

Abstract
Genetic lesions affecting a number of kinases and other elements within the epidermal growth factor receptor (EGFR) signaling pathway have been implicated in the pathogenesis of human non–small-cell lung cancer (NSCLC). We performed mutational profiling of a large cohort of lung adenocarcinomas to uncover other potential somatic mutations in genes of this pathway that could contribute to lung tumorigenesis. We have identified in 2 of 207 primary lung tumors a somatic activating mutation in exon 2 of MEK1 (i.e., mitogen-activated protein kinase kinase 1 or MAP2K1) that substitutes asparagine for lysine at amino acid 57 (K57N) in the nonkinase portion of the kinase. Neither of these two tumors harbored known mutations in other genes encoding components of the EGFR signaling pathway (i.e., EGFR, HER2, KRAS, PIK3CA, and BRAF). Expression of mutant, but not wild-type, MEK1 leads to constitutive activity of extracellular signal–regulated kinase (ERK)-1/2 in human 293T cells and to growth factor–independent proliferation of murine Ba/F3 cells. A selective MEK inhibitor, AZD6244, inhibits mutant-induced ERK activity in 293T cells and growth of mutant-bearing Ba/F3 cells. We also screened 85 NSCLC cell lines for MEK1 exon 2 mutations; one line (NCI-H1437) harbors a Q56P substitution, a known transformation-competent allele of MEK1 originally identified in rat fibroblasts, and is sensitive to treatment with AZD6244. MEK1 mutants have not previously been reported in lung cancer and may provide a target for effective therapy in a small subset of patients with lung adenocarcinoma.
Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide (1). Mutational profiling studies have revealed that at least five genes encoding epidermal growth factor receptor (EGFR) signaling pathway components are mutated in non–small-cell lung cancers (NSCLC). Whereas EGFR and KRAS mutations are detected in ∼10% and 20% of tumors, respectively, somatic mutations have also been identified in HER2/ERBB2 (∼2%; exons 19 and 20), the lipid kinase PIK3CA (∼4%; exons 9 and 20), and the serine/threonine kinase BRAF (∼2%; exons 11 and 15; ref. 2). All alterations have been shown to confer gain-of-function properties in comparison with their respective wild-type counterparts. Except for PIK3CA alterations (3), mutations in EGFR, HER2, BRAF, and KRAS rarely occur in the same tumor, suggesting that they may have functionally equivalent roles in lung tumorigenesis. These mutations are found predominantly in tumors with adenocarcinoma histology.
The mitogen-activated protein kinase (MAPK) pathway plays a major role in the EGFR signaling cascade. After activation of EGFR signaling, key downstream steps involve phosphorylation by RAF1 kinase of two distinct serine residues on both MEK1 and MEK2 (4). The MEK proteins share 80% sequence homology and encode dual-specificity kinases of the STE kinase family (homologues of yeast sterile-7, -11, and -20; ref. 5). MEK1/2 subsequently phosphorylate specific threonine and tyrosine residues in the activation loops of ERK1/2.
Altered MEK proteins have been implicated in cancer. A transformation-competent mutant of MEK1 with a Q56P substitution in the nonkinase portion of the kinase has been described in rat fibroblasts (6), and engineered mutants (with alterations of the key regulatory serine residues) can transform NIH 3T3 cells (7). Recently, a human ovarian cancer cell line, ES-2, was found to harbor an activating mutation in MEK1 that substitutes asparagine for aspartic acid at position 67 (D67N; ref. 8). Moreover, small-molecule inhibitors of MEK seem to be promising as antitumor agents (9). However, to our knowledge, somatic mutations in MEK1/2 have not yet been reported in lung cancer.9
Here, we report identification of a novel somatic mutation in MEK1 in human lung tumors, identified via mutational profiling of genes encoding EGFR signaling pathway proteins in a large cohort of lung adenocarcinomas (10). We determine the functional consequences of this genetic alteration in two separate cell systems. We also examine a large collection of primary tumors and cell lines for the presence of this and other MEK1 exon 2 mutations.
