

Abstract
Alpha (α)-enolase (e), a glycolytic enzyme, has an alternative role as a surface receptor of several bacteria mediating plasminogen (pg) binding. It is also recognized as a virulence factor of some pathogenic bacteria facilitating plasminogen activation and host cell invasion. A mycoplasmal α-enolase is also a plasminogen binding protein. Molecular interactions of enolase from Mycoplasma pneumoniae with host plasminogen would be useful for exploring the pathogen-host interaction. In an attempt to identify plasminogen binding sites of M. pneumoniae enolase, homology modeling and docking studies were conducted to obtain modeled structures of the M. pneumoniae enolase-plasminogen complex. The refined model was validated further by standard methods. Molecular docking revealed hydrogen bonding of eLys70-pgTyr50, eAsn165-pgThr66, eAla168-pgGlu21, eAsp17-pgLys70, and eAsn213-pgPro68/pgAsn69. Substantial decreases in accessible surface area (ASA) were observed and in concurrence with hydrogen bond pattern. These findings provide a detailed prediction of key residues that interact at the protein-protein interface. Our theoretical prediction is consistent with known biochemical data. The predicted interaction complex can be of great assistance in understanding structural insights, which is necessary to pathogen and host-component interaction. The ability of M. pneumoniae enolase to bind plasminogen may be indicative of an important role in invasion of this pathogen to host.
Keywords: Mycoplasma pneumoniae enolase, human plasminogen, homology modeling, molecular docking
Background
Mycoplasma pneumoniae is one of the most commonly encountered pathogenic bacteria causing particular human diseases. Serious infections requiring hospitalization occur in both young adults and children. This pathogen infects not only pulmonary but also multiple organ systems. Extrapulmonary complications can occur in association with M. pneumoniae infection as a result of direct invasion especially central nervous system as encephalitis. Encephalitis manifestations are greater severity and more clinical importance than the primary respiratory infection. Over the past decades, the incidence of this bacterium invading CNS has increased dramatically [1,2]. This trend may be partially due to several factors that contribute to the mistreatment, misdiagnosis, or mistreatment of patients as well as patient compliance, misuse of antimicrobial agents, and longer pathogen survival in the immuno-compromised host through antigenic mutations. Patients who suffer from persistent M. pneumoniae infection are a continuous transmission source to others in their community [3]. In fact, a number of recently developed therapies are no longer effective treatments [4]. Indeed, very little is known about the mechanisms underlying the virulence of this pathogen. A study of the mechanisms adopted by M. pneumoniae to invade and survive within host cells was carried out using confocal laser scanning microscopy indicating that M. pneumoniae has advanced mechanisms, which are not phagocytosis, for entering host cells [5]. It was suggested that Mycoplasma species invaded host cells by attaching to the surface of host cells and later cross the membrane into host cells [6]. A proposed mechanism, based on several studies, involved in pathogenesis is the interaction of bacterial cells with the human plasminogen system [7,8]. A study demonstrated that Mycoplasma fermentans was able to bind and activate plasminogen [9]. Similarly, plasminogen binding and activation were observed in M. pneumoniae [8]. Host-derived plasmin activity is capable of dissolving fibrin meshes and extracellular matrices of the host cell, which facilitates bacterial entry [10]. Studies of α-enolase-plasminogen complexes have clearly shown that receptor-bound plasminogen is more readily converted to plasmin than free plasminogen [11,12]. Enolase was characterized, by using competitive plasminogen binding assays and cross-linking studies with 125I-labeled plasminogen and intact streptococci, as a plasminogen receptor on the surface of streptococci [11]. This investigation clearly showed that enolase significantly contributed to overall ability of Streptococcus to bind plasminogen. In a manner similar to that of streptococci, enolase mediated the binding of Candida albicans to plasminogen, which increased ability of cells in crossing in vitro human brain microvascular endothelial cells [7]. Recently, there have been a number of reports indicating that enolase enhances the virulence of some pathogens [6]. The microbial enolase is captured by human plasminogen, and its subsequent conversion to plasmin provides a mechanism to augment virulence, favoring host tissue invasion [13,14,15]. For such action, enolase protein must be located on the surface of microbial pathogens [11,15]. The matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) analysis of the peptide sequence of Streptococcus mutans enolase revealed possibility of phosphorylation site(s) [15]. These findings suggested that enolase phosphorylation might occur and lead to its translocation to pathogen's cell surface. In addition, immunogold bead staining supported the MALDI-TOF MS finding that enolase was detected on the surface of S. mutans [15]. The secreted enolase reassociates with the bacterial cell surface was confirmed by direct binding of radiolabeled recombinant protein to pneumococci and by field emission scanning electron microscopic analysis [13]. The M. fermentans α-enolase binding to plasminogen has recently been reported [16]. In light of the above findings, this work is an attempt to predict molecular interaction of M. pneumoniae enolase with human plasminogen which would be useful for further investigation of the mechanism of M. pneumoniae invasion to human brain.
