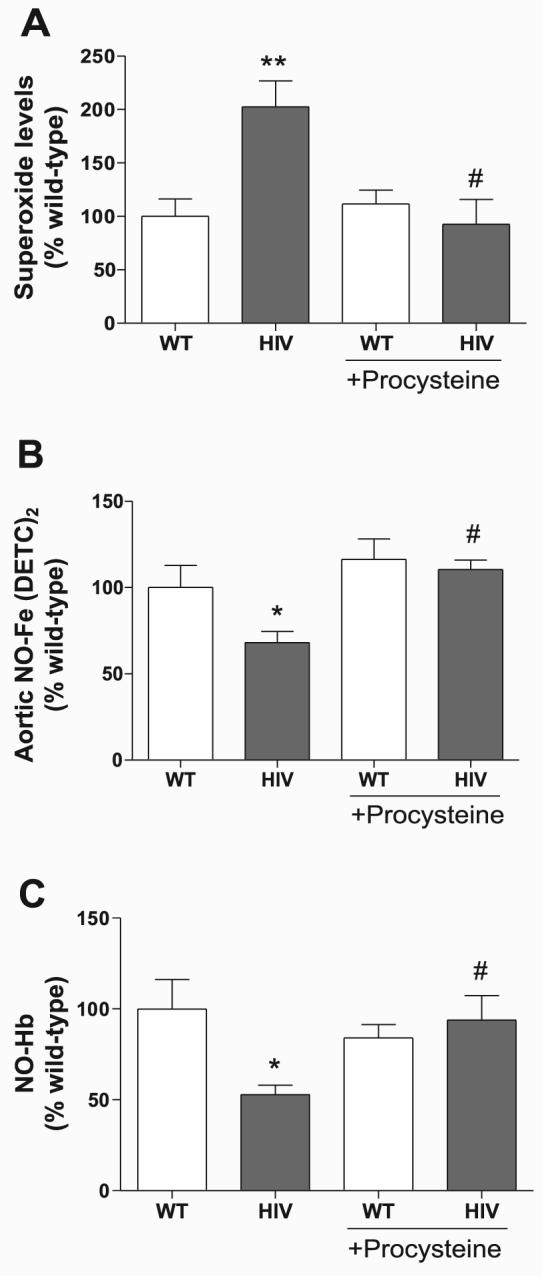
Treatment with the GSH precursor procysteine reduces aortic superoxide and restores NO in HIV-1 transgenic rats. Rats were given 0.35% procysteine for 3 wk in their drinking water. Isolated aortas were incubated with the superoxide spin probe CMH (A; n = 4–9) or the NO colloid Fe(DETC)2 (B; n = 3–10), normalized to the dry weight of aorta, and expressed as a percentage of WT control. C: whole blood was processed for NO-Hb (n = 5–12), and samples were analyzed by ESR. The data from animals not treated with procysteine represent basal values from Figs. 1 and 4. *P < 0.05; **P < 0.01 compared with WT. #P < 0.05 compared with HIV.
Fig. 9.
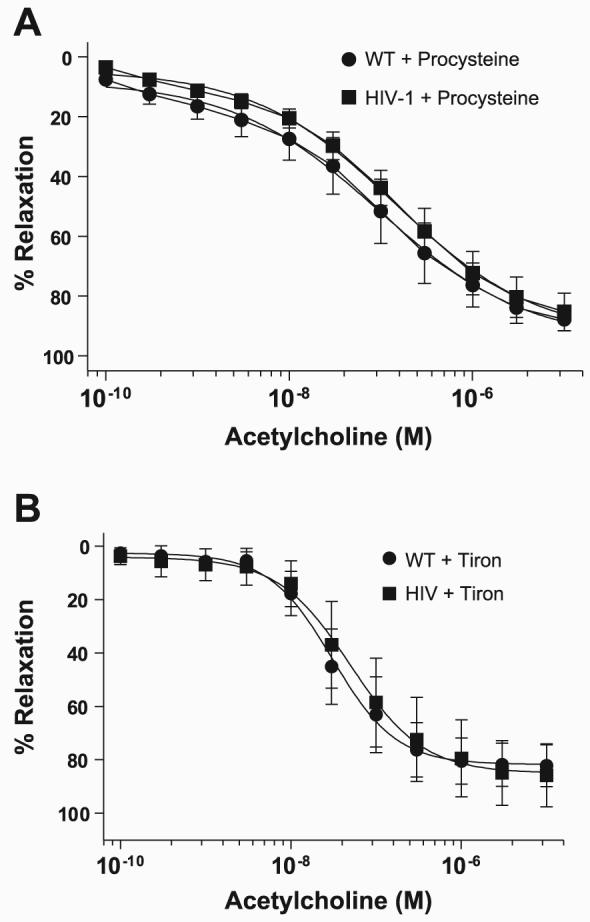
Superoxide scavenging improves endothelium-dependent vasorelaxation in HIV-1 transgenic rats. A: rats were given 0.35% procysteine for 3 wk in their drinking water. Force measurements were measured in isolated WT and HIV-1 transgenic rat aortas. Aortas were incubated with phenylephrine to 80% maximum contraction and treated with increasing concentrations of the endothelium-dependent vasorelaxant acetylcholine (n = 7–8). B: endothelium-dependent relaxation was measured in isolated aortas from vehicle-treated rats that were preincubated with the SOD mimetic tiron (n = 3–4).
DISCUSSION
In the current study, we used a transgenic rat model to examine the relationship among HIV-1, oxidative stress, and vascular homeostasis. We have demonstrated for the first time decreased vascular NO and elevated vascular superoxide as a result of HIV-1 protein expression. More importantly, we have shown that these HIV-1 protein-mediated effects are reversed when rats are given the GSH precursor procysteine, thus highlighting the importance of antioxidant depletion and oxidative stress in HIV-1-associated vascular dysfunction.
