

Abstract
The chromosome 10q region has recently received a great deal of attention in late-onset Alzheimer’s disease (LOAD), given the growing evidence of linkage to LOAD, or to A-beta levels, reported by several groups. In a recent paper we reported evidence of linkage in this region in our subset of the NIMH AD Genetics Initiative pedigrees, approaching genome-wide significance (non-parametric LOD score = 3.27), when only families with maternal disease origin were analyzed[1]. We have now extended this work, using an independent subset of NIMH AD pedigrees from the University of Alabama at Birmingham (UAB), and show further evidence of linkage using parent-of-origin information. As in our Hopkins sample, maternal but not paternal pedigrees show significantly increased linkage in the chromosome 10q region compared to the unstratified sample. Combining data from our previous fine-mapping work on this region and five new markers genotyped in all pedigrees results in a non-parametric LOD score of 3.73 in the same region, a value that reaches genome wide significance for linkage, with an empirical p value = 0.003. These results support our earlier findings and narrow the region of interest. In combination with findings from other groups, these results provide further evidence that this chromosome 10 region harbors a gene implicated in LOAD, and our use of parent-of-origin information has been useful in further narrowing the region of interest.
Keywords: linkage, Alzheimer’s disease, human chromosome 10, genetic imprinting
Recent genome-wide scans in samples of late-onset Alzheimer’s disease (LOAD) families have reported linkage to LOAD or A-beta levels in a region close to the centromere of chromosome 10q, roughly 60 to 90 cM from the p-terminus, [2–7] although no gene has been definitively identified. Efforts to replicate and further fine-map this LOAD locus may be limited by the existence of heterogeneity in LOAD family samples. With this in mind, we have focused on potentially important phenotypes to improve linkage and fine-mapping signals. Given reports of excess maternal transmission of Alzheimer’s disease as well as differential effects for mitochondrial haplogroups, [8–11] one such characteristic of interest is parental origin of disease. In a previous report, we showed that evidence of linkage to this chromosome 10q region is specific to families with an affected mother in our set of JHU families initially collected as part of the 3-site NIMH Genetics Initiative. Those with an affected father showed no evidence of linkage to this region[1]. This parent-of-origin stratified analysis provided much stronger evidence of linkage to the chromosome 10q region, and a narrower 1-LOD interval, than analysis of the unstratified JHU data set, or the overall 3-site NIMH sample, and the results continued to improve upon fine-mapping (non-parametric LOD score = 3.27). This encouraged the use of parent-of-origin information in pursuing this chromosome 10 AD locus.
In an attempt to further localize this LOAD locus and replicate the maternal origin effect, we have now examined linkage using parent of origin information for an independent sample of LOAD families from the NIMH set for which parental affection information was available, those collected at the University of Alabama (UAB). This UAB sample consists of 140 families, with parental affection established through multiple family member reports and medical records where available. Included are 20 families with report of an affected mother, 16 families reporting an affected father, 40 families with parental affection information but lacking a sib pair (34) or having both parents affected (6), and finally, 63 families with no information. All analyses presented here incorporate the 12 markers previously included in our JHU fine-mapping project (4 from the original scan and 8 initial follow-up markers), plus an additional 5 markers, resulting in a total of 17 markers in the region between D10S1228 and D10S1225, providing an average spacing of 1.8cM. Markers were chosen according to heterozygosity and location. Updated phenotype information for the JHU families was incorporated, resulting in the number of JHU families and sib-pairs reported here being slightly different than the numbers reported in the original publication. There was no difference in the age of onset between the UAB and JHU samples, nor was there any difference in onset age between the maternal or paternal families. The sample is over two-thirds female and over ninety percent Caucasian. Sample characteristics are presented in Table 1.
Table 1.
