

Abstract
With the exponential rise in the number of viable novel drug targets, computational methods are being increasingly applied to accelerate the drug discovery process. Virtual High Throughput Screening (vHTS) is one such established methodology to identify drug candidates from large collection of compound libraries. Although it complements the expensive and time consuming High Throughput Screening (HTS) of compound libraries, vHTS possess inherent challenges. The successful vHTS requires the careful implementation of each phase of computational screening experiment right from target preparation to hit identification and lead optimization. This article discusses some of the important considerations that are imperative for designing a successful vHTS experiment.
Keywords: virtual high throughput screening, receptor based and ligand based screening, homology models, chemical databases, ADME filters, toxicity filters
Background
The discovery of novel drug targets has increased exponentially in recent years due to advances in genomic and molecular biology techniques. Experimental and computational methods are effectively applied to accelerate the process of lead identification and optimization. The chemical leads are small potential drug like molecules which are capable of modulating the function of the target proteins that are further optimized to act as a therapeutic drug against a targeted disease. Conventional experimental methods like High Throughput Screening (HTS) continue to be the best method for rapid identification of drug leads. HTS identifies lead molecules by performing individual biochemical assays with over millions of compounds. However, the huge cost and time consumed with this technology has lead to the integration of cheaper and effective computational methodology namely virtual High Throughput Screening (vHTS). vHTS is a computational screening method which is widely applied to screen insilico collection of compound libraries to check the binding affinity of the target receptor with the library compounds [1]. This is achieved by using a scoring function which computes the complementarity of the target receptor with the compounds. HTS and vHTS are complementary methods [2] and vHTS has been shown to reduce false positives in HTS [3]. Several vHTS strategies have been practiced [4] and the technique is being continuously optimized for better performance.
Methodology
Based on the availability of structural data, vHTS is carried out using receptor based or ligand based screening methods. Receptor based method involves usage of 3D structure of the target receptors to search for potential candidate compounds that can modulate the target receptor function. Each of the database compounds is docked into the receptor binding site and best electrostatic fit is predicted. Successful application of receptor based method has been reported in many targets [5]. In case where structural information of the target is unavailable, ligand based method make use of the information provided by known inhibitors. Structures similar to the known inhibitors are identified from chemical databases by variety of methods, including similarity and substructure searching, pharmacophore matching and 3D shape matching. Numerous successful applications of ligand based methods have also been reported [2]. In both the methods, the compounds are ranked using an appropriate scoring function based on either complementarity or similarity and the top ranking compounds are taken to the next step of experimental assays. General workflow of vHTS experiment is shown in Figure 1.
Figure 1.
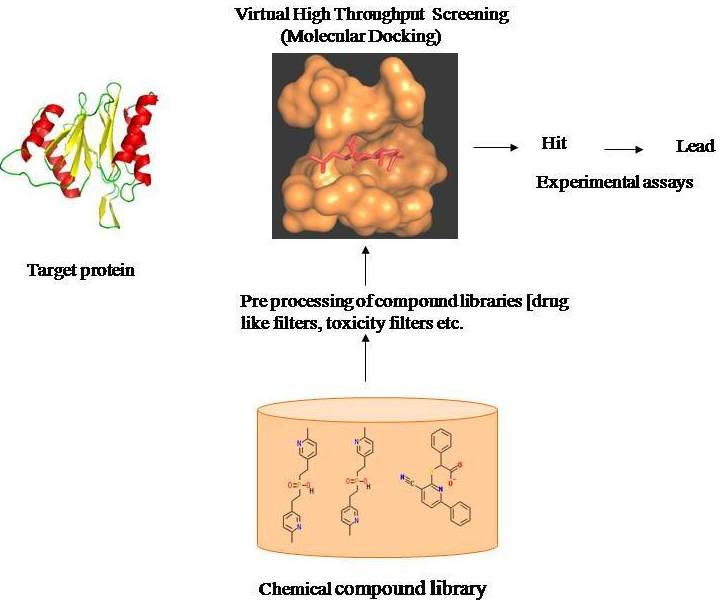
Virtual high throughput screening workflow.