Materials and Methods
Tissue procurement and mutational profiling
All specimens were obtained with patients' consent via a protocol approved by the Memorial Sloan-Kettering Cancer Center Institutional Review Board (see ref. 10 for details). Cell lines were obtained from American Type Culture Collection and Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ).
Mutation verification
See ref. 10 for details. All mutations were verified by bidirectional sequence analysis of an independent PCR product. Variants were deemed somatic if they were found to be absent in matched normal tissue. A combination of dideoxynucleotide sequencing and/or mass spectrometry–based genotyping (Sequenom) was used to screen additional tumor samples for MEK1 exon 2 mutations. Primers used for MEK1 exon 2 sequencing were 5′-TTTCTTTCCATGATAGGAGT-3′ and 5′-ATCAGTCTTCCTTCTACCCT-3′.
Expression constructs
The full-length human MEK1 cDNA cloned into the pDNR-Dual vector was obtained from the Harvard Institute of Proteomics. Specific mutations were generated using a QuikChange Site-Directed Mutagenesis Kit (Stratagene) with the following primers: 5′-TTACCCAGAATCAGAAGGTGG-3′ and 5′-CCACCTTCTGATTCTGGGTAA-3′ to create MEK1 K57N, and 5′-GCTGGCGTCTAGGGAAGCTTT-3′ and 5′-AAAGCTTCCCTAGACGCCAGC-3′ to recreate a stop codon. MEK1WT and MEK1K57N cDNAs were cloned into pLP-CMVneo plasmid using the Creator DNA Cloning Kit (Clontech). Expression plasmids encoding mutant BRAFV600E or KRASG12V were kind gifts from J. Fagin (Memorial Sloan-Kettering Cancer Center, New York, NY) and M. Phillips (New York University, New York, NY), respectively.
The full-length human ERBB4 cDNA was obtained in pCMV6-XL5 from Origene. The N181S mutation was generated using the QuikChange Kit and primers 5′-GTGTCAACAAGTGGTAGTTCA-3′ and 5′-TGAACTACCACTTGTTGACAC-3′.
Cell culture
293T cells, maintained as per established protocols, were transfected with pLP-CMVneo vector alone, pLP-CMVneo MEK1WT, pLP-CMV-neo MEK1K57N, pCMV6-XL5 ERBB4WT, or pCMV6-XL5 ERBB4N181S using FuGENE 6 Transfection Reagent (Roche Diagnostics).
Ba/F3 cells, maintained as per established protocols, were transfected with expression plasmids using Nucleofector II and the Cell Line Optimization Kit V (Amaxa Biosystems). Stable clones were then derived by 14-d selection in 1.25 mg/mL G418. To obtain interleukin-3 (IL-3)–independent Ba/F3 clones, IL-3 was removed from the cell culture media. Ten days after the first round of IL-3 withdrawal, ∼17% and 4% of cells harboring mutant and wild-type cDNAs, respectively, were viable. These cells were then recultured in the presence of IL-3, and after a second round of IL-3 withdrawal, ∼50% and 2% of cells harboring mutant and wild-type cells, respectively, were viable (data not shown). After a third round of IL-3 withdrawal, the majority of cells harboring mutant MEK1 cDNAs survived and were able to proliferate in the absence of IL-3. No cells harboring wild-type MEK1 cDNAs were alive. Comparable results were obtained from an independent transfection of parental Ba/F3 cells. All IL-3–independent clones were verified by direct sequencing to harbor mutant cDNAs.
Immunoblotting
Cell lysates were examined using established protocols. Total MEK1, phospho-ERK1/2 (Thr202/Tyr204), total ERK1/2, and secondary horseradish peroxidase–conjugated antirabbit antibodies were from Cell Signaling Technology.
Cellular assays
To assess cellular proliferation, G418-resistant Ba/F3 cells carrying either MEK1WT or MEK1K57N, respectively, were plated in the absence of IL-3 on day 0, along with parental Ba/F3 cells cultured with or without IL-3. Viable cells were counted by trypan blue staining each day for 4 consecutive days. Values reported were calculated as percent of viable cells compared with those obtained from day 0. All assays were done at least two independent times.
