

Abstract
The Escherichia coli MG1655 genome has been completely sequenced. The annotated sequence, biochemical information, and other information were used to reconstruct the E. coli metabolic map. The stoichiometric coefficients for each metabolic enzyme in the E. coli metabolic map were assembled to construct a genome-specific stoichiometric matrix. The E. coli stoichiometric matrix was used to define the system's characteristics and the capabilities of E. coli metabolism. The effects of gene deletions in the central metabolic pathways on the ability of the in silico metabolic network to support growth were assessed, and the in silico predictions were compared with experimental observations. It was shown that based on stoichiometric and capacity constraints the in silico analysis was able to qualitatively predict the growth potential of mutant strains in 86% of the cases examined. Herein, it is demonstrated that the synthesis of in silico metabolic genotypes based on genomic, biochemical, and strain-specific information is possible, and that systems analysis methods are available to analyze and interpret the metabolic phenotype.
Keywords: bioinformatics, metabolism, genotype-phenotype relation, flux balance analysis
The complete genome sequence for a number of microorganisms has been established (The Institute for Genomic Research at www.tigr.org). The genome sequencing efforts and the subsequent bioinformatic analyses have defined the molecular “parts catalogue” for a number of living organisms. However, it is evident that cellular functions are multigeneic in nature, thus one must go beyond a molecular parts catalogue to elucidate integrated cellular functions based on the molecular cellular components (1). Therefore, to analyze the properties and the behavior of complex cellular networks, one needs to use methods that focus on the systemic properties of the network. Approaches to analyze, interpret, and ultimately predict cellular behavior based on genomic and biochemical data likely will involve bioinformatics and computational biology and form the basis for subsequent bioengineering analysis.
In moving toward the goal of developing an integrated description of cellular processes, it should be recognized that there exists a history of studying the systemic properties of metabolic networks (2) and many mathematical methods have been developed to carry out such studies. These methods include approaches such as metabolic control analysis (3, 4), flux balance analysis (FBA) (5–7), metabolic pathway analysis (8–11, 69), cybernetic modeling (12), biochemical systems theory (13), temporal decomposition (14), and so on. Although many mathematical methods and approaches have been developed, there are few comprehensive metabolic systems for which detailed kinetic information is available and where such detailed analysis can be carried out (see refs. 15–17 for a few noteworthy exceptions).
To analyze, interpret, and predict cellular behavior, each individual step in a biochemical network must be described, normally with a rate equation that requires a number of kinetic constants. Unfortunately, it currently is not possible to formulate this level of description of cellular processes on a genome scale. The kinetic parameters cannot be estimated from the genome sequence and these parameters are not available in the literature. In the absence of kinetic information, it is, however, still possible to assess the theoretical capabilities of one integrated cellular process, namely metabolism, and examine the feasible metabolic flux distributions under a steady-state assumption. The steady-state analysis is based on the constraints imposed on the metabolic network by the stoichiometry of the metabolic reactions, which basically represent mass balance constraints. The steady-state analysis of metabolic networks based on the mass balance constraints is known as FBA (7, 18, 19). This analysis differs from detailed kinetic modeling of cellular processes, in that it does not attempt to predict the exact behavior of metabolic networks. Rather it uses known constraints on the integrated function of multiple enzymes to separate the states that a system can reach from those that it cannot. Then within the domain of allowable behavior one can study the genotype-phenotype relation, such as the stoichiometric optimal growth performance in a defined environment.
In this manuscript, we have used the biochemical literature, the annotated genome sequence data, and strain-specific information, to formulate an organism scale in silico representation of the Escherichia coli MG1655 metabolic capabilities. FBA then was used to assess metabolic capabilities subject to these constraints leading to qualitative predictions of growth performance.
Materials and Methods
Definition of the E. coli MG1655 Metabolic Map.
An in silico representation of E. coli metabolism has been constructed. We have used the biochemical literature (20), genomic information (21), and the metabolic databases (22–24). Because of the long history of E. coli research, there was biochemical or genetic evidence for every metabolic reactions included in the in silico representation, and in most cases, there was both genetic and biochemical evidence (Table 1). The complete list of genes included in the in silico analysis is shown in Table 1, and the metabolic reactions catalyzed by these genes can be found on the web (http://gcrg.ucsd.edu/downloads.html). The stoichiometric coefficients for each metabolic reaction within this list were used to form the stoichiometric matrix S.