Sample Description.
| JHU
|
UAB
|
JHU + UAB
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| All | Mat | Pat | All | Mat | Pat | All | Mat | Pat | |
| # Families | 147 | 48 | 24 | 140 | 20 | 16 | 287 | 68 | 40 |
| Affected/unaffected sibs | 357/136 | 117/29 | 55/19 | 325/165 | 47/47 | 35/9 | 682/301 | 164/76 | 90/28 |
| Affected females/males | 258/99 | 81/36 | 46/9 | 246/79 | 36/11 | 27/8 | 504/178 | 117/47 | 73/17 |
| Affected sib pairs | 293 | 98 | 43 | 242 | 35 | 22 | 535 | 133 | 65 |
| Mean age of onset (sd) | 72.2 (7.2) | 70.9 (6.6) | 71.5 (7.5) | 72.3 (7.0) | 71.1 (6.3) | 73.9 (7.0) | 72.2 (7.1) | 71.0 (7.4) | 72.4 (7.4) |
| % Caucasian | 93.6 | 92.3 | 96.4 | 92.6 | 95.7 | 100 | 93.1 | 93.3 | 97.8 |
We first attempted to replicate the maternal origin effect in this independent sample by comparing the evidence for linkage to this 10q region among all UAB families, to evidence among the 20 maternal families and 16 paternal families. Non-parametric linkage analyses were carried out as in our original paper [1]. MLS (non-parametric LOD) scores developed by Kong and Cox [12] were calculated as implemented in the ASM software. This is based on the Sall statistic implemented in Genehunter-Plus software [12;13]. Allele frequencies were calculated using the entire set of families and all members with genotype information, regardless of affection status.
Although this small UAB maternal sample provided relatively low linkage scores, the change in LOD score (0.79 LOD) within our 1-LOD interval was significant, increasing from 0.11 in the overall sample to 0.90 among the maternal families (Table 2). To evaluate whether such a difference in linkage signal among a subset of 20 families is likely due to chance, we performed a simulation study taking randomly chosen sets of 20 families from the total 140 UAB families, and derived an empirical distribution of 10q signals among 5000 replicates of random subsets. Only 268/5000 replicates had a LOD exceeding the observed LOD=0.90 anywhere within the 1-LOD interval on 10q, providing an empirical p value estimate of 0.054 that this maternal increase among the UAB families was due to chance. The paternal UAB sample also showed an increase, but of only 0.48 LOD units, which was not statistically significant. This taken together with the decrease in LOD scores observed in the paternal JHU families [1] suggests that the change in score is not due to familial loading.
Table 2.
Nonparametric LOD scores for overall and maternal-only analyses of individual sites and combined data sets.
| JHU
|
UAB
|
JHU + UAB
|
|||||
|---|---|---|---|---|---|---|---|
| ALL | MAT | ALL | MAT | ALL | MAT | ||
| Marker | cM | N=147 | N=48 | N=140 | N=20 | N=287 | N=68 |
| D10S1228 | 57.4 | 0.50 | 0.44 | 0.00 | 0.00 | 0.11 | 0.23 |
| D10S1426 | 59 | 1.02 | 1.11 | 0.00 | 0.00 | 0.18 | 0.52 |
| D10S507 | 59.8 | 1.02 | 1.62 | 0.00 | 0.00 | 0.18 | 0.91 |
| D10S1169 | 60.6 | 1.06 | 1.97 | 0.00 | 0.00 | 0.19 | 1.23 |
| D10S1208 | 63.3 | 2.02 | 2.91 | 0.00 | 0.00 | 0.47 | 2.16 |
| D10S1247 | 63.3 | 2.07 | 2.99* | 0.00 | 0.00 | 0.51 | 2.24 |
| D10S1217 | 63.83 | 1.91 | 2.71 | 0.00 | 0.01 | 0.48 | 2.12 |
| D10S1233 | 66.5 | 1.42 | 2.73 | 0.00 | 0.22 | 0.52 | 2.81 |
| D10S1413 | 67.39 | 1.29 | 2.69 | 0.00 | 0.32 | 0.54 | 2.92 |
| D10S1724 | 70.23 | 1.34 | 2.38 | 0.04 | 0.81 | 0.98 | 3.20 |
| D10S1220 | 70.2 | 1.26 | 2.45 | 0.04 | 0.83 | 0.94 | 3.28 |
| D10S1784 | 72.9 | 1.08 | 2.56 | 0.11 | 0.90** | 0.98 | 3.46 |
| D10S539 | 72.9 | 1.10 | 2.59 | 0.07 | 0.60 | 0.89 | 3.16 |
| D10S1642 | 74.5 | 1.04 | 2.89 | 0.08 | 0.66 | 0.94 | 3.51 |
| D10S1221 | 75.6 | 1.11 | 2.90 | 0.17 | 0.85 | 1.11 | 3.73*** |
| D10S464 | 79 | 0.85 | 1.75 | 0.33 | 0.92 | 0.12 | 2.66 |
| D10S1225 | 80.8 | 0.53 | 0.85 | 0.29 | 0.86 | 0.81 | 1.63 |
Shading corresponds to the original 1LOD interval defined by the JHU maternal families.(Bassett, Avramopoulos, and Fallin 2002)
Empirical sig of change in score, JHU ALL to JHU MAT; p=0.021
Empirical sig of change in score, UAB ALL to UAB MAT, within the 1-LOD interval; p=0.054.