Perhaps the most striking observation in the HIV-1 transgenic rat is the dramatic decrease in aortic tissue NO and NO metabolites in the blood. These findings support a previous study by Dickie et al. (24), who demonstrated lower NO production in macrophages isolated from a murine HIV-1 transgenic model. Decreased NO levels are associated with increased endothelial cell permeability, activation, and dys-function in a number of experimental systems. Endothelium-dependent relaxation data from this study provide compelling evidence for endothelial dysfunction in HIV-1 transgenic rats. We observed similar impairment of endothelium-dependent relaxation in a mouse model expressing the same NL4-3Δ gag/pol HIV-1 transgene (unpublished results), suggesting that this is not a species-specific observation. Furthermore, we found that endothelium-dependent relaxation was not impaired in 3-mo-old transgenic rats (data not shown), implying that the impaired vascular response occurs over time due to increased exposure to HIV-1 proteins. Although other groups have demonstrated endothelial dysfunction using a murine leukemia retrovirus model (5) or Tat-incubated porcine arteries (73), our study has the advantage of measuring endothelial dysfunction in response to actual HIV-1 regulatory and structural proteins in circulation. NO depletion and endothelial dysfunction are clearly associated with HIV-1 transgene expression and support the transgenic rat as a useful model to study HIV-1-associated vascular effects.
Endothelial dysfunction promotes the development of cardiovascular disease in otherwise healthy individuals (37); however, it remains to be determined whether lower NO levels increase cardiovascular risk in HIV-1-positive patients. Studies have shown that HIV-1-infected patients have significantly less exhaled NO than uninfected individuals (59, 75). In addition, NO deficiencies have been reported in HIV-1 patients with pulmonary tuberculosis (80) and toxoplasmic encephalitis (87). Although it is tempting to speculate that NO deficiencies in the clinical setting predispose HIV-1 patients to cardiovascular risk, it is also important to note that other factors such as disease stage, antiretroviral treatment, and secondary infections may complicate the drawing of definitive conclusions. For example, the protease inhibitor (PI) indinavir reduces endothelial NO and enhances endothelial dysfunction in healthy HIV-1-negative men (83). Patients receiving a PI-based therapy also demonstrate significantly greater carotid intima media thickness than patients on a non-PI-based therapy or HIV-1-negative volunteers (85). Nevertheless, endothelial dysfunction, as determined by impaired flow-mediated dilation (FMD) of the brachial artery, has been reported in some HIV-1-infected patients with no associations between FMD and antiretroviral drug regimens (9, 10, 84). These clinical findings argue for a potential viral effect on endothelial dysfunction in vivo. Our current study provides compelling evidence that HIV-1 protein expression in the transgenic rat model causes endothelial dys-function, as determined by NO depletion and impaired endothelium-dependent relaxation. These findings support clinical studies implicating viral proteins in the development of cardiovascular complications in HIV-1-positive patients.
In assessing NO biomarkers, we determined that HIV-1 transgenic rats have lower levels of NO-Hb and serum nitrite. ESR measurement of NO-Hb has proven to be a highly quantitative and specific method for detecting endothelium-derived NO bioavailability in vivo (26). Similarly, although NO decomposition produces both nitrate and nitrite, circulating nitrite levels positively correlate with endothelial-specific NOS activity (48, 55). Studies utilizing eNOS−/− mice or specific pharmacological inhibitors have demonstrated that eNOS is the source of >70% of circulating nitrite and that serum nitrate levels reflect NO production from other NOS isoforms or from dietary nitrate intake (48, 89). Therefore, we decided to determine whether the effects of HIV-1 transgene expression on NO levels are the result of alterations in eNOS. Interestingly, we did not detect any differences in total eNOS expression when measuring eNOS protein levels. In addition, no significant differences in phosphorylation of eNOS-Ser1177 or BH4 levels were detected. Although the lack of an effect on BH4 suggests that eNOS is likely not uncoupled in the HIV-1 transgenic rats, it is possible that decreased l-arginine or increased asymmetric dimethylarginine (ADMA) could lead to eNOS uncoupling (34). However, our data collectively suggest that differences in eNOS regulation, as measured by eNOS-Ser1177 phosphorylation and BH4 availability, do not explain the decreased NO levels in HIV-1 transgenic rats.
We next examined ROS, particularly superoxide, in aortas from wild-type and transgenic rats, because increased superoxide is associated with various cardiovascular diseases. Moreover, when superoxide is present in high enough concentrations, reaction rates favor the interaction of superoxide with NO (7). This reaction depletes NO levels and concomitantly creates the damaging oxidant peroxynitrite. Although our DHE fluorescence data qualitatively indicate that HIV-1 protein expression induced vascular oxidative stress, ESR experiments in intact vascular sections unequivocally demonstrated increased superoxide, which was restored to wild-type levels upon preincubation with the superoxide scavenger PEG-SOD. ESR analysis using the spin probe CMH has certain technical advantages over other methods of superoxide detection, including high sensitivity and specificity, low cellular toxicity, long half-life and stability, and resistance to auto-oxidation (27). Moreover, peroxynitrite generation was higher in the transgenic rat aortas according to our ELISA data for 3-nitrotyrosine, a footprint of peroxynitrite damage. These data support findings from Bauer's group that show increased 3-nitrotyrosine immunostaining in aorta and coronary vessels from a murine LP-BM5 retrovirus-infection model, as well as increased 3-nitrotyrosine immunostaining in left ventricular myocardium of HIV-1-positive patients (5). Our group (44) has also previously demonstrated increased nitrotyrosine levels in the lungs of HIV-1 transgenic mice challenged with endotoxin. Thus these data show that aortas from HIV-1 transgenic rats are in a state of oxidative stress. The reduction in NO in the transgenic rats most likely arises not through alterations in eNOS expression or regulation, but rather from enhanced superoxide-mediated NO degradation.