Table 1.
The genes included in the E. coli metabolic genotype (21)
| Central metabolism (EMP, PPP, TCA cycle, electron transport) | aceA, aceB, aceE, aceF, ackA, acnA, acnB, acs, adhE, agp, appB, appC, atpA, atpB, atpC, atpD, atpE, atpF, atpG, atpH, atpI, cydA, cydB, cydC, cydD, cyoA, cyoB, cyoC, cyoD, dld, eno, fba, fbp, fdhF, fdnG, fdnH, fdnI, fdoG, fdoH, fdoI, frdA, frdB, frdC, frdD, fumA, fumB, fumC, galM, gapA, gapC_1, gapC_2, glcB, glgA, glgC, glgP, glk, glpA, glpB, glpC, glpD, gltA, gnd, gpmA, gpmB, hyaA, hyaB, hyaC, hybA, hybC, hycB, hycE, hycF, hycG, icdA, lctD, ldhA, lpdA, malP, mdh, ndh, nuoA, nuoB, nuoE, nuoF, nuoG, nuoH, nuoI, nuoJ, nuoK, nuoL, nuoM, nuoN, pckA, pfkA, pfkB, pflA, pflB, pflC, pflD, pgi, pgk, pntA, pntB, ppc, ppsA, pta, purT, pykA, pykF, rpe, rpiA, rpiB, sdhA, sdhB, sdhC, sdhD, sfcA, sucA, sucB, sucC, sucD, talB, tktA, tktB, tpiA, trxB, zwf, pgl (30), maeB (30) |
| Alternative carbon source | adhC, adhE, agaY, agaZ, aldA, aldB, aldH, araA, araB, araD, bglX, cpsG, deoB, fruK, fucA, fucI, fucK, fucO, galE, galK, galT, galU, gatD, gatY, glk, glpK, gntK, gntV, gpsA, lacZ, manA, melA, mtlD, nagA, nagB, nanA, pfkB, pgi, pgm, rbsK, rhaA, rhaB, rhaD, srlD, treC, xylA, xylB |
| Amino acid metabolism | adi, aldH, alr, ansA, ansB, argA, argB, argC, argD, argE, argF, argG, argH, argI, aroA, aroB, aroC, aroD, aroE, aroF, aroG, aroH, aroK, aroL, asd, asnA, asnB, aspA, aspC, avtA, cadA, carA, carB, cysC, cysD, cysE, cysH, cysI, cysJ, cysK, cysM, cysN, dadA, dadX, dapA, dapB, dapD, dapE, dapF, dsdA, gabD, gabT, gadA, gadB, gdhA, glk, glnA, gltB, gltD, glyA, goaG, hisA, hisB, hisC, hisD, hisF, hisG, hisH, hisI, ilvA, ilvB, ilvC, ilvD, ilvE, ilvG_1, ilvG_2, ilvH, ilvI, ilvM, ilvN, kbl, ldcC, leuA, leuB, leuC, leuD, lysA, lysC, metA, metB, metC, metE, metH, metK, metL, pheA, proA, proB, proC, prsA, putA, sdaA, sdaB, serA, serB, serC, speA, speB, speC, speD, speE, speF, tdcB, tdh, thrA, thrB, thrC, tnaA, trpA, trpB, trpC, trpD, trpE, tynA, tyrA, tyrB, ygjG, ygjH, alaB (42), dapC (43), pat (44), prr (44), sad (45), methylthioadenosine nucleosidase (46), 5-methylthioribose kinase (46), 5-methylthioribose-l-phosphate isomerase (46), adenosyl homocysteinase (47), l-cysteine desulfhydrase (44), glutaminase A (44), glutaminase B (44) |
| Purine & pyrimidine metabolism | add, adk, amn, apt, cdd, cmk, codA, dcd, deoA, deoD, dgt, dut, gmk, gpt, gsk, guaA, guaB, guaC, hpt, mutT, ndk, nrdA, nrdB, nrdD, nrdE, nrdF, purA, purB, purC, purD, purE, purF, purH, purK, purL, purM, purN, purT, pyrB, pyrC, pyrD, pyrE, pyrF, pyrG, pyrH, pyrI, tdk, thyA, tmk, udk, udp, upp, ushA, xapA, yicP, CMP glycosylase (48) |