Methodology
The protein sequence of M. pneumonaie enolase sequence was obtained from GenBank (Accession number P75189). Modeling templates were searched by using BLAST. In order to analyze modeled complex of protein-protein interaction, sequence identity of 40-50% between target and template may be required. Multiple sequence alignment was performed by using ClustalW. The atomic coordinates of Escherichia coli enolase was retrieved from Protein Data Bank, (PDB ID: 1E9I). Molecular structures of M. pneumonaie enolase were modeled by using restraint-based modeling implemented in the program MODELLER [17]. Several models were generated and were then energy minimized using the molecular dynamics and simulation procedure CHARMM [18] in program MODELLER. The quality and stereochemistry of the models were evaluated using the program PROCHECK [19]. The final model was selected based on stereochemical quality. The main-chain conformations for 98.80% amino acid residues were within the favored or allowed regions of the Ramachandran plot and the overall G-factor was -0.11, indicating that molecular geometry of the model is of good quality. The selected model was then added Mg2+ and further refined by energy minimization by the NAMD program (http://www.ks.uiuc.edu/Research/namd/) by 2,000 steps of conjugate gradient minimization until the energy gradient RMS was < 0.05 kcal (mol Å)-1. Structural models were visualized by PyMol™ Molecular Graphics System version 0.97 (DeLano Scientific LLC, San Carlos, CA, USA, http://www.pymol.org). The VMD program was used to superimpose structure of M. pneumoniae enolase model with crystal structures of enolases from E. coli (PDB ID: 1E9I) [20] and S. pneumoniae (PDB ID: 1W6T) [13]. M. pneumoniae enolase model was docked to human plasminogen using Hex 4.5 (http: //www.csd.abdn. ac.uk/hex/). The atomic coordinates of human plasminogen was retrieved from PDB (PDB ID: 1B2I) [21]. Automate energy minimization was applied to each docking solution. The Chimera program (http: //www.cgl.ucsf. edu/chimera/) was exploited to identify hydrogen bonds using default parameters and geometric criteria described previously [22]. Mark Gerstein's calc-surface program, which is implemented in the program Chimera, was used to calculate the solvent accessibility at the interface of M. pneumoniae enolase and human plasminogen before and after docking.
Results and discussion
Template identification and model quality
Crystal structures of enolases from many organisms, including those from bacteria, have already been determined and available in PDB -13,20]. Based on sequence similarity analysis, M. pneumoniae enolase shows 55% amino acid sequence identity with Escherichia coli enolase (Figure 1). It is one of top ranks with high degree of sequence identities between M. pneumoniae enolase and other enolases with known structures. Practically, at this level of sequence identity, it is good enough to use crystallographic structures of E. coli enolase as a template, in order to obtain high quality alignment for structure prediction by homology modeling. An E. coli enolase crystal structure 1E9I [20] was specifically selected on the basis of BLAST result and was utilized as a template for M. pneumoniae enolase structure modeling. Structural models for M. pneumoniae enolase were built by MODELLER program [17] based on atomic coordinates of 1E9I and were then energy minimized. The model with the lowest objective function (348.7318), which was considered as the best one, was selected and subjected to quality evaluation. The PROCHECK Ramachandran plot analysis shows that the main-chain conformations for 98.80% of amino acid residues are within the most favored or allowed regions (Figure 2). The structural model of M. pneumoniae enolase is shown in Figure 3a. The G-factors, indicating the quality of covalent and bond-angle distance, were -0.07 for dihedrals, -0.22 for covalent, and overall -0.11. The overall main-chain and side-chain parameters, as evaluated by PROCHECK, are all very favorable. The comparable Ramachandran plot characteristic and G-factors confirm the quality of predicted model. The M. pneumoniae enolase model consists of typical two domains. The N-terminal one contains three-stranded antiparallel β-sheets, followed by six α-helices. The C-terminal domain composes of eleven-stranded mixed α/β-barrel with connectivity. The first α-helix within the C-terminal domain, H7, and the second β-strand, S7, of the barrel domain were arranged in an antiparallel fashion compared to all other α-helices and β-strands (Figure 3a). The RMSD (Root Mean Square Deviation) between predicted model and template is 0.4 Å (Angstrom). The overall conformation of M. pneumoniae enolase model is similar to those of E. coli enolase and S. pneumoniae enolase crystal structures [13,20] as observed by the superposition analysis shown in Figure 3b. The Mg2+, a metal ion cofactor, was encircled by Asp256, Glu310, and Asp337 which located in active site of enolase (Figure 3a). Although Mg2+ is required for catalytic activity of the enzyme, it may also play a role in stabilizing enolase conformation [20].