Increased superoxide levels arise from an increase in superoxide-generating systems and/or impairment of superoxide scavenging. Others have reported that HIV-1-Tat activates NADPH oxidase in human umbilical vein endothelial cells in culture (94). Our current study does not demonstrate upregulation of NADPH oxidase subunits, which may be explained by inherent differences between in vitro and in vivo experimental systems. However, we believe the HIV-1 transgenic model more accurately mimics the effects of viral proteins on the vasculature in the clinical setting, given that it takes into account the effects of circulating HIV-1 protein levels, as well as the effects of multiple HIV-1 proteins (i.e., Nef, gp120) rather than Tat alone (78). Our data also do not allow us to conclusively exclude a potential contribution of mitochondria to total cellular superoxide levels. However, the lack of an effect on Mn-SOD, which is the primary line of defense against mitochondrial superoxide, suggests that the elevation of superoxide in the HIV-1 transgenic aortas is likely not due to changes in mitochondrial ROS generation. Although we did not detect differences in XO or NADPH oxidase subunit expression or NADPH oxidase activity, we present data showing decreased expression and activity of the cytosolic isoform of SOD, which is the enzyme responsible for the dismutation of superoxide to hydrogen peroxide. This decrease in Cu/Zn-SOD activity is likely to be physiologically significant, given that Cu/Zn-SOD is the major isoform in the rodent vasculature, accounting for 50–80% of total SOD activity (30). In addition, aortic levels of the antioxidant GSH were significantly depressed in the HIV-1 transgenic rats, suggesting a diminished capacity to detoxify other free radicals in the vasculature.
Reductions in SOD are reported in many models of vascular disease but are often believed to be a consequence of enhanced oxidative stress from other sources. However, genetic studies in mice show that Cu/Zn-SOD deficiency directly increases vascular oxidative stress and impairs endothelium-dependent relaxation (22, 25). Moreover, depleting GSH by administering the GSH synthase inhibitor buthionine sulfoximine (BSO) has been used as a model to cause impaired endothelium-dependent vasorelaxation (56) as well as severe hypertension in rats (91). Thus reductions in antioxidant capacity, although likely not the most common means of primarily elevating vascular oxidative stress, have been causatively associated with the development of vascular disease in various models. It is possible that this unusual condition in the HIV-1 transgenic rats is due to the activity of unique HIV-1 proteins. For example, the viral protein Tat, which is expressed and functional in our HIV-1 transgenic rats (78), is a known transcriptional transactivator of many host cellular genes and has been shown to directly repress SOD mRNA transcripts in nonvascular cell types (33). It is possible that Tat directly regulates the Cu/Zn-SOD mRNA transcript in the endothelium or vascular smooth muscle cells in the HIV-1 transgenic rats. Alternatively, Tat may activate signaling pathways in endothelial cells that inhibit sod1 promoter activity. Incubation of endothelial cells with recombinant Tat increases tumor necrosis factor (TNF)-α production (1), and TNF-α inhibits Cu/Zn-SOD promoter activity through activation of the JNK pathway (2). Tat-induced activation of TNF-α and JNK signaling may inhibit vascular Cu/Zn-SOD gene transcription, decrease Cu/Zn-SOD protein and activity, and increase superoxide levels. This may provide a potential positive feedback loop in the context of HIV-1 infection as viral replication, and thus Tat production, are increased by the activation of the redox-sensitive transcription factor NF-κB (63). Tat-mediated reduction in Cu/Zn-SOD may increase superoxide levels, which may then stimulate HIV-1 replication and viral protein production in a continuous cycle. Clearly, more research is warranted to determine the mechanism by which HIV-1 protein expression in the transgenic rat decreases Cu/Zn-SOD, as well as the potential role that GSH depletion might have on this observation.
Interestingly, HIV-1 patients have lower levels of the antioxidants ascorbic acid, tocopherol, carotenoids, SOD, and GSH (13, 31, 76). Our data show that HIV-1 protein expression in the transgenic rat decreased antioxidant levels, which likely accounts for the observed increase in vascular superoxide. This concept is further reinforced by our data showing that procysteine treatment of transgenic rats lowered aortic superoxide, increased aortic NO-Fe(DETC)2, and restored NO-Hb to wild-type levels while improving acetylcholine-mediated vasorelaxation. Although procysteine supplementation is most likely not a cure-all, this finding begs the question of whether antioxidant therapy in patients could at least ameliorate some HIV-1-associated complications. Administration of the GSH precursor N-acetyl-cysteine to HIV-1 patients is associated with restored GSH levels and decreased mortality (42). Dual vitamin C and E supplementation reduces plasma lipid peroxidation and oxidative stress in HIV-1-positive individuals (3), and treatment with multivitamins (containing vitamins B, C, and E) significantly reduces the incidence of hypertension in HIV-1-positive pregnant women (64). Similarly, twice daily supplementation of HIV-1 patients with either selenium or β-carotene for 1 yr prevented an increase in serum von Willebrand factor and soluble thrombomodulin (21), two common markers of endothelial dysfunction. By therapeutically targeting the source of HIV-1-induced oxidative stress, through either antioxidant supplementation or target-based drug development, we may be able to help prevent or ameliorate the progression of cardiovascular disease in HIV-1 patients.
The HIV-1 protein(s) that increase cardiovascular risk are unknown, as is the exact mechanism by which this occurs. More than likely, HIV-1-associated cardiovascular disease progression is a multifactorial process, resulting from a combination of viral proteins interacting through distinct pathways. Recently, Mujawar et al. (68) demonstrated that Nef inhibits cholesterol efflux from ABCA1 transporters in macrophages, which are responsible for lipidation of apoA-1 and the production of HDL. In the context of HIV-1 infection, Nef may reroute host cholesterol transport to support viral replication and thereby lower circulating HDL levels. This decrease in HDL, coupled with an increase in lipid-laden macrophages, may contribute to increased cardiovascular risk in HIV-1-positive patients. In addition to altered lipid metabolism, Nef and Tat are associated with ROS generation in monocytes and microglia (50, 71, 92), and transgenic mouse studies have shown that Tat decreases liver and cardiac GSH (20, 77). Tat and gp120 induce expression of cell adhesion molecules (79) and lead to increased endothelial cell permeability (4, 11, 79) and dysfunction (73). Protein expression of Nef, Tat, and gp120 has been reported previously in the spleens of HIV-1 transgenic rats (78). Furthermore, gp120 is also present in transgenic rat macrophages, B cells, T cells, and in the serum at a concentration of 141 pg/ml (78). These levels are lower than reported circulating levels in HIV-1 patients (36, 70), suggesting that our results may actually underestimate the direct effects of HIV-1 proteins on the vasculature in humans.