To assess cell growth inhibition, cells were seeded in 96-well plates (6 × 104−8 × 104 per well) in triplicate and treated with different concentrations of AZD6244. Cell growth inhibition was measured at 48 h posttreatment using CellTiter Blue Reagent (Promega). All assays were done at least two independent times.
Results
We analyzed genomic DNA from a total of 261 resected, clinically annotated NSCLC specimens (10). We screened the coding sequences of 39 genes encoding mostly components of the EGFR signaling pathway for somatic mutations via high-throughput dideoxynucleotide sequencing of PCR-amplified gene products. Sequencing of 9MB of tumor sequence identified 239 putative nonsynonymous sequence variations that differed from reference sequences listed in the National Center for Biotechnology Information (RefSeq) database for each respective gene. We previously reported the examination of 22 sequence variations found in RAS family genes and 135 sequence variations localized to exons encoding the protein kinase domains (10). In that study, we identified a total of 37 nonsynonymous somatic mutations, found collectively in EGFR, HER2, KRAS, BRAF, PIK3CA, and FGFR4 (10).
Here, we examined the remaining putative genetic variants occurring in exons encoding domains outside of the kinase regions of their respective kinases. Of 82 putative nonkinase domain sequence variations, representing 69 distinct types of mutations, we confirmed the existence of 27 (18 distinct types) on sequencing a second independent PCR product from tumor DNA. We found 22 of the 27 (12 types) in corresponding matched normal tissue, suggesting that they were single nucleotide polymorphisms. Two variants were of unknown significance (ERBB4: P572L, FGFR4: C56S) because we were unable to obtain a PCR product from matched normal samples.
Of the remaining three confirmed somatic mutations, one involved an A→G change at nucleotide 542 in exon 4 of ERBB4, which substitutes serine for asparagine at position 181 (data not shown). This mutation was found in 1 of 171 lung tumor samples. The N181S substitution does not seem to grossly affect kinase activity; (a) according to the polymorphism phenotyping bioinformatics tools PolyPhen and SIFT, an N181S substitution is predicted to be “benign” and “tolerated,” respectively; and (b) lysates from 293T cells transiently transfected with cDNAs encoding wild-type or N181S ERBB4 do not display major differences by immunoblotting in phosphotyrosine reactivity of ERBB4 protein (data not shown).
The remaining two confirmed somatic mutations involved the same G→T transversion in exon 2 at nucleotide 171 of MEK1, found initially in 2 of 93 independent lung tumor samples (of which, 89, including the two with MEK1 changes, were of adenocarcinoma histology; Fig. 1A). This change substitutes asparagine for lysine at position 57. K57 is highly conserved among various species (Fig. 1B) and is located between the nuclear export signal (amino acids 33–44) and kinase domain (amino acids 68–271) of MEK1 (Fig. 1C). Consistent with the G→T mutation, a type of transversion known to be smoking related, both samples harboring the MEK1K57N mutation were from former smokers. Neither patient had prior chemotherapy or exposure to asbestos or radiation. Both patients presented with stage IA disease and are alive with nearly 4 years of follow-up. Notably, neither of these two tumors harbored mutations in EGFR, HER2, KRAS, BRAF, PIK3CA, or FGFR4.
Figure 1.