| Vitamin & cofactor metabolism | acpS, bioA, bioB, bioD, bioF, coaA, cyoE, cysG, entA, entB, entC, entD, entE, entF, epd, folA, folC, folD, folE, folK, folP, gcvH, gcvP, gcvT, gltX, glyA, gor, gshA, gshB, hemA, hemB, hemC, hemD, hemE, hemF, hemH, hemK, hemL, hemM, hemX, hemY, ilvC, lig, lpdA, menA, menB, menC, menD, menE, menF, menG, metF, mutT, nadA, nadB, nadC, nadE, ntpA, pabA, pabB, pabC, panB, panC, panD, pdxA, pdxB, pdxH, pdxJ, pdxK, pncB, purU, ribA, ribB, ribD, ribE, ribH, serC, thiC, thiE, thiF, thiG, thiH, thrC, ubiA, ubiB, ubiC, ubiG, ubiH, ubiX, yaaC, ygiG, nadD (49), nadF (49), nadG (49), panE (50), pncA (49), pncC (49), thiB (51), thiD (51), thiK (51), thiL (51), thiM (51), thiN (51), ubiE (52), ubiF (52), arabinose-5-phosphate isomerase (22), phosphopantothenate-cysteine ligase (50), phosphopantothenate-cysteine decarboxylase (50), phospho-pantetheine adenylyltransferase (50), dephosphoCoA kinase (50), NMN glycohydrolase (49) |
| Lipid metabolism | accA, accB, accD, atoB, cdh, cdsA, cls, dgkA, fabD, fabH, fadB, gpsA, ispA, ispB, pgpB, pgsA, psd, pssA, pgpA (53) |
| Cell wall metabolism | ddlA, ddlB, galF, galU, glmS, glmU, htrB, kdsA, kdsB, kdtA, lpxA, lpxB, lpxC, lpxD, mraY, msbB, murA, murB, murC, murD, murE, murF, murG, murI, rfaC, rfaD, rfaF, rfaG, rfaI, rfaJ, rfaL, ushA, glmM (54), lpcA (55), rfaE (55), tetraacyldisaccharide 4′ kinase (55), 3-deoxy-d-manno-octulosonic-acid 8-phosphate phosphatase (55) |
| Transport processes | araE, araF, araG, araH, argT, aroP, artI, artJ, artM, artP, artQ, brnQ, cadB, chaA, chaB, chaC, cmtA, cmtB, codB, crr, cycA, cysA, cysP, cysT, cysU, cysW, cysZ, dctA, dcuA, dcuB, dppA, dppB, dppC, dppD, dppF, fadL, focA, fruA, fruB, fucP, gabP, galP, gatA, gatB, gatC, glnH, glnP, glnQ, glpF, glpT, gltJ, gltK, gltL, gltP, gltS, gntT, gpt, hisJ, hisM, hisP, hisQ, hpt, kdpA, kdpB, kdpC, kgtP, lacY, lamB, livF, livG, livH, livJ, livK, livM, lldP, lysP, malE, malF, malG, malK, malX, manX, manY, manZ, melB, mglA, mglB, mglC, mtlA, mtr, nagE, nanT, nhaA, nhaB, nupC, nupG, oppA, oppB, oppC, oppD, oppF, panF, pheP, pitA, pitB, pnuC, potA, potB, potC, potD, potE, potF, potG, potH, potI, proP, proV, proW, proX, pstA, pstB, pstC, pstS, ptsA, ptsG, ptsI, ptsN, ptsP, purB, putP, rbsA, rbsB, rbsC, rbsD, rhaT, sapA, sapB, sapD, sbp, sdaC, srlA_1, srlA_2, srlB, tdcC, tnaB, treA, treB, trkA, trkG, trkH, tsx, tyrP, ugpA, ugpB, ugpC, ugpE, uraA, xapB, xylE, xylF, xylG, xylH, fruF (56), gntS (57), metD (43), pnuE (49), scr (56) |
The in silico E. coli MG1655 metabolic genotype used herein is available on the web: http://gcrg.ucsd.edu/downloads.html.
Determining the Capabilities of the E. coli Metabolic Network.
The theoretical metabolic capabilities of E. coli were assessed by FBA (5–7). The metabolic capabilities of the in silico metabolic genotype were partially defined by mass balance constraints; mathematically represented by a matrix equation:
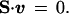 |
1 |
The matrix S is the mxn stoichiometric matrix, where m is the number of metabolites and n is the number of reactions in the network. The E. coli stoichiometric matrix was 436 × 720. The vector v represents all fluxes in the metabolic network, including the internal fluxes, transport fluxes, and the growth flux. The optimal v vector was determined and defined the steady-state metabolic flux distribution.