Our study adds to the growing body of evidence linking HIV-1 infection to cardiovascular disease. We have shown that in the absence of antiretroviral therapy, HIV-1-protein expression reduces vascular and systemic NO bioavailability. Furthermore, HIV-1 transgenic rat aortas demonstrate significantly less maximum relaxation to acetylcholine, indicative of endothelial dysfunction. NO depletion likely does not occur through regulation of eNOS, but rather through enhanced superoxide-mediated degradation. The reduction of vascular antioxidant capacity allows for elevated superoxide levels to react with NO, resulting in NO depletion and the production of peroxynitrite. The increase in vascular ROS coupled with NO deficiency predisposes the aorta to cardiovascular risk. Procysteine administration reverses these effects, highlighting the importance of antioxidant reduction in HIV-1-associated cardiovascular disease. These findings further the state of the field in three major ways. First, they show that HIV-1 proteins, in the absence of antiretroviral therapy, likely contribute to increased cardiovascular risk by inducing oxidative stress in HIV-1 patients. Second, our findings validate use of the HIV-1 transgenic rat as a safe and appropriate small-animal model for studying HIV-1-induced cardiovascular disease. Finally, our findings point to the importance of antioxidant deficiencies in causing HIV-1-associated cardiovascular disease and suggest the possibility of pharmacologically targeting antioxidant systems for future drug development in this growing population.
ACKNOWLEDGMENTS
We acknowledge Erik Walp and Robert Raynor for assistance in animal experiments and Alana Reed and Kristi Porter for critical discussion of the manuscript.
GRANTS
This research was funded by National Heart, Lung, and Blood Institute Grant R01 HL070892 (to R. L. Sutliff).
REFERENCES
- 1.Acheampong E, Mukhtar M, Parveen Z, Ngoubilly N, Ahmad N, Patel C, Pomerantz RJ. Ethanol strongly potentiates apoptosis induced by HIV-1 proteins in primary human brain microvascular endothelial cells. Virology. 2002;304:222–234. doi: 10.1006/viro.2002.1666. [DOI] [PubMed] [Google Scholar]
- 2.Afonso V, Santos G, Collin P, Khatib AM, Mitrovic DR, Lomri N, Leitman DC, Lomri A. Tumor necrosis factor-alpha down-regulates human Cu/Zn superoxide dismutase 1 promoter via JNK/AP-1 signaling pathway. Free Radic Biol Med. 2006;41:709–721. doi: 10.1016/j.freeradbiomed.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 3.Allard JP, Aghdassi E, Chau J, Tam C, Kovacs CM, Salit IE, Walmsley SL. Effects of vitamin E and C supplementation on oxidative stress and viral load in HIV-infected subjects. AIDS. 1998;12:1653–1659. doi: 10.1097/00002030-199813000-00013. [DOI] [PubMed] [Google Scholar]
- 4.Annunziata P, Cioni C, Toneatto S, Paccagnini E. HIV-1 gp120 increases the permeability of rat brain endothelium cultures by a mechanism involving substance P. AIDS. 1998;12:2377–2385. doi: 10.1097/00002030-199818000-00006. [DOI] [PubMed] [Google Scholar]
- 5.Baliga RS, Chaves AA, Jing L, Ayers LW, Bauer JA. AIDS-related vasculopathy: evidence for oxidative and inflammatory pathways in murine and human AIDS. Am J Physiol Heart Circ Physiol. 2005;289:H1373–H1380. doi: 10.1152/ajpheart.00304.2005. [DOI] [PubMed] [Google Scholar]
- 6.Baruchel S, Wainberg MA. The role of oxidative stress in disease progression in individuals infected by the human immunodeficiency virus. J Leukoc Biol. 1992;52:111–114. doi: 10.1002/jlb.52.1.111. [DOI] [PubMed] [Google Scholar]
- 7.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and the ugly. Am J Physiol Cell Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 8.Benov L, Sztejnberg L, Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med. 1998;25:826–831. doi: 10.1016/s0891-5849(98)00163-4. [DOI] [PubMed] [Google Scholar]
- 9.Blum A, Hadas V, Burke M, Yust I, Kessler A. Viral load of the human immunodeficiency virus could be an independent risk factor for endothelial dysfunction. Clin Cardiol. 2005;28:149–153. doi: 10.1002/clc.4960280311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonnet D, Aggoun Y, Szezepanski I, Bellal N, Blanche S. Arterial stiffness and endothelial dysfunction in HIV-infected children. AIDS. 2004;18:1037–1041. doi: 10.1097/00002030-200404300-00012. [DOI] [PubMed] [Google Scholar]
- 11.Bragardo M, Buonfiglio D, Feito MJ, Bonissoni S, Redoglia V, Rojo JM, Ballester S, Portoles P, Garbarino G, Malavasi F, Dianzani U. Modulation of lymphocyte interaction with endothelium and homing by HIV-1 gp120. J Immunol. 1997;159:1619–1627. [PubMed] [Google Scholar]
- 12.Brown LA, Ping XD, Harris FL, Gauthier TW. Glutathione availability modulates alveolar macrophage function in the chronic ethanol-fed rat. Am J Physiol Lung Cell Mol Physiol. 2007;292:L824–L832. doi: 10.1152/ajplung.00346.2006. [DOI] [PubMed] [Google Scholar]