Empirical sig of change in score, JHU+UAB ALL to JHU+UAB MAT; p=0.002; Empirical significance of observed linkage; p=0.005.
Once the value of restricting our linkage focus to maternal families had been validated in the UAB sample, we sought to achieve the best fine-mapping evidence and location by combining the JHU and UAB maternal LOAD families for further analysis. The combined set includes 287 families - 147 JHU families and 140 UAB families. Of these, 68 families (133 ASPs) were considered ‘maternal’ - 48 of the JHU families and 20 of the UAB families. The combined maternal pedigrees provide a non-parametric LOD score of 3.73 in the same 10q region, compared to a LOD=1.1 in the overall combined set (mean IBD sharing = 0.590 for maternal, 0.506 for paternal and 0.514 overall). The empirical p value for this change in LOD was p=0.0024 (see Figure 1).
Figure 1.
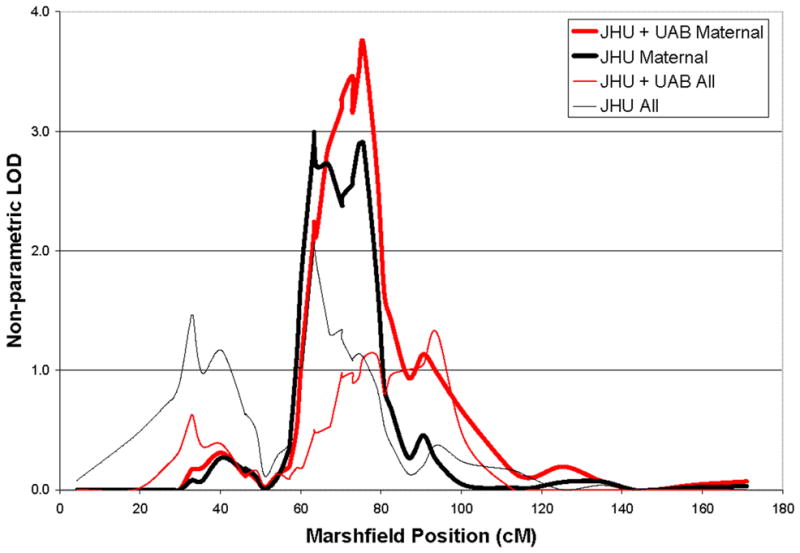
Chromosome 10 nonparametric LOD scores for overall and maternal-only analyses for individual and combined data sets.
The linkage signal (LOD=3.73) exceeds the conservative threshold of 3.6 for genome wide significance[14]. In order to exclude the possibility that the LOD score was inflated due to the use of sex-averaged as opposed to sex-specific linkage maps, we used the linkage analysis package ASPEX [15] and performed both types of analyses. We found that both sex-average and sex-specific maps provided very similar results (LOD 3.71 and 3.75 respectively). We then performed simulations to estimate the empirical significance of this linkage finding using Merlin[16] to assign genotypes to our pedigrees under the assumption of no linkage, while keeping marker distances and missing data constant. We performed 1000 genome simulations and determined that for this specific study, genome-wide significance (5 false positives per 100 studies) is reached at a LOD of 2.8 and our signal corresponds to p=0.005. Further, this maternal stratification has decreased the 1-LOD interval to 15 cM (24.6 Mb).
Our results strongly suggest the presence of linkage in this already established LOAD linkage region on 10q, and add further support for a maternal origin effect of the underlying LOAD variants through this additional genome-wide significant finding. The maternal origin finding requires further evaluation of the potential mechanisms in LOAD. Possibilities include sex-specific trinucleotide repeat expansions, interactions with the maternally inherited mitochondrial genome, or genomic imprinting. Each of these has some precedent in developmental or brain disorders.