For the E. coli metabolic network, the number of fluxes was greater than the number of mass balance constraints; thus, there was a plurality of feasible flux distributions that satisfied the mass balance constraints (defined in Eq. 1), and the solutions (or feasible metabolic flux distributions) were confined to the nullspace of the matrix S.
In addition to the mass balance constraints, we imposed constraints on the magnitude of each individual metabolic flux.
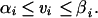 |
2 |
The linear inequality constraints were used to enforce the reversibility/irreversibility of metabolic reactions and the maximal metabolic fluxes in the transport reactions. The intersection of the nullspace and the region defined by the linear inequalities formally defined a region in flux space that we will refer to as the feasible set. The feasible set defined the capabilities of the metabolic network subject to the subset of cellular constraints, and all feasible metabolic flux distributions lie within the feasible set (see Fig. 1). However, every vector v within the feasible set is not reachable by the cell under a given condition because of other constraints not considered in the analysis (i.e., maximal internal fluxes and gene regulation). The feasible set can be further reduced by imposing additional constraints, and if all of the necessary details to describe metabolic dynamics are known, then the feasible set may reduce to a small region or even a single point (see Fig. 1).
Figure 1.

The feasible solution set for a hypothetical metabolic reaction network. (A) The steady-state operation of the metabolic network is restricted to the region within a cone, defined as the feasible set (8). The feasible set contains all flux vectors that satisfy the physicochemical constrains (Eqs. 1 and 2). Thus, the feasible set defines the capabilities of the metabolic network. All feasible metabolic flux distributions lie within the feasible set, and (B) in the limiting case, where all constraints on the metabolic network are known, such as the enzyme kinetics and gene regulation, the feasible set may be reduced to a single point. This single point must lie within the feasible set.
For the analysis presented herein, we defined αi = 0 for irreversible internal fluxes, and αi = −∞ for reversible internal fluxes. The reversibility of the metabolic reactions was determined from the biochemical literature and is identified for each reaction on the web site. The transport flux for inorganic phosphate, ammonia, carbon dioxide, sulfate, potassium, and sodium was unrestrained (αi = −∞ and βi = ∞). The transport flux for the other metabolites, when available in the in silico medium, was constrained between zero and the maximal level (0 < vi < vimax). However, when the metabolite was not available in the medium, the transport flux was constrained to zero. The transport flux for metabolites that were capable of leaving the metabolic network (i.e., acetate, ethanol, lactate, succinate, formate, pyruvate, etc.) always was unconstrained in the outward direction.
A particular metabolic flux distribution within the feasible set was found by using linear programming (LP). A commercially available LP package was used (lindo, Lindo Systems, Chicago). LP identified a solution that minimized a particular metabolic objective (subject to the imposed constraints) (5, 25, 26), and was formulated as shown. Minimize −Z, where
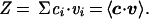 |
3 |
The vector c was used to select a linear combination of metabolic fluxes to include in the objective function (27). Herein, c was defined as the unit vector in the direction of the growth flux, and the growth flux was defined in terms of the biosynthetic requirements:
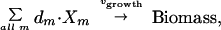 |
4 |
where dm is the biomass composition of metabolite Xm (defined from the literature; ref. 28), and the growth flux is modeled as a single reaction that converts all of the biosynthetic precursors into biomass.
Results
FBA was used to examine the change in the metabolic capabilities caused by gene deletions. To simulate a gene deletion, the flux through the corresponding enzymatic reaction was restricted to zero. Genes that code for isozymes or genes that code for components of same enzyme complex were simultaneously removed (i.e., aceEF, sucCD). The optimal value of the objective (Zmutant) was compared with the “wild-type” objective (Z) to determine the systemic effect of the gene deletion. The ratio of optimal growth yields (Zmutant/Z) was calculated (Fig. 2).
Figure 2.