- 13.Buhl R, Jaffe HA, Holroyd KJ, Wells FB, Mastrangeli A, Saltini C, Cantin AM, Crystal RG. Systemic glutathione deficiency in symptom-free HIV-seropositive individuals. Lancet. 1989;2:1294–1298. doi: 10.1016/s0140-6736(89)91909-0. [DOI] [PubMed] [Google Scholar]
- 14.Cai H, Dikalov S, Griendling KK, Harrison DG. Detection of reactive oxygen species and nitric oxide in vascular cells and tissues: comparison of sensitivity and specificity. Methods Mol Med. 2007;139:293–312. doi: 10.1007/978-1-59745-571-8_20. [DOI] [PubMed] [Google Scholar]
- 15.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 16.Carr A. Cardiovascular risk factors in HIV-infected patients. J Acquir Immune Defic Syndr. 2003;34(Suppl 1):S73–S78. doi: 10.1097/00126334-200309011-00011. [DOI] [PubMed] [Google Scholar]
- 17.Castro L, Rodriguez M, Radi R. Aconitase is readily inactivated by peroxynitrite, but not by its precursor, nitric oxide. J Biol Chem. 1994;269:29409–29415. [PubMed] [Google Scholar]
- 18.Cebrian M, Miro O, Font C, Coll-Vinent B, Grau JM. HIV-related vasculitis. AIDS Patient Care STDS. 1997;11:245–258. doi: 10.1089/apc.1997.11.245. [DOI] [PubMed] [Google Scholar]
- 19.Chang HC, Samaniego F, Nair BC, Buonaguro L, Ensoli B. HIV-1 Tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycans through its basic region. AIDS. 1997;11:1421–1431. doi: 10.1097/00002030-199712000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Choi J, Liu RM, Kundu RK, Sangiorgi F, Wu W, Maxson R, Forman HJ. Molecular mechanism of decreased glutathione content in human immunodeficiency virus type 1 Tat-transgenic mice. J Biol Chem. 2000;275:3693–3698. doi: 10.1074/jbc.275.5.3693. [DOI] [PubMed] [Google Scholar]
- 21.Constans J, Seigneur M, Blann AD, Renard M, Resplandy F, Amiral J, Guerin V, Boisseau MR, Conri C. Effect of the antioxidants selenium and beta-carotene on HIV-related endothelium dysfunction. Thromb Haemost. 1998;80:1015–1017. [PubMed] [Google Scholar]
- 22.Cooke CL, Davidge ST. Endothelial-dependent vasodilation is reduced in mesenteric arteries from superoxide dismutase knockout mice. Cardiovasc Res. 2003;60:635–642. doi: 10.1016/j.cardiores.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 23.Dickie P, Felser J, Eckhaus M, Bryant J, Silver J, Marinos N, Notkins AL. HIV-associated nephropathy in transgenic mice expressing HIV-1 genes. Virology. 1991;185:109–119. doi: 10.1016/0042-6822(91)90759-5. [DOI] [PubMed] [Google Scholar]
- 24.Dickie P, Roberts A, Lee R. A defect in HIV-1 transgenic murine macrophages results in deficient nitric oxide production. J Leukoc Biol. 2001;70:592–600. [PubMed] [Google Scholar]
- 25.Didion SP, Ryan MJ, Didion LA, Fegan PE, Sigmund CD, Faraci FM. Increased superoxide and vascular dysfunction in CuZnSOD-deficient mice. Circ Res. 2002;91:938–944. doi: 10.1161/01.res.0000043280.65241.04. [DOI] [PubMed] [Google Scholar]
- 26.Dikalov S, Fink B. ESR techniques for the detection of nitric oxide in vivo and in tissues. Methods Enzymol. 2005;396:597–610. doi: 10.1016/S0076-6879(05)96052-7. [DOI] [PubMed] [Google Scholar]
- 27.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HH, Owens GK, Lambeth JD, Griendling KK. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 29.El-Sadr WM, Lundgren JD, Neaton JD, Gordin F, Abrams D, Arduino RC, Babiker A, Burman W, Clumeck N, Cohen CJ, Cohn D, Cooper D, Darbyshire J, Emery S, Fatkenheuer G, Gazzard B, Grund B, Hoy J, Klingman K, Losso M, Markowitz N, Neuhaus J, Phillips A, Rappoport C. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. 2006;355:2283–2296. doi: 10.1056/NEJMoa062360. [DOI] [PubMed] [Google Scholar]
- 30.Faraci FM, Didion SP. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol. 2004;24:1367–1373. doi: 10.1161/01.ATV.0000133604.20182.cf. [DOI] [PubMed] [Google Scholar]
- 31.Favier A, Sappey C, Leclerc P, Faure P, Micoud M. Antioxidant status and lipid peroxidation in patients infected with HIV. Chem Biol Interact. 1994;91:165–180. doi: 10.1016/0009-2797(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 32.Fenster BE, Tsao PS, Rockson SG. Endothelial dysfunction: clinical strategies for treating oxidant stress. Am Heart J. 2003;146:218–226. doi: 10.1016/S0002-8703(02)94796-4. [DOI] [PubMed] [Google Scholar]
- 33.Flores SC, Marecki JC, Harper KP, Bose SK, Nelson SK, McCord JM. Tat protein of human immunodeficiency virus type 1 represses expression of manganese superoxide dismutase in HeLa cells. Proc Natl Acad Sci USA. 1993;90:7632–7636. doi: 10.1073/pnas.90.16.7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 35.Gewaltig MT, Kojda G. Vasoprotection by nitric oxide: mechanisms and therapeutic potential. Cardiovasc Res. 2002;55:250–260. doi: 10.1016/s0008-6363(02)00327-9. [DOI] [PubMed] [Google Scholar]