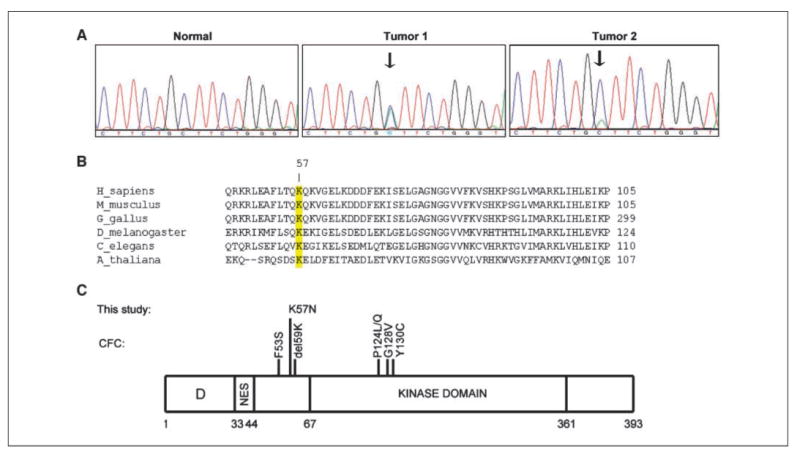
Identification of a MEK1K57N mutation in human lung adenocarcinoma. A, reverse sequencing chromatograms display presence of a G→T mutation at position 171 in exon 2 of MEK1 in two tumor samples. This mutation was absent from matched normal control samples. (Forward sequence chromatograms not shown due to space constraints.) B, protein sequence alignment of MEK1 from various species shows that K57 is a highly conserved residue. Numbers indicate amino acid positions. C, K57N is located in between the nuclear export signal (NES) and the kinase domain of MEK1. D, docking domain. Shown are mutations found in patients with cardio-facio-cutaneous (CFC) syndrome.
To determine if a K57N substitution in MEK1 affected MAPK signaling, we assayed the ability of the mutant to induce ERK phosphorylation in 293T cells. Extracts from cells transiently transfected with plasmids encoding wild-type or mutant MEK1 cDNAs displayed higher levels of total MEK1 protein than did control-transfected cells, but only cells transfected with MEK1K57N cDNAs displayed enhanced levels of ERK phosphorylation (Fig. 2A). We observed similar levels of induced ERK phosphorylation in lysates from cells transfected with cDNAs encoding two well-characterized mutants, BRAFV600E and KRASG12V, known to activate the ERK pathway (Fig. 2A).
Figure 2.

Functional characterization of the MEK1K57N mutant. A, 293T cells were transiently transfected with expression plasmids encoding various cDNAs, and corresponding lysates from cells maintained in serum were subjected to immunoblotting with the indicated antibodies. Lysates from cells harboring MEK1 cDNAs displayed higher levels of total MEK1 protein compared with control-transfected cells, but only cells transfected with MEK1K57N cDNAs displayed enhanced phospho-ERK expression, at levels comparable to those induced by mutants BRAF and KRAS. B, Ba/F3 cells were stably transfected with vectors encoding wild-type or MEK1K57N cDNAs. The resulting cells were then cultured in the absence of IL-3. Numbers of live cells were counted daily for 4 d. Parental Ba/F3 cells grown in the absence or presence of IL-3 serve as controls. C, lysates from IL-3–independent K57N Ba/F3 cells display high levels of pERK and tMEK1 compared with parental Ba/F3 cells grown in the absence of IL-3. Immunoblotting was done with the indicated antibodies.
To characterize additional functional consequences of the K57N change, we generated stable polyclonal populations of Ba/F3 cells expressing wild-type or mutant MEK1. Ba/F3 cells are a murine pro-B cell line that is normally dependent on IL-3 for growth, but they can be rendered IL-3 independent by introduction of transforming tyrosine kinases such as BCR-ABL (11). For some oncogenic proteins, very high levels of mutant kinase are required to derive growth factor independence (12). In two independent assays, we derived Ba/F3 cells harboring cDNAs encoding MEK1K57N that grew in the absence of IL-3. By contrast, we could not derive IL-3–independent Ba/F3 cells expressing the wild-type kinase (Fig. 2B). The growth rate of IL-3–independent Ba/F3 cells expressing mutant MEKK57N was similar to that of parental Ba/F3 cells cultured in the presence of IL-3 (Fig. 2B). Immunoblotting of lysates from the cells harboring mutant MEK1 revealed very high levels of total MEK1 protein and ERK phosphorylation compared with parental Ba/F3 controls (Fig. 2C). Thus, overexpression of mutant, but not wild-type, MEK1 is able to convert Ba/F3 cells to cytokine-independent growth.