Gene deletions in E. coli MG1655 central intermediary metabolism; maximal biomass yields on glucose for all possible single gene deletions in the central metabolic pathways. The optimal value of the mutant objective function (Zmutant) compared with the “wild-type” objective function (Z), where Z is defined in Eq. 3. The ratio of optimal growth yields (Zmutant/Z). The results were generated in a simulated aerobic environment with glucose as the carbon source. The transport fluxes were constrained as follows: βglucose = 10 mmol/g-dry weight (DW) per h; βoxygen = 15 mmol/g-DW per h. The maximal yields were calculated by using FBA with the objective of maximizing growth. The biomass yields are normalized with respect to the results for the full metabolic genotype. The yellow bars represent gene deletions that reduced the maximal biomass yield to less than 95% of the in silico wild type.
Gene Deletions.
E. coli MG1655 in silico was subjected to deletion of each individual gene product in the central metabolic pathways [glycolysis, pentose phosphate pathway (PPP), tricarboxylic acid (TCA) cycle, respiration processes], and the maximal capability of each in silico mutant metabolic network to support growth was assessed with FBA. The simulations were performed under an aerobic growth environment on minimal glucose medium.
The results identified the essential (required for growth) central metabolic genes (Fig. 2). For growth on glucose, the essential gene products were involved in the three-carbon stage of glycolysis, three reactions of the TCA cycle, and several points within the PPP. The remainder of the central metabolic genes could be removed and E. coli in silico maintained the potential to support cellular growth. This result was related to the interconnectivity of the metabolic reactions. The in silico gene deletion results suggest that a large number of the central metabolic genes can be removed without eliminating the capability of the metabolic network to support growth under the conditions considered.
Are the in Silico Redundancy Results Consistent with Mutant Data?
The in silico gene deletion study results were compared with growth data from known mutants. The growth characteristics of a series of E. coli mutants on several different carbon sources were examined and compared with the in silico deletion results (Table 2). From this analysis, 86% (68 of 79 cases) of the in silico predictions were consistent with the experimental observations.
Table 2.
Comparison of the predicted mutant growth characteristics from the gene deletion study to published experimental results with single mutants
| Gene | glc | gl | succ | ac | Reference |
|---|---|---|---|---|---|
| aceA | +/+ | +/+ | −/− | (58) | |
| aceB | −/− | (58) | |||
| aceEF* | −/+ | (60) | |||
| ackA | +/+ | (61) | |||
| acn | −/− | −/− | (58) | ||
| acs | +/+ | (61) | |||
| cyd | +/+ | (62) | |||
| cyo | +/+ | (62) | |||
| eno† | −/+ | −/+ | −/− | −/− | (30) |
| fba∥ | −/+ | (30) | |||
| fbp | +/+ | −/− | −/− | −/− | (30) |
| frd | +/+ | +/+ | +/+ | (60) | |
| gap | −/− | −/− | −/− | −/− | (30) |
| glk | +/+ | (30) | |||
| gltA | −/− | −/− | (58) | ||
| gnd | +/+ | (30) | |||
| idh | −/− | −/− | (58) | ||
| mdh†† | +/+ | +/+ | +/+ | (63) | |
| ndh | +/+ | +/+ | (59) | ||
| nuo | +/+ | +/+ | (59) | ||
| pfk† | −/+ | (30) | |||
| pgi‡ | +/+ | +/− | +/− | (30) | |
| pgk | −/− | −/− | −/− | −/− | (30) |
| pgl | +/+ | (30) | |||
| pntAB | +/+ | +/+ | +/+ | (29) | |
| ppc§ | ±/+ | −/+ | +/+ | (63, 64) | |
| pta | +/+ | (61) | |||
| pts | +/+ | (30) | |||
| pyk | +/+ | (30) | |||
| rpi | −/− | −/− | −/− | −/− | (30) |
| sdhABCD | +/+ | −/− | −/− | (58) | |
| sucAB | +/+ | −/+ | −/+ | (60) | |
| tktAB | −/− | (30) | |||
| tpi** | −/+ | −/− | −/− | −/− | (30) |
| unc | +/+ | ±/+ | −/− | (66–68) | |
| zwf | +/+ | +/+ | +/+ | (30) |
Results are scored as + or − meaning growth or no growth determined from in vivo/in silico data. The ± indicates that suppressor mutations have been observed that allow the mutant strain to grow. In 68 of 79 cases the in silico behavior is the same as the experimentally observed behavior. glc, glucose; ac, acetate; gl, glycerol; succ, succinate.
The in vivo aceAE strain is able to grow under anaerobic growth conditions by using the pyruvate formate lyase.