- 36.Gilbert M, Kirihara J, Mills J. Enzyme-linked immunoassay for human immunodeficiency virus type 1 envelope glycoprotein 120. J Clin Micro-biol. 1991;29:142–147. doi: 10.1128/jcm.29.1.142-147.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glasser SP, Selwyn AP, Ganz P. Atherosclerosis: risk factors and the vascular endothelium. Am Heart J. 1996;131:379–384. doi: 10.1016/s0002-8703(96)90370-1. [DOI] [PubMed] [Google Scholar]
- 38.Greenwood AJ, Hughes J, Wallace G, Seed P, Stanford MR, Graham EM. Soluble intercellular adhesion molecule-1 (sICAM-1) and vascular cell adhesion molecule-1 (sVCAM-1) in patients with HIV/AIDS does not appear to correlate with cytomegalovirus retinitis. Int J STD AIDS. 1998;9:713–714. [PubMed] [Google Scholar]
- 39.Grunfeld C, Pang M, Doerrler W, Shigenaga JK, Jensen P, Feingold KR. Lipids, lipoproteins, triglyceride clearance, and cytokines in human immunodeficiency virus infection and the acquired immunodeficiency syndrome. J Clin Endocrinol Metab. 1992;74:1045–1052. doi: 10.1210/jcem.74.5.1373735. [DOI] [PubMed] [Google Scholar]
- 40.Hamilton CA, Miller WH, Al-Benna S, Brosnan MJ, Drummond RD, McBride MW, Dominiczak AF. Strategies to reduce oxidative stress in cardiovascular disease. Clin Sci (Lond) 2004;106:219–234. doi: 10.1042/CS20030379. [DOI] [PubMed] [Google Scholar]
- 41.Hausladen A, Fridovich I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J Biol Chem. 1994;269:29405–29408. [PubMed] [Google Scholar]
- 42.Herzenberg LA, De Rosa SC, Dubs JG, Roederer M, Anderson MT, Ela SW, Deresinski SC, Herzenberg LA. Glutathione deficiency is associated with impaired survival in HIV disease. Proc Natl Acad Sci USA. 1997;94:1967–1972. doi: 10.1073/pnas.94.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hung CC, Chang SC. Impact of highly active antiretroviral therapy on incidence and management of human immunodeficiency virus-related opportunistic infections. J Antimicrob Chemother. 2004;54:849–853. doi: 10.1093/jac/dkh438. [DOI] [PubMed] [Google Scholar]
- 44.Jacob BA, Porter KM, Elms SC, Cheng PY, Jones DP, Sutliff RL. HIV-1-induced pulmonary oxidative and nitrosative stress: exacerbated response to endotoxin administration in HIV-1 transgenic mouse model. Am J Physiol Lung Cell Mol Physiol. 2006;291:L811–L819. doi: 10.1152/ajplung.00468.2005. [DOI] [PubMed] [Google Scholar]
- 45.Johnson MA, Gathe JC, Jr, Podzamczer D, Molina JM, Naylor CT, Chiu YL, King MS, Podsadecki TJ, Hanna GJ, Brun SC. A once-daily lopinavir/ritonavir-based regimen provides noninferior antiviral activity compared with a twice-daily regimen. J Acquir Immune Defic Syndr. 2006;43:153–160. doi: 10.1097/01.qai.0000242449.67155.1a. [DOI] [PubMed] [Google Scholar]
- 46.Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 47.Joshi VV, Pawel B, Connor E, Sharer L, Oleske JM, Morrison S, Marin-Garcia J. Arteriopathy in children with acquired immune deficiency syndrome. Pediatr Pathol. 1987;7:261–275. doi: 10.1080/15513818709177129. [DOI] [PubMed] [Google Scholar]
- 48.Kleinbongard P, Dejam A, Lauer T, Rassaf T, Schindler A, Picker O, Scheeren T, Godecke A, Schrader J, Schulz R, Heusch G, Schaub GA, Bryan NS, Feelisch M, Kelm M. Plasma nitrite reflects constitutive nitric oxide synthase activity in mammals. Free Radic Biol Med. 2003;35:790–796. doi: 10.1016/s0891-5849(03)00406-4. [DOI] [PubMed] [Google Scholar]
- 49.Kopp JB, Klotman ME, Adler SH, Bruggeman LA, Dickie P, Marinos NJ, Eckhaus M, Bryant JL, Notkins AL, Klotman PE. Progressive glomerulosclerosis and enhanced renal accumulation of basement membrane components in mice transgenic for human immunodeficiency virus type 1 genes. Proc Natl Acad Sci USA. 1992;89:1577–1581. doi: 10.1073/pnas.89.5.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lachgar A, Sojic N, Arbault S, Bruce D, Sarasin A, Amatore C, Bizzini B, Zagury D, Vuillaume M. Amplification of the inflammatory cellular redox state by human immunodeficiency virus type 1-immunosuppressive tat and gp160 proteins. J Virol. 1999;73:1447–1452. doi: 10.1128/jvi.73.2.1447-1452.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lafeuillade A, Alessi MC, Poizot-Martin I, Boyer-Neumann C, Zandotti C, Quilichini R, Aubert L, Tamalet C, Juhan-Vague I, Gastaut JA. Endothelial cell dysfunction in HIV infection. J Acquir Immune Defic Syndr. 1992;5:127–131. [PubMed] [Google Scholar]
- 52.Lam CF, Peterson TE, Richardson DM, Croatt AJ, d'Uscio LV, Nath KA, Katusic ZS. Increased blood flow causes coordinated upregulation of arterial eNOS and biosynthesis of tetrahydrobiopterin. Am J Physiol Heart Circ Physiol. 2006;290:H786–H793. doi: 10.1152/ajpheart.00759.2005. [DOI] [PubMed] [Google Scholar]
- 53.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lau YE, Galligan JJ, Kreulen DL, Fink GD. Activation of ETB receptors increases superoxide levels in sympathetic ganglia in vivo. Am J Physiol Regul Integr Comp Physiol. 2006;290:R90–R95. doi: 10.1152/ajpregu.00505.2005. [DOI] [PubMed] [Google Scholar]