We next assessed whether a small-molecule MEK inhibitor, AZD6244 (13), could affect biological properties induced by the MEK1 mutant. AZD6244 treatment of 293T cells transiently transfected with plasmids encoding MEK1K57N cDNAs readily inhibited the appearance of ERK phosphorylation (Fig. 3A). Moreover, compared with parental Ba/F3 cells growing in IL-3, IL-3–independent MEK1K57N-harboring Ba/F3 cells were more sensitive to the MEK inhibitor (Fig. 3B). Because the administration of IL-3 to cultures of cytokine-independent MEK1K57N-harboring Ba/F3 cells prevented AZD6244-induced growth inhibition, these data suggest that MEK inhibitors may effectively target cancer cells that depend on the activity of mutant MEK1 for survival.
Figure 3.
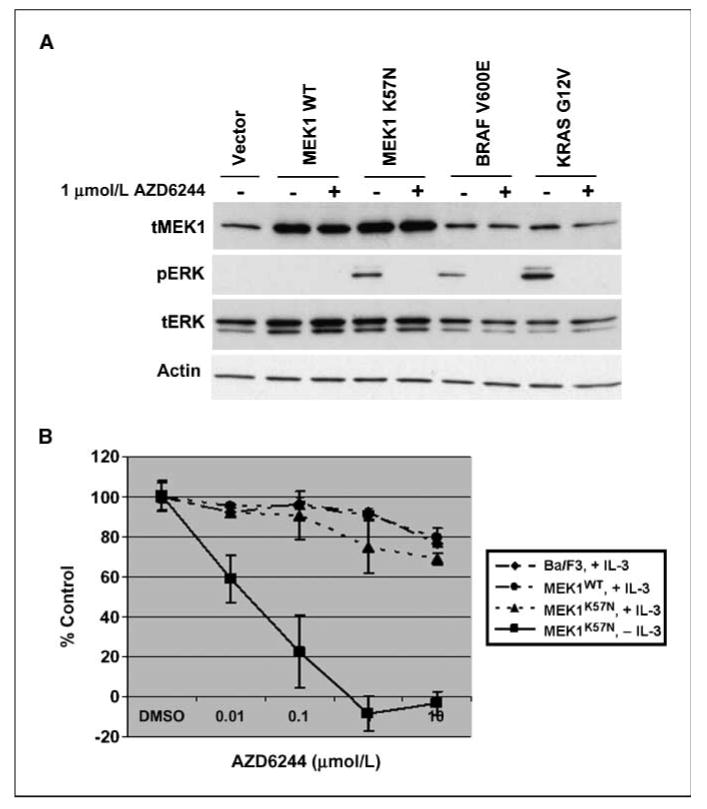
MEK1K57N-induced activity is sensitive to MEK inhibition by AZD6244. A, 293T cells were transiently transfected with expression plasmids encoding various cDNAs, and corresponding lysates were subjected to immunoblotting with the indicated antibodies. Lysates from cells harboring mutant MEK1K57N are sensitive to AZD6244 as shown by inhibition of pERK expression. MAPK signaling is also inhibited by drug treatment in the cells expressing BRAFV600E and KRASG12V mutant constructs. B, Ba/F3 cells were stably transfected with vectors encoding wild-type or MEK1K57N cDNAs. The resulting cells were then cultured for 48 h with AZD6244 in the absence or presence of IL-3. Parental Ba/F3 cells cultured in the presence of IL-3 were also treated with the drug. Growth inhibition assays were done using CellTiter Blue Reagent as described in Materials and Methods.