Figure 1.

Sequence alignment of Mycoplasma pneumoniae enolase (P75189) with Escherichia coli enolase (PDB ID: 1E9I) using CLUSTAL W.
Figure 2.
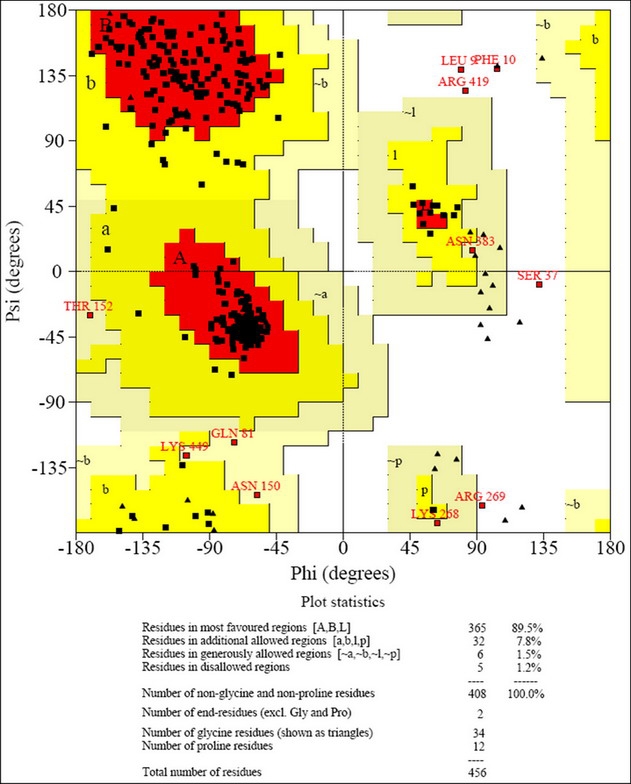
Ramachandran plot for theoretical model of M. pneumoniae enolase.
Figure 3.

Theoretical model of M. pneumoniae enolase. This model was conducted by MODELLER program. (a) Overall structure of M. pneumoniae enolase with containing Mg2+ cofactor. (b) Comparison of enolase structure among three diferrent organisms.
Interaction of M. pneumoniae enolase and human plasminogen
Based on rigid-body docking using HEX 4.5, both proteins were analyzed for shape complementary, hydrophobic effects resulting from a decrease in the solvent accessible surface, and electrostatic interactions. The key amino acid residues within the docking complex model involved in the interaction between enolase (e) and human plasminogen (pg) are eLys70-pgTyr50, eAsn165-pgThr66, eAla168-pgGlu21, eAsp17-pgLys70, and eAsn213-pgPro68/pgAsn69 (Figure 4a). These residues were determined based on intermolecular hydrogen bond lengths of amino acid residues interacted between human plasminogen and M. pneumoniae enolase. All hydrogen bond lengths appear to be shorter than 3.4 Å (Table 1 under supplementary material). This suggests that hydrogen bonds can be plausibly formed. The docking result indicated that the complex could be stabilized by hydrogen bonding. Electrostatic potential surface area showed that 10 amino acid residues of M. pneumoniae enolase appeared to be available for making contact with plasminogen (Figure 4b). These also include eArg24, eLys70, and eLys216, positive charge residues located at the opposite end of the binding pocket. On human plasminogen molecule, several charge amino acid residues, including pgGlu21, pgAsp26, and pgAsp67, appeared in electrostatic potential surface area. Moreover, the decreases of accessible surface area (DASA) of the docking complex were observed Table 2a and Table 2b (see supplementary material). Considering M. pneumoniae enolase, significant change of accessible surface area of eLys70 occurred. Noticeably, hydrogen bonding was observed on this residue. For human plasminogen on the complex, the model showed large decrease in accessible surface area involving residues pgGlu21, pgTyr50, pgAsp67, pgPro68, pgAsn69 and pgLys70. Some of these appeared to form hydrogen bonds with corresponding residues of M. pneumoniae enolase. The results suggest that model of the interaction complex between M. pneumoniae enolase and human plasminogen can be practicable. Our model of M. pneumoniae enolase-plasminogen complex was in agreement with the previous study lysine binding site (LBS) of plasminogen in terms of charge characteristics [23]. In addition, the proposed interaction between human plasminogen and M. pneumoniae enolase agreed with previous experimental investigations [11,12,13]. The interaction between human plasminogen and M. pneumoniae enolase proposed in this study is useful for understanding the possible mechanism used by M. pneumoniae to invade human brain tissue. For instance, the interaction between human plasminogen and M. pneumoniae enolase might provide a vehicle for targeting cells. This line of work may lead to insight into host-pathogen interaction and provide valuable information for prophylactic strategies in combating infections at a very early stage.