Large increases or expansions in the number of trinucleotide tandem repeats are involved in a number of inherited degenerative conditions and these increases appear to be specific to gender of the transmitting parent. For example, Huntington’s disease and a number of the spinocerebellar ataxias, characterized by increased numbers of CAG repeats, are known to be paternally transmitted[17;18]. Recent work in HD examining the paternal transmission of expansions in transgenic mice finds that the expansions occur equally in both X and Y chromosomes in parental sperm and thus gender dependent expansion are likely to occur post-zygotically, suggesting the embryonic environment may induce these expansions[19].
There is growing evidence of mitochondrial dysfunction in AD, with a number of studies reporting decreased mitochondrial enzyme activity, including deficiencies of cytochrome c oxidase (COX) activity, particularly in the CA1 region of the hippocampus, as well as in non-degenerating AD tissue[20–22]. This dysfunction produces an increase in reactive oxygen species (ROS) and it appears that females have a greater vulnerability to oxidative damage, showing greater upregulation of antioxidant defense[23]. In addition, it has recently been shown that Aβ interacts with ABAD in the mitochondria of AD patients and that inhibition of this interaction suppresses Aβ induced apoptosis and free-radical generation in neurons, providing a direct link between mitochondria and AD[24]. In NT2 cells expressing mtDNA from AD subjects (AD cybrids), exposure to Aβ produces excessive mitochondrial membrane potential depolarization, increased cytoplasmic cytochrome c, and elevated caspase-3 activity compared to normal cybrids. Thus the induction of multiple constituents of the mitochondrial apoptosis pathway by Aβ is dependent on the mitochondria carried by the cell[25]. Finally, there has been an examination of the distribution of mtDNA haplogroups in AD cases and controls with one study reporting a protective effect for APOE e4 carriers with the K or U haplogroup, while another reported no relationship to APOE status but rather differential risk according to haplogroup and sex[9;10]. The combination of a defect in the maternally inherited mitochondrial genome and a defect in a related gene on chromosome 10 could explain the parent-of-origin effect we observe in our data.
Finally, imprinting mechanisms, where there is differential expression or silencing of genes based on the sex of transmitting parent, are also known to be involved in embryonic development[26] and in gene expression in the brain (see Keverne 1997[27]for review) and can provide a direct explanation for our finding. Methylation is an important mechanism of imprinting [28] and there are findings that suggest this process may be altered in AD patients when compared to controls[29]. However, there is no evidence of the existence of imprinted genes in our region of interest. One report of uniparental disomy for chromosome 10 with apparently normal phenotype[30] makes extensive imprinting unlikely, yet it does not rule out imprinting of one or a few genes, a possibility that has not yet been investigated. It is also worth noting here that the recent work in HD mentioned above suggests a type of epigenetic regulation influenced by the sex of the embryo rather than the sex of the transmitting parent[19;31].
In summary, our work provides compelling evidence for a LOAD locus on chromosome 10q, consistent with the growing evidence of linkage to LOAD in this region reported by other groups. Importantly, our efforts to reduce heterogeneity through the use of specific phenotypes has led to the finding that this 10q signal is specific to families with an affected mother. This has now been shown in two independent family samples, and suggests that this type of stratification may be important for further localization of this LOAD gene. This finding also suggests mechanisms involving a parent-of-origin effect, like genomic imprinting, or interaction with mitochondrial genes, should be considered in greater detail as a mechanism in AD pathogenesis.
Acknowledgments
This work has been supported by NIA through a grant to Dr. Bassett (AG021804) and by NINDS through a grant to Dr. Go (NS45934). Original sample collections were supported by NIMH through grants to Dr. Bassett (MH46290) and Dr. Go (MH46373).