Finally, we screened an additional 114 NSCLCs (including 33 squamous cell carcinomas) for the MEK1K57N change or other mutations in exon 2 of MEK1. No tumors harbored the MEK1K57N mutation. We also examined an additional 85 NSCLC cell lines. Whereas no lines carried the K57N mutation, we did find that NCI-H1437 cells harbor a mutation encoding a Q56P MEK1 variant (data not shown). These cells were originally derived from a male smoker. They do not carry mutations in EGFR, KRAS, BRAF, or PIK3CA but do display an inferred copy number of up to 3 at the genomic region of MEK1 on chromosome 15.10 H1437 cells also display sensitivity in the nanomolar range to AZD6244 (data not shown).11 We did not detect any other MEK1 exon 2 mutations in 19 primary samples from patients with chronic myelomonocytic leukemia (with wild-type KRAS) or in 54 additional tumor cell lines (14 melanomas, 13 colon carcinomas, 13 breast carcinomas, 6 neuroblastoma/neuroepithelial tumors, 4 prostate carcinomas, and 4 pancreas carcinomas).
Discussion
We report the identification of a novel somatic mutation in MEK1 in human lung adenocarcinoma. The same heterozygous mutation, K57N, was found in 2 of 207 NSCLCs, neither of which harbored mutations in other genes encoding components of the EGFR signaling pathway. Functional characterization of this mutant in vitro indicates that its expression in 293T cells leads to constitutive activation of downstream signaling components (i.e., phospho-ERK activity) and in Ba/F3 cells confers IL-3 independence. Collectively, these results indicate that, in two separate in vitro systems, MEK1K57N mutants display gain-of-function properties compared with wild-type protein.
K57 is located in a region between the nuclear export signal and catalytic domain of MEK1. How the K57N substitution affects the structure of MEK1 is unclear, as the solved crystal structure of MEK1 analyzed only amino acids 62 to 393 (14). Preliminary experiments using green fluorescent protein–tagged wild-type and mutant proteins suggest that the mutation does not affect the cytoplasmic localization of the protein (data not shown).
Nonetheless, multiple functional studies involving amino acids in this region of the protein show that it seems to be critical for the regulation of MEK1 activity. For example, in the study that originally identified the Q56P mutation, MEK1Q56P-glutathione-S-transferase fusion proteins were ∼170-fold more active than the wild-type counterparts in terms of their ability to phosphorylate recombinant ERK in vitro (6). In a separate study, deletion of a predicted α-helix encompassing residues 32 to 51 resulted in a mutant (ΔN3) with basal activity 45 times greater than that of the wild-type enzyme (7). Similarly, the K57N substitution, shown here, and the D67N substitution (8) both lead to constitutive activation of the MAPK pathway in vitro.
Whereas somatic alterations in MEK1 have not yet been reported in human lung cancers, recent studies have identified germ-line MEK1 mutations in patients with cardio-facio-cutaneous (CFC) syndrome, a complex developmental disorder involving the heart, face, and skin (15). Mutations in affected individuals include F53S, del59K, P124L/Q, G128V, and Y130C (Fig. 1C), the latter two of which occur in the kinase domain (15, 16). The F53S mutant, like the Y130C mutant, is more active than wild type protein in stimulating ERK phosphorylation (15). It is not yet clear if CFC patients are at an increased risk for cancer, but three affected individuals have developed neoplasms (i.e., rhabdomyosarcoma, hepatoblastoma, and acute lymphoblastic leukemia; refs. 17–19). In future studies, it will be interesting to compare the functional consequences of germ-line versus somatic mutations in MEK1. Notably, germ-line mutations in KRAS and BRAF, associated with Noonan and CFC syndromes, respectively, are also different from their somatic counterparts found in human cancers (15, 20).
Finally, we show that the downstream effects induced by the MEK1K57N mutant can be inhibited by treatment with a selective MEK inhibitor. Taken together, our results suggest that mutant MEK1 may provide a target for anticancer therapy in a small subset of patients with lung adenocarcinoma.
Acknowledgments
Grant support: The Carmel Hill Fund, The Doris Duke Charitable Foundation (W. Pao); Joan's Legacy: The Joan Scarangello Foundation to Conquer Lung Cancer (W. Pao and Y. Gong); the Long Island League to Abolish Cancer (W. Pao); K08-CA097980 (W. Pao), R01-CA121210 (W. Pao), funds from the Miner Family (W. Pao), and the Anbinder Fund (M. Ladanyi). R.K. Thomas is a fellow of the International Association for the Study of Lung Cancer.