- 55.Lauer T, Preik M, Rassaf T, Strauer BE, Deussen A, Feelisch M, Kelm M. Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc Natl Acad Sci USA. 2001;98:12814–12819. doi: 10.1073/pnas.221381098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laursen JB, Boesgaard S, Trautner S, Rubin I, Poulsen HE, Alder-shvile J. Endothelium-dependent vasorelaxation in inhibited by in vivo depletion of vascular thiol levels: role of endothelial nitric oxide synthase. Free Radic Res. 2001;35:387–394. doi: 10.1080/10715760100300901. [DOI] [PubMed] [Google Scholar]
- 57.Lewis W, Copeland WC, Day BJ. Mitochondrial DNA depletion, oxidative stress, and mutation: mechanisms of dysfunction from nucleoside reverse transcriptase inhibitors. Lab Invest. 2001;81:777–790. doi: 10.1038/labinvest.3780288. [DOI] [PubMed] [Google Scholar]
- 58.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 59.Loveless MO, Phillips CR, Giraud GD, Holden WE. Decreased exhaled nitric oxide in subjects with HIV infection. Thorax. 1997;52:185–186. doi: 10.1136/thx.52.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lyle AN, Griendling KK. Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology (Bethesda) 2006;21:269–280. doi: 10.1152/physiol.00004.2006. [DOI] [PubMed] [Google Scholar]
- 61.Mandell BF, Calabrese LH. Infections and systemic vasculitis. Curr Opin Rheumatol. 1998;10:51–57. doi: 10.1097/00002281-199801000-00008. [DOI] [PubMed] [Google Scholar]
- 62.Maniker AH, Hunt CD. Cerebral aneurysm in the HIV patient: a report of six cases. Surg Neurol. 1996;46:49–54. doi: 10.1016/0090-3019(96)00030-4. [DOI] [PubMed] [Google Scholar]
- 63.Mellors JW, Griffith BP, Ortiz MA, Landry ML, Ryan JL. Tumor necrosis factor-alpha/cachectin enhances human immunodeficiency virus type 1 replication in primary macrophages. J Infect Dis. 1991;163:78–82. doi: 10.1093/infdis/163.1.78. [DOI] [PubMed] [Google Scholar]
- 64.Merchant AT, Msamanga G, Villamor E, Saathoff E, O'Brien M, Hertzmark E, Hunter DJ, Fawzi WW. Multivitamin supplementation of HIV-positive women during pregnancy reduces hypertension. J Nutr. 2005;135:1776–1781. doi: 10.1093/jn/135.7.1776. [DOI] [PubMed] [Google Scholar]
- 65.Miller FJ, Jr, Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298–1305. doi: 10.1161/01.res.82.12.1298. [DOI] [PubMed] [Google Scholar]
- 66.Miriyala S, Gongora Nieto MC, Mingone C, Smith D, Dikalov S, Harrison DG, Jo H. Bone morphogenic protein-4 induces hypertension in mice: role of noggin, vascular NADPH oxidases, and impaired vasorelaxation. Circulation. 2006;113:2818–2825. doi: 10.1161/CIRCULATIONAHA.106.611822. [DOI] [PubMed] [Google Scholar]
- 67.Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol. 2007;42:271–279. doi: 10.1016/j.yjmcc.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 68.Mujawar Z, Rose H, Morrow MP, Pushkarsky T, Dubrovsky L, Mukhamedova N, Fu Y, Dart A, Orenstein JM, Bobryshev YV, Bukrinsky M, Sviridov D. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol. 2006;4:e365. doi: 10.1371/journal.pbio.0040365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nath A. Human immunodeficiency virus (HIV) proteins in neuropatho-genesis of HIV dementia. J Infect Dis. 2002;186(Suppl 2):S193–S198. doi: 10.1086/344528. [DOI] [PubMed] [Google Scholar]
- 70.Oh SK, Cruikshank WW, Raina J, Blanchard GC, Adler WH, Walker J, Kornfeld H. Identification of HIV-1 envelope glycoprotein in the serum of AIDS and ARC patients. J Acquir Immune Defic Syndr. 1992;5:251–256. [PubMed] [Google Scholar]
- 71.Olivetta E, Pietraforte D, Schiavoni I, Minetti M, Federico M, Sanchez M. HIV-1 Nef regulates the release of superoxide anions from human macrophages. Biochem J. 2005;390:591–602. doi: 10.1042/BJ20042139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pace GW, Leaf CD. The role of oxidative stress in HIV disease. Free Radic Biol Med. 1995;19:523–528. doi: 10.1016/0891-5849(95)00047-2. [DOI] [PubMed] [Google Scholar]
- 73.Paladugu R, Fu W, Conklin BS, Lin PH, Lumsden AB, Yao Q, Chen C. Hiv Tat protein causes endothelial dysfunction in porcine coronary arteries. J Vasc Surg. 2003;38:549–556. doi: 10.1016/s0741-5214(03)00770-5. [DOI] [PubMed] [Google Scholar]
- 74.Palella FJ, Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338:853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 75.Palm J, Lidman C, Graf P, Alving K, Lundberg J. Nasal nitric oxide is reduced in patients with HIV. Acta Otolaryngol. 2000;120:420–423. doi: 10.1080/000164800750000676. [DOI] [PubMed] [Google Scholar]
- 76.Peterhans E. Reactive oxygen species and nitric oxide in viral diseases. Biol Trace Elem Res. 1997;56:107–116. doi: 10.1007/BF02778986. [DOI] [PubMed] [Google Scholar]