† The in silico pfk strain is able to grow by increasing the PPP flux ≈ 5× and using the pps gene product to overcome PEP deficiency.
‡ The in silico pgi strain is unable to grow with glycerol or succinate as the carbon source because it is unable to synthesize glycogen and one carbohydrate component in the lipopolysaccharide. These are likely nonessential components of the biomass.
§ The grow on glycerol and glucose is possible through the utilization of the glyoxylate bypass. Constitutive mutations in the glyoxylate bypass can suppress the ppc phenotype.
¶ The in silico eno strain is able to grow by the synthesis and degradation of serine.
∥ There is evidence that fba has an inhibitory effect on stable RNA synthesis (65). Such an inhibition cannot be predicted by FBA.
The inability of tpi mutants to grow on glucose may be related to the accumulation of dihydroxyacetone phosphate, which leads to the formation of the bactericidal compound methylglyoxal (30).
†† Very slow growth on glycerol and succinate.
How Are Cellular Fluxes Redistributed?
The potential of many in silico deletion strains to support growth led to questions regarding how the E. coli metabolic genotype deals with the loss of metabolic functions. The answer involves the degree of stoichiometric connectivity of key metabolites. For illustration, the flux redistributions to optimally support growth of a single mutant and a double mutant were investigated.
The optimal metabolic flux distribution for the in silico wild type was calculated (Fig. 3). The constraints used in the LP problem are defined in the figure legend. The in silico results suggest that optimally the oxidative branch of the PPP was used to generate a large fraction of the NADPH (66% in silico: 20–50% reported in the literature, ref. 29), and the TCA cycle produced NADH. The optimal flux distribution also suggested that the majority of the high-energy phosphate bonds were generated via oxidative phosphorylation and acetate secretion because of limitations of the oxygen supply.
Figure 3.

Rerouting of metabolic fluxes. (Black) Flux distribution for the complete gene set. (Red) zwf- mutant. Biomass yield is 99% of the results for the full metabolic genotype. (Blue) zwf- pnt- mutant. Biomass yield is 92% of the results for the full metabolic genotype (see text). The solid lines represent enzymes that are being used, with the corresponding flux value noted. The fluxes [substrates converted/h per g-dry weight (DW)] were calculated by using FBA with the input parameters of glucose uptake rate (βglucose = 6.6 mmol glucose/h per g-DW) and oxygen uptake rate (βoxygen = 12.4 mmol oxygen/h per g-DW) (41).
The in silico gene deletion results predicted that the optimal biomass yield of the zwf- (glucose-6-phosphate dehydrogenase) in silico strain was slightly less than the wild type. The optimal flux distribution of the zwf- in silico strain (Fig. 2) was calculated, and the NADPH was optimally generated through the transhydrogenase reaction and an elevated TCA cycle flux. The PPP biosynthetic precursors were generated in the nonoxidative branch. This metabolic flux rerouting resulted in an optimal biomass yield that was 99% of the in silico wild type.
The transhydrogenase (pnt) also was deleted in silico, creating an in silico double deletion mutant and eliminating an alternate source of NADPH. The double mutant still maintained growth potential. The optimal flux distribution (Fig. 2) used the isocitrate dehydrogenase and the malic enzyme to produce NADPH. The optimal biomass yield of the double mutant was 92% of the in silico wild type. The FBA results were consistent with the experimental observations that the zwf- strain (30) and the pnt- strain (29) are able to grow at near wild-type yields. Furthermore, the zwf- pnt- double mutant strain also has been shown to grow (μmutant/μwild type = 57%) (29).
Discussion
Extensive information about the molecular composition and function of several single-cellular organisms has become available. A next important step will be to incorporate the available information to generate whole-cell models with interpretative and predictive capability. Herein, we have taken a step in that direction by using a set of constraints on cellular metabolism on the whole-cell level to analyze the metabolic capabilities of the extensively studied bacterium E. coli. We have calculated the optimal metabolic network utilization with a FBA. The in silico results, based only on stoichiometric and capacity constraints, were consistent with experimental data for the wild type and many of the mutant strains examined.