We thank H. Varmus for critical reading of the manuscript, M. Balak and A. Lash for their contributions, and P. Smith and AstraZeneca for providing unpublished data and AZD6244.
Footnotes
K. Michel and R. Thomas, unpublished data.
P. Smith (Astra Zeneca), personal communication.
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2006;118:257–62. doi: 10.1002/ijc.21496. [DOI] [PubMed] [Google Scholar]
- 3.Kawano O, Sasaki H, Endo K, et al. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer. 2006;54:209–15. doi: 10.1016/j.lungcan.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Kyriakis JM, App H, Zhang XF, et al. Raf-1 activates MAP kinase-kinase. Nature. 1992;358:417–21. doi: 10.1038/358417a0. [DOI] [PubMed] [Google Scholar]
- 5.Crews CM, Alessandrini A, Erikson RL. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science. 1992;258:478–80. doi: 10.1126/science.1411546. [DOI] [PubMed] [Google Scholar]
- 6.Bottorff D, Stang S, Agellon S, Stone JC. RAS signalling is abnormal in a c-raf1 MEK1 double mutant. Mol Cell Biol. 1995;15:5113–22. doi: 10.1128/mcb.15.9.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mansour SJ, Matten WT, Hermann AS, et al. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–70. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 8.Estep AL, Palmer C, McCormick F, Rauen KA. Mutation analysis of BRAF, MEK1 and MEK2 in 15 ovarian cancer cell lines: implications for therapy. PLoS ONE. 2007;2:e1279. doi: 10.1371/journal.pone.0001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sebolt-Leopold JS, Dudley DT, Herrera R, et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999;5:810–6. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 10.Marks JL, McLellan MD, Zakowski MF, et al. Mutational analysis of EGFR and related signaling pathway genes in lung adenocarcinomas identifies a novel somatic kinase domain mutation in FGFR4. PLoS ONE. 2007;2:e426. doi: 10.1371/journal.pone.0000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proc Natl Acad Sci U S A. 1988;85:9312–6. doi: 10.1073/pnas.85.23.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu X, Huang LJ, Lodish HF. Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J Biol Chem. 2008;283:5258–66. doi: 10.1074/jbc.M707125200. [DOI] [PubMed] [Google Scholar]
- 13.Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–83. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 14.Ohren JF, Chen H, Pavlovsky A, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11:1192–7. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez-Viciana P, Tetsu O, Tidyman WE, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–90. doi: 10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- 16.Schulz AL, Albrecht B, Arici C, et al. Mutation and phenotypic spectrum in patients with cardio-facio-cutaneous and Costello syndrome. Clin Genet. 2008;73:62–70. doi: 10.1111/j.1399-0004.2007.00931.x. [DOI] [PubMed] [Google Scholar]
- 17.Bisogno G, Murgia A, Mammi I, Strafella MS, Carli M. Rhabdomyosarcoma in a patient with cardio-facio-cutaneous syndrome. J Pediatr Hematol Oncol. 1999;21:424–7. doi: 10.1097/00043426-199909000-00016. [DOI] [PubMed] [Google Scholar]
- 18.Al-Rahawan MM, Chute DJ, Sol-Church K, et al. Hepatoblastoma and heart transplantation in a patient with cardio-facio-cutaneous syndrome. Am J Med Genet A. 2007;143:1481–8. doi: 10.1002/ajmg.a.31819. [DOI] [PubMed] [Google Scholar]
- 19.Makita Y, Narumi Y, Yoshida M, et al. Leukemia in cardio-facio-cutaneous (CFC) syndrome: a patient with a germline mutation in BRAF proto-oncogene. J Pediatr Hematol Oncol. 2007;29:287–90. doi: 10.1097/MPH.0b013e3180547136. [DOI] [PubMed] [Google Scholar]
- 20.Schubbert S, Zenker M, Rowe SL, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331–6. doi: 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]