Figure 4.

Model of M. pneumoniae enolase-human plasminogen complex interaction. (a) H-bond represent in red line. (b) Electrostatic surfaces, a color ramp was calculated, which positive potential charges in blue, negative values in red, and intermediate values in white by using PyMol (http://pymol.sourceforge.net/).
Conclusion
The rationale in building a M. pneumoniae enolase model and performing a binding study with human plasminogen is to gain details of interaction between the two proteins. M. pneumoniae enolase modeling was conducted using homology modeling. Comparison of the obtained model with experimentally derived crystal structures of E. coli enolase and S. pneumoniae enolase revealed that they were all basically similar. The docking studies revealed the important residues involving in the interaction of M. pneumoniae enolase with human plasminogen. Analyses of the interaction model between human plasminogen and M. pneumoniae enolase, based on distances of hydrogen bonds, changes of solvent accessible surface, electrostatic potentials, showed that this binding complex was reliable. Our theoretical prediction may lead to the establishment of prophylactic and therapeutic approaches.
Supplementary material
Acknowledgments
This research was supported by the Thailand Graduate Institute of Science and Technology (TGIST) scholarship TGIST 01-48-039 from the National Science and Technology Development Agency (NSTDA). We would like to acknowledge Dr. Stefano O. Casalotti and Duangpon Wanapun for critical reading of the manuscript and Dr. Sudsanguan Chusacultanachai and Somchai Pongpattakitchote for helpful discussions.
References
- 1.Coelho M, et al. Clin Neurol Neurosurg. 2004;106:129. doi: 10.1016/j.clineuro.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Bitnun A, et al. Clin Infect Dis. 2001;32:1674. doi: 10.1086/320748. [DOI] [PubMed] [Google Scholar]
- 3.Wattanathum A, et al. Chest. 2003;123:1512. doi: 10.1378/chest.123.5.1512. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki S, et al. Antimicrob Agents Chemother. 2006;50:709. doi: 10.1128/AAC.50.2.709-712.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yavlovich A, et al. FEMS Microbiol Lett. 2004;233:241. doi: 10.1016/j.femsle.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 6.Pancholi V, Chhatwal GS. J Med Microbiol. 2003;293:391. doi: 10.1078/1438-4221-00283. [DOI] [PubMed] [Google Scholar]
- 7.Jong AY, et al. J Med Microbiol. 2003;52:615. doi: 10.1099/jmm.0.05060-0. [DOI] [PubMed] [Google Scholar]
- 8.Yavlovich A, et al. Infect Immunol. 2004;72:5004. doi: 10.1128/IAI.72.9.5004-5011.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yavlovich A, et al. Infect Immunol. 2001;69:1977. doi: 10.1128/IAI.69.4.1977-1982.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lottenberg R, et al. Trend Microbiol. 1994;2:20. doi: 10.1016/0966-842x(94)90340-9. [DOI] [PubMed] [Google Scholar]
- 11.Pancholi V, Fischetti VA. J Biol Chem. 1998;273:14503. doi: 10.1074/jbc.273.23.14503. [DOI] [PubMed] [Google Scholar]
- 12.Redlitz A, et al. Eur J Biochem. 1995;227:407. doi: 10.1111/j.1432-1033.1995.tb20403.x. [DOI] [PubMed] [Google Scholar]
- 13.Bergmann S, et al. Mol Microbiol. 2001;40:1273. doi: 10.1046/j.1365-2958.2001.02448.x. [DOI] [PubMed] [Google Scholar]
- 14.Fox D, Smulian AG. Med Mycol. 2001;39:495. doi: 10.1080/mmy.39.6.495.507. [DOI] [PubMed] [Google Scholar]
- 15.Ge J, et al. Infect Immun. 2004;72:6748. doi: 10.1128/IAI.72.11.6748-6752.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yavlovich A, et al. Infect Immunol. 2007;75:5716. doi: 10.1128/IAI.01049-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sali A, et al. Proteins. 1995;23:318. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- 18.Sali A, Blundell TL. J Mol Biol. 1993;234:779. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 19.Laskowski RA, et al. J Appl Cryst. 1993;26:283. [Google Scholar]
- 20.Fox K, Kühnel BF. J Mol Biol. 2001;313:583. doi: 10.1006/jmbi.2001.5065. [DOI] [PubMed] [Google Scholar]
- 21.Marti DN, et al. Biochemistry. 1999;38:15741. doi: 10.1021/bi9917378. [DOI] [PubMed] [Google Scholar]
- 22.Mills JE, Dean PM. J Comput Aided Mol Des. 1996;10:607. doi: 10.1007/BF00134183. [DOI] [PubMed] [Google Scholar]
- 23.Gong Y, et al. J Biol Chem. 2001;276:19078. doi: 10.1074/jbc.M101387200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.