Reference List
- 1.Bassett SS, Avramopoulos D, Fallin D. Evidence for parent of origin effect in late-onset Alzheimer disease. Am J Med Genet. 2002;114(6):679–686. doi: 10.1002/ajmg.10648. [DOI] [PubMed] [Google Scholar]
- 2.Kehoe P, Wavrant-DeVrieze F, Crook R, Wu W, Holmans P, Fenton I, Spurlock G, Norton N, Williams H, Williams N, Lovestone S, Perez-Tur J, Hutton M, Chartier-Harlin MC, Shears S, Roehl K, Booth J, Vorst WV, Ramic D, Williams J, Goate A, Hardy J, Owen MJ. A full genome scan for late onset Alzheimer’s disease. Hum Mol Genet. 1999;8(2):237–245. doi: 10.1093/hmg/8.2.237. [DOI] [PubMed] [Google Scholar]
- 3.Myers A, Wavrant-DeVrieze F, Holmans P, Hamshere M, Crook R, Compton D, Marshall H, Meyer D, Shears S, Booth J, Ramic D, Knowles H, Morris JC, Williams N, Norton N, Abraham R, Kehoe P, Williams H, Rudrasingham V, Rice F, Giles P, Tunstall N, Jones L, Lovestone S, Williams J, Owen MJ, Hardy J, Goate A. Full genome screen for Alzheimer disease: Stage II analysis. Am J Med Genet. 2002;114:235–244. doi: 10.1002/ajmg.10183. [DOI] [PubMed] [Google Scholar]
- 4.Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, Adamson J, Ronald J, Blangero J, Hutton M, Younkin SG. Linkage of plasma Abeta42 to a quantitative locus on chromosome 10 in late-onset Alzheimer’s disease pedigrees. Science. 2000;290(5500):2303–2304. doi: 10.1126/science.290.5500.2303. [DOI] [PubMed] [Google Scholar]
- 5.Lendon C, Craddock N. Susceptibility gene(s) for Alzheimer’s disease on chromosome 10. Trends Neurosci. 2001;24(10):557–559. doi: 10.1016/s0166-2236(00)01912-3. [DOI] [PubMed] [Google Scholar]
- 6.Kamboh MI. Molecular genetics of late-onset Alzheimer’s disease. Ann Hum Genet. 2004;68(Pt 4):381–404. doi: 10.1046/j.1529-8817.2004.00110.x. [DOI] [PubMed] [Google Scholar]
- 7.Bertram L, Tanzi RE. The current status of Alzheimer’s disease genetics: what do we tell the patients? Pharmacol Res. 2004;50(4):385–396. doi: 10.1016/j.phrs.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 8.Edland SD, Silverman JM, Peskind ER, Tsuang D, Wijsman E, Morris JC. Increased risk of dementia in mothers of Alzheimer’s disease cases: evidence for maternal inheritance. Neurology. 1996;47(1):254–256. doi: 10.1212/wnl.47.1.254. [DOI] [PubMed] [Google Scholar]
- 9.van der Walt JM, Dementieva YA, Martin ER, Scott WK, Nicodemus KK, Kroner CC, Welsh-Bohmer KA, Saunders AM, Roses AD, Small GW, Schmechel DE, Murali DP, Gilbert JR, Haines JL, Vance JM, Pericak-Vance MA. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci Lett. 2004;365(1):28–32. doi: 10.1016/j.neulet.2004.04.051. [DOI] [PubMed] [Google Scholar]
- 10.Carrieri G, Bonafe M, De Luca M, Rose G, Varcasia O, Bruni A, Maletta R, Nacmias B, Sorbi S, Corsonello F, Feraco E, Andreev KF, Yashin AI, Franceschi C, De Benedictis G. Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer’s disease. Hum Genet. 2001;108(3):194–198. doi: 10.1007/s004390100463. [DOI] [PubMed] [Google Scholar]
- 11.Klunemann HH, Kloiber S, Wurster HW, Klein HE. Familial plaque-only Alzheimer’s disease in the rottal-inn and passau counties. Psychiatr Prax. 2004;31(Suppl 1):61–63. doi: 10.1055/s-2004-828439. [DOI] [PubMed] [Google Scholar]
- 12.Kong A, Cox NJ. Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet. 1997;61(5):1179–1188. doi: 10.1086/301592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996;58(6):1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 14.Lander ES, Schork NJ. Genetic dissection of complex traits. Science. 1994;265(5181):2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- 15.Hinds D, Risch N. The ASPEX package: affected sib-pair mapping. 1996. Ref Type: Unpublished Work. [Google Scholar]