- 77.Raidel SM, Haase C, Jansen NR, Russ RB, Sutliff RL, Velsor LW, Day BJ, Hoit BD, Samarel AM, Lewis W. Targeted myocardial transgenic expression of HIV Tat causes cardiomyopathy and mitochondrial damage. Am J Physiol Heart Circ Physiol. 2002;282:H1672–H1678. doi: 10.1152/ajpheart.00955.2001. [DOI] [PubMed] [Google Scholar]
- 78.Reid W, Sadowska M, Denaro F, Rao S, Foulke J, Jr, Hayes N, Jones O, Doodnauth D, Davis H, Sill A, O'Driscoll P, Huso D, Fouts T, Lewis G, Hill M, Kamin-Lewis R, Wei C, Ray P, Gallo RC, Reitz M, Bryant J. An HIV-1 transgenic rat that develops HIV-related pathology and immunologic dysfunction. Proc Natl Acad Sci USA. 2001;98:9271–9276. doi: 10.1073/pnas.161290298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ren Z, Yao Q, Chen C. HIV-1 envelope glycoprotein 120 increases intercellular adhesion molecule-1 expression by human endothelial cells. Lab Invest. 2002;82:245–255. doi: 10.1038/labinvest.3780418. [DOI] [PubMed] [Google Scholar]
- 80.Schon T, Gebre N, Sundqvist T, Aderaye G, Britton S. Effects of HIV co-infection and chemotherapy on the urinary levels of nitric oxide metabolites in patients with pulmonary tuberculosis. Scand J Infect Dis. 1999;31:123–126. doi: 10.1080/003655499750006137. [DOI] [PubMed] [Google Scholar]
- 81.Seigneur M, Constans J, Blann A, Renard M, Pellegrin JL, Amiral J, Boisseau M, Conri C. Soluble adhesion molecules and endothelial cell damage in HIV infected patients. Thromb Haemost. 1997;77:646–649. [PubMed] [Google Scholar]
- 82.Sessa WC, Garcia-Cardena G, Liu J, Keh A, Pollock JS, Bradley J, Thiru S, Braverman IM, Desai KM. The Golgi association of endothelial nitric oxide synthase is necessary for the efficient synthesis of nitric oxide. J Biol Chem. 1995;270:17641–17644. doi: 10.1074/jbc.270.30.17641. [DOI] [PubMed] [Google Scholar]
- 83.Shankar SS, Dube MP. Clinical aspects of endothelial dysfunction associated with human immunodeficiency virus infection and antiretroviral agents. Cardiovasc Toxicol. 2004;4:261–269. doi: 10.1385/ct:4:3:261. [DOI] [PubMed] [Google Scholar]
- 84.Solages A, Vita JA, Thornton DJ, Murray J, Heeren T, Craven DE, Horsburgh CR., Jr Endothelial function in HIV-infected persons. Clin Infect Dis. 2006;42:1325–1332. doi: 10.1086/503261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sudano I, Spieker LE, Noll G, Corti R, Weber R, Luscher TF. Cardiovascular disease in HIV infection. Am Heart J. 2006;151:1147–1155. doi: 10.1016/j.ahj.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 86.Tarpey MM, Wink DA, Grisham MB. Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol. 2004;286:R431–R444. doi: 10.1152/ajpregu.00361.2003. [DOI] [PubMed] [Google Scholar]
- 87.Torre D, Zeroli C, Ferrario G, Pugliese A, Speranza F, Orani A, Casari S, Bassi P, Poggio A, Carosi GP, Fiori GP. Levels of nitric oxide, gamma interferon and interleukin-12 in AIDS patients with toxoplasmic encephalitis. Infection. 1999;27:218–220. doi: 10.1007/BF02561533. [DOI] [PubMed] [Google Scholar]
- 88.Triggle CR, Hollenberg M, Anderson TJ, Ding H, Jiang Y, Ceroni L, Wiehler WB, Ng ES, Ellis A, Andrews K, McGuire JJ, Pannirselvam M. The endothelium in health and disease—a target for therapeutic intervention. J Smooth Muscle Res. 2003;39:249–267. doi: 10.1540/jsmr.39.249. [DOI] [PubMed] [Google Scholar]
- 89.Tsikas D, Gutzki FM, Stichtenoth DO. Circulating and excretory nitrite and nitrate as indicators of nitric oxide synthesis in humans: methods of analysis. Eur J Clin Pharmacol. 2006;62(Suppl 1):51–59. [Google Scholar]
- 90.Van der Valk M, Kastelein JJ, Murphy RL, van Leth F, Katlama C, Horban A, Glesby M, Behrens G, Clotet B, Stellato RK, Molhuizen HO, Reiss P. Nevirapine-containing antiretroviral therapy in HIV-1 infected patients results in an anti-atherogenic lipid profile. AIDS. 2001;15:2407–2414. doi: 10.1097/00002030-200112070-00008. [DOI] [PubMed] [Google Scholar]
- 91.Vaziri ND, Wang XQ, Oveisi F, Rad B. Induction of oxidative stress by glutathione depletion causes severe hypertension in normal rats. Hypertension. 2000;36:142–146. doi: 10.1161/01.hyp.36.1.142. [DOI] [PubMed] [Google Scholar]
- 92.Vilhardt F, Plastre O, Sawada M, Suzuki K, Wiznerowicz M, Kiyokawa E, Trono D, Krause KH. The HIV-1 Nef protein and phagocyte NADPH oxidase activation. J Biol Chem. 2002;277:42136–42143. doi: 10.1074/jbc.M200862200. [DOI] [PubMed] [Google Scholar]
- 93.Wink DA, Mitchell JB. Chemical biology of nitric oxide: Insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 94.Wu RF, Gu Y, Xu YC, Mitola S, Bussolino F, Terada LS. Human immunodeficiency virus type 1 Tat regulates endothelial cell actin cytoskeletal dynamics through PAK1 activation and oxidant production. J Virol. 2004;78:779–789. doi: 10.1128/JVI.78.2.779-789.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, Taylor WR, Griendling KK. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998;32:488–495. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]