Important considerations for vHTS
Incorporating target flexibility in molecular docking
Proteins undergo conformational change upon ligand binding and due to this rigid protein docking may be inadequate for computational drug designing applications [6,7]. Sampling the intrinsic flexibility of the protein binding site remains a great challenge in flexible protein docking. Nevertheless this problem is being increasingly addressed by several advanced methods which aim at accurate representation of protein flexibility and its ligand recognition [8]. Most of the attempts to incorporate protein flexibility make use of an ensemble of protein structures. A crystallographic structure of the same target bound to different ligands provides a good starting point to obtain ensemble of protein structures. Ensemble of structures derived from NMR studies has been used to incorporate protein flexibility in docking studies of HIV-1 protease that has demonstrated good docking accuracy [9]. The study has also shown that docking against NMR structures provide an efficient alternative method for standard docking against individual X-ray crystal structures. In the absence of experimental studies, conformational ensemble generated by Molecular Dynamics (MD) simulations has also been used to dock ligands to flexible receptors [10]. The choice of appropriate docking tool which considers reasonable flexibility in protein conformation enhances the reliability of prediction of protein binding affinity.
vHTS using Homology models
In the absence of crystal structures, homology models have helped in lead identification process of drug targets like falcipain-3 [11] and GPCR [12]. Recently, we have used a homology model of a novel drug target of Plasmodium falciparum PfHslV [13], an ATP dependant protease and this model has helped in the identification of several hit compounds (unpublished data). Database of homology models like ModWeb [14] and organism specific database like plasmodium model database [15] which are being experimentally validated can be a good source to obtain the models of target proteins. It has been shown that models based on templates with greater than 50% sequence identity has yielded 5-fold better enrichment than one with less sequence identity [16]. On the other hand, small structural errors, such as incorrect assignment of few side chain rotamers have been the cause of poor performance. Hence successful vHTS using a homology model depends on the quality of template crystal structures.
Pre - processing chemical databases
Wide collections of both publicly and commercially available compound libraries are screened in the lead identification process. The compound collection for screening is usually derived from commercial corporate collection or from in silico libraries generated by combinatorial methods. Many factors contribute to the selection of an appropriate chemical database for vHTS. Various kinds of filters are being applied to the compound libraries prior to screening in order to: i) increase the probability of finding hits with drug like characteristics (ADME properties) or lead like characteristics (with physico chemical profile of a lead), ii) to ensure the meaningful composition of the library to be screened, there by saving computational demand while screening.
ADME property filters and toxicity filters
Most important and widely applied filter is the drug-likeness of the compound which represent ADME profile (Adsorption, Distribution, Metabolism and Excretion) as determined by Lipinski’s rule of five, which is a set of rules based on molecular weight, lipophilicity and hydrophobicity, H bond donors and H bond acceptors [17]. The compounds which are labeled as drug-like (Figure 2) resembles the existing drug molecules and exhibit the following property cut-off values: MW ≤ 500, LogP ≤ 5, H bond acceptors ≤ 10 and H bond donors ≤ 5. The compounds exceeding the cut-off values tend to have solubility and permeability problems which would lead to poor oral bioavailabity. In addition to the rule of five there are other properties which help in distinguishing drug and non drug-like compounds. The extensions include polar surface area ≤ 140 Å and rotatable bonds ≤ 10. Prediction of toxicity before the synthesis of compounds ensures the removal of compounds with potential toxic effects. A number of in silico systems for toxicity prediction are available, which help in the classification of toxic and non-toxic compounds [18]. The above mentioned filters are essential to discard compounds without any drug like characteristics and undesirable toxic effects before screening. Such in silico filters are essential to minimize expensive drug failures in the later stage of drug development.
Figure 2.

Drug-like compounds [21].
Meaningful composition of library
Tautomer enumeration is also an essential step that has been shown to enhance the hit rates in the vHTS [19]. Several studies have shown that specific tautomeric states of molecules are present which interact with the active site residues of the target [20]. Tautomers are often recognized as different structures by computational applications, leading to different results. Hence chemical databases need to be enriched with correct bioactive tautomers to ensure reliable results. Removal of compounds known to interact with anti targets e.g., hERG (human Ether-a-go-go Related Gene) channel can be useful [21]. In addition to various factors it is important to confirm the compound availability or the synthetic feasibility of the compound before considering them for vHTS.