- 16.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30(1):97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 17.Ikeuchi T, Igarashi S, Takiyama Y, Onodera O, Oyake M, Takano H, Koide R, Tanaka H, Tsuji S. Non-Mendelian transmission in dentatorubral-pallidoluysian atrophy and Machado-Joseph disease: the mutant allele is preferentially transmitted in male meiosis. Am J Hum Genet. 1996;58(4):730–733. [PMC free article] [PubMed] [Google Scholar]
- 18.Kremer B, Almqvist E, Theilmann J, Spence N, Telenius H, Goldberg YP, Hayden MR. Sex-dependent mechanisms for expansions and contractions of the CAG repeat on affected Huntington disease chromosomes. Am J Hum Genet. 1995;57(2):343–350. [PMC free article] [PubMed] [Google Scholar]
- 19.Kovtun IV, Welch G, Guthrie HD, Hafner KL, McMurray CT. CAG repeat lengths in X- and Y-bearing sperm indicate that gender bias during transmission of Huntington’s disease gene is determined in the embryo. J Biol Chem. 2004;279(10):9389–9391. doi: 10.1074/jbc.M313080200. [DOI] [PubMed] [Google Scholar]
- 20.Bonilla E, Tanji K, Hirano M, Vu TH, DiMauro S, Schon EA. Mitochondrial involvement in Alzheimer’s disease. Biochim Biophys Acta. 1999;1410(2):171–182. doi: 10.1016/s0005-2728(98)00165-0. [DOI] [PubMed] [Google Scholar]
- 21.Orth M, Schapira AH. Mitochondria and degenerative disorders. Am J Med Genet. 2001;106(1):27–36. doi: 10.1002/ajmg.1425. [DOI] [PubMed] [Google Scholar]
- 22.Swerdlow RH, Kish SJ. Mitochondria in Alzheimer’s disease. Int Rev Neurobiol. 2002;53:341–385. doi: 10.1016/s0074-7742(02)53013-0. [DOI] [PubMed] [Google Scholar]
- 23.Schuessel K, Leutner S, Cairns NJ, Muller WE, Eckert A. Impact of gender on upregulation of antioxidant defence mechanisms in Alzheimer’s disease brain. J Neural Transm. 2004;111(9):1167–1182. doi: 10.1007/s00702-004-0156-5. [DOI] [PubMed] [Google Scholar]
- 24.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304(5669):448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 25.Cardoso SM, Santana I, Swerdlow RH, Oliveira CR. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Abeta toxicity. J Neurochem. 2004;89(6):1417–1426. doi: 10.1111/j.1471-4159.2004.02438.x. [DOI] [PubMed] [Google Scholar]
- 26.Kelly TL, Trasler JM. Reproductive epigenetics. Clin Genet. 2004;65(4):247–260. doi: 10.1111/j.0009-9163.2004.00236.x. [DOI] [PubMed] [Google Scholar]
- 27.Keverne EB. Genomic imprinting in the brain. Curr Opin Neurobiol. 1997;7(4):463–468. doi: 10.1016/s0959-4388(97)80023-2. [DOI] [PubMed] [Google Scholar]
- 28.Holliday R. DNA methylation and epigenetic mechanisms. Cell Biophys. 1989;15(1–2):15–20. doi: 10.1007/BF02991575. [DOI] [PubMed] [Google Scholar]
- 29.Payao SL, Smith MD, Bertolucci PH. Differential chromosome sensitivity to 5-azacytidine in Alzheimer’s disease. Gerontology. 1998;44(5):267–271. doi: 10.1159/000022023. [DOI] [PubMed] [Google Scholar]
- 30.Jones C, Booth C, Rita D, Jazmines L, Spiro R, McCulloch B, McCaskill C, Shaffer LG. Identification of a case of maternal uniparental disomy of chromosome 10 associated with confined placental mosaicism. Prenat Diagn. 1995;15(9):843–848. doi: 10.1002/pd.1970150909. [DOI] [PubMed] [Google Scholar]
- 31.Kovtun IV, Therneau TM, McMurray CT. Gender of the embryo contributes to CAG instability in transgenic mice containing a Huntington’s disease gene. Hum Mol Genet. 2000;9(18):2767–2775. doi: 10.1093/hmg/9.18.2767. [DOI] [PubMed] [Google Scholar]