The construction of comprehensive in silico metabolic maps provided a framework to study the consequences of alterations in the genotype and to gain insight into the genotype-phenotype relation. The stoichiometric matrix and FBA were used to analyze the consequences of the loss of a gene product function on the metabolic capabilities of E. coli. The results demonstrated an important property of the E. coli metabolic network, namely that there are relatively few critical gene products in central metabolism. The nonessential genes in several organisms have been found experimentally on a genome scale (31, 32), which opens up the opportunity to critically test the in silico predictions. The in silico analysis also suggests that although the ability to grow in one defined environment is only slightly altered the ability to adjust to different environments may be diminished (33). Therefore, the in silico analysis provides a methodology for relating the specific biochemical function of the metabolic enzymes to the integrated properties of the metabolic network.
The in silico analysis presented herein is not the typical metabolic modeling; more appropriately, the analysis can be thought of as a constraining approach. This approach defines the “best” the cell can do and identifies what the cell cannot do, rather than attempting to predict how the cell actually will behave under a given set of conditions. To accomplish this, we have used a set of physicochemical constraints for which there is reliable information available, in particular the stoichiometric properties. FBA does not directly consider regulation or the regulatory constraints on the metabolic network.
The results of FBA can be interpreted in a qualitative or a quantitative sense. At the first level we can ask whether a cell is able to grow under given circumstances and how a loss of the function of a gene product influences this ability. The results presented herein fall into this category. Quantitative predictions would hold true if the cell optimized its growth under the growth conditions considered. Therefore, when applying LP to predict quantitatively the optimal metabolic pathway utilization, it is assumed that the cell has found an “optimal solution” for survival through natural selection, and we have equated survival with growth. Although E. coli may grow optimally in defined media, one should not expect that optimizing growth is the governing objective of the cell under all growth conditions. For example, the regulatory mechanisms can only evolve to stoichiometric optimality in a condition to which the cell has been exposed. Furthermore, the growth behavior of mutant strains is unlikely to be optimal. However, FBA can still be used to delineate the metabolic capabilities of mutant cells based on constraining features, because both wild-type and mutant cells must obey the physicochemical constraints imposed.
The constraints on the system accurately reflect the steady-state capabilities of the metabolic network, but does the calculated optimal flux vector in the feasible set accurately reflect the behavior of the actual metabolic network? It has been shown that in a minimal media the metabolic behavior of wild-type E. coli is consistent with stoichiometric optimality (34). Furthermore, more detailed and critical experimental results are consistent with the hypothesis that E. coli does optimize its growth in acetate or succinate minimal media (33). Taken together these results call for critical experimental investigation to evaluate the hypothesis that stoichiometric and capacity constraints are the principal constraints that limit E. coli maximal growth. Even though growth and metabolic behavior in minimal media are consistent with FBA results, one still must determine the generality of optimal performance. The call for critical experimentation is particularly timely, given the increasing number of genome scale measurements that are now possible through two-dimensional gels (35, 36) and DNA array technology (37, 38). Furthermore, the ability to precisely remove ORFs can be used to design critical experiments (39). The in silico model can be used to choose the most informative knockouts and to design growth experiments with the knockouts.
At the present time, the annotation of the E. coli genome is incomplete, and about one-third of its ORFs do not have a functional assignment. Thus, the metabolic genotype studied here may lack some metabolic capabilities that E. coli possesses. The biochemical literature also was used to define the in silico metabolic genotype, and given the long history of E. coli metabolic research (20), a large percentage of the E. coli metabolic capabilities likely have been identified. However, if additional metabolic capabilities are discovered (40), the E. coli stoichiometric matrix can be updated, leading to an iterative model building process. Additionally, the in silico analysis can help identify missing or incorrect functional assignments by identifying sets of metabolic reactions that are not connected to the metabolic network by the mass balance constraints.
The ability to analyze, interpret, and ultimately predict cellular behavior has been a long sought-after goal. The genome sequencing projects are defining the molecular components within the cell, and describing the integrated function of these molecular components will be a challenging task. The results presented herein suggest that it may be possible to analyze cellular metabolism based on a subset of the constraining features. Continued prediction and experimental verification will be an integral part in the further development of in silico strains. Deciphering the complex relation between the genotype and the phenotype will involve the biological sciences, computer science, and quantitative analysis, all of which must be included in the bioengineering of the 21st century.
Acknowledgments
We thank Ramprasad Ramakrishna, George Church, and Christophe Schilling for critical advice and input. National Institutes of Health Grant GM 57089 and National Science Foundation Grant MCB 9873384 supported this research.