Chemical diversity of chemical databases
Chemical database with wide chemical space and structural diversity offers an ideal solution for lead discovery. The basis behind such an analysis is that, similar compounds exhibit similar physico-chemical and biological properties. A chemical database enriched with the representatives of dissimilar compounds or diverse chemical structures shall increase the probability of finding different leads with similar biological activities. It also ensures that database is small but diverse enough to be readily managed. Diversity of chemical databases is evaluated by examining how much the compounds within the library differ in terms of the distribution of their properties. Different kinds of diversity quantification are carried out: Distance based methods, Cell-partitioning methods and clustering methods. At this juncture, it will be useful to mention the study on the major chemical databases demonstrating the diversity presented in these databases and also the overlapping structures in these databases [22]. Such studies provide a view of the diverse nature of the database and helps in selecting an appropriate chemical database.
Conclusion
vHTS is cost-effective and reliable technique that can be applied to identify potential leads and avoid undesirable compounds that would otherwise result in expensive and time consuming experimental methods. However, vHTS often requires careful preparation of both target and compound library, use of optimal parameters as well as careful analysis of the results. It should be noted that experience and knowledge about the target are very crucial in identifying true positives in such experiments. We have discussed some of the essential considerations in designing vHTS experiment focusing the pre-processing of chemical databases that are used in vHTS.
References
- 1.Shoichet BK. Nature. 2004;432:862. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bajorath J. Nat Rev Drug Discov. 2002;1:882. doi: 10.1038/nrd941. [DOI] [PubMed] [Google Scholar]
- 3.Jenkins JL, et al. Proteins. 2003;50:81. doi: 10.1002/prot.10270. [DOI] [PubMed] [Google Scholar]
- 4.McInnes C. Curr Opin Chem Biol. 2007;5:494. doi: 10.1016/j.cbpa.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 5.Kitchen DB, et al. Nat Rev Drug Discov. 2004;3:935. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 6.Carlson HA. Curr Opin Chem Biol. 2002;6:447. doi: 10.1016/s1367-5931(02)00341-1. [DOI] [PubMed] [Google Scholar]
- 7.Teague SJ. Nat Rev Drug Discov. 2003;2:527. doi: 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
- 8.Teodoro ML, Kavraki LE. Curr Pharm Des. 2003;9:1635. doi: 10.2174/1381612033454595. [DOI] [PubMed] [Google Scholar]
- 9.Huang SY, Zou X. Protein Sci. 2007;16:43. doi: 10.1110/ps.062501507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong CF, et al. Proteins. 2005;61:850. doi: 10.1002/prot.20688. [DOI] [PubMed] [Google Scholar]
- 11.Desai PV, et al. J Med Chem. 2004;47:6609. doi: 10.1021/jm0493717. [DOI] [PubMed] [Google Scholar]
- 12.Bissantz C, et al. Proteins. 2003;50:5. doi: 10.1002/prot.10237. [DOI] [PubMed] [Google Scholar]
- 13.Ramasamy G, et al. Mol Biochem Parasitol. 2007;152:139. doi: 10.1016/j.molbiopara.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Pieper U, et al. Nuc Acid Research. 2006;34:291. [Google Scholar]
- 15.Ramasamy G, et al. Bioinformation. 2007;5:50. [Google Scholar]
- 16.Oshiro C, et al. J Med Chem. 2004;47:764. doi: 10.1021/jm0300781. [DOI] [PubMed] [Google Scholar]
- 17.Lipinski CA, et al. Adv Drug Deliv Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 18.Aronov AM. Drug Discov Today. 2005;10:149. doi: 10.1016/S1359-6446(04)03278-7. [DOI] [PubMed] [Google Scholar]
- 19.Dearden JC. J Comput Aided Mol Des. 2003;17:119. doi: 10.1023/a:1025361621494. [DOI] [PubMed] [Google Scholar]
- 20.Oellien F, et al. J Chem Inf Model. 2006;46:2342. doi: 10.1021/ci060109b. [DOI] [PubMed] [Google Scholar]
- 21.Keller TH, et al. Curr Opin Chem Biol. 2006;10:357. doi: 10.1016/j.cbpa.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 22.Krier M, et al. J Chem Inf Model. 2006;46:512. doi: 10.1021/ci050352v. [DOI] [PubMed] [Google Scholar]