Abbreviations
- FBA
flux balance analysis
- LP
linear programming
- TCA
tricarboxylic acid
- PPP
pentose phosphate pathway
References
- 1.Weng G, Bhalla U S, Iyengar R. Science. 1999;284:92–96. doi: 10.1126/science.284.5411.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey J E. Biotechnol Prog. 1998;14:8–20. doi: 10.1021/bp9701269. [DOI] [PubMed] [Google Scholar]
- 3.Kacser H, Burns J A. Symp Soc Exp Biol. 1973;27:65–104. [PubMed] [Google Scholar]
- 4.Fell D. Understanding the Control of Metabolism. London: Portland; 1996. [Google Scholar]
- 5.Varma A, Palsson B O. Bio/Technology. 1994;12:994–998. [Google Scholar]
- 6.Edwards J, Palsson B. J Biol Chem. 1999;274:17410–17416. doi: 10.1074/jbc.274.25.17410. [DOI] [PubMed] [Google Scholar]
- 7.Bonarius H P J, Schmid G, Tramper J. Trends Biotechnol. 1997;15:308–314. [Google Scholar]
- 8.Schilling C H, Schuster S, Palsson B O, Heinrich R. Biotechnol Prog. 1999;15:296–303. doi: 10.1021/bp990048k. [DOI] [PubMed] [Google Scholar]
- 9.Liao J C, Hou S Y, Chao Y P. Biotechnol Bioeng. 1996;52:129–140. doi: 10.1002/(SICI)1097-0290(19961005)52:1<129::AID-BIT13>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 10.Schuster S, Dandekar T, Fell D A. Trends Biotechnol. 1999;17:53–60. doi: 10.1016/s0167-7799(98)01290-6. [DOI] [PubMed] [Google Scholar]
- 11.Mavrovouniotis M, Stephanopoulos G. Comput Chem Eng. 1992;16:605–619. [Google Scholar]
- 12.Kompala D S, Ramkrishna D, Jansen N B, Tsao G T. Biotechnol Bioeng. 1986;28:1044–1056. doi: 10.1002/bit.260280715. [DOI] [PubMed] [Google Scholar]
- 13.Savageau M A. J Theor Biol. 1969;25:365–369. doi: 10.1016/s0022-5193(69)80026-3. [DOI] [PubMed] [Google Scholar]
- 14.Palsson B O, Joshi A, Ozturk S S. Fed Proc. 1987;46:2485–2489. [PubMed] [Google Scholar]
- 15.Shu J, Shuler M L. Biotechnol Bioeng. 1989;33:1117–1126. doi: 10.1002/bit.260330907. [DOI] [PubMed] [Google Scholar]
- 16.Lee I-D, Palsson B O. Biomed Biochim Acta. 1991;49:771–789. [PubMed] [Google Scholar]
- 17.Tomita M, Hashimoto K, Takahashi K, Shimizu T S, Matsuzaki Y, Miyoshi F, Saito K, Tanida S, Yugi K, Venter J C, et al. Bioinformatics. 1999;15:72–84. doi: 10.1093/bioinformatics/15.1.72. [DOI] [PubMed] [Google Scholar]
- 18.Edwards J S, Ramakrishna R, Schilling C H, Palsson B O. In: Metabolic Engineering. Lee S Y, Papoutsakis E T, editors. New York: Dekker; 1999. pp. 13–57. [Google Scholar]
- 19.Sauer U, Cameron D C, Bailey J E. Biotechnol Bioeng. 1998;59:227–238. [PubMed] [Google Scholar]
- 20.Neidhardt F C, editor. Escherichia coli and Salmonella: Cellular and Molecular Biology. Washington, DC: Am. Soc. Microbiol.; 1996. [Google Scholar]
- 21.Blattner F R, Plunkett G, 3rd, Bloch C A, Perna N T, Burland V, Riley M, Collado-Vides J, Glasner J D, Rode C K, Mayhew G F, et al. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 22.Karp P D, Riley M, Saier M, Paulsen I T, Paley S M, Pellegrini-Toole A. Nucleic Acids Res. 2000;28:56–59. doi: 10.1093/nar/28.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Selkov E, Jr, Grechkin Y, Mikhailova N, Selkov E. Nucleic Acids Res. 1998;26:43–45. doi: 10.1093/nar/26.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogata H, Goto S, Fujibuchi W, Kanehisa M. Biosystems. 1998;47:119–128. doi: 10.1016/s0303-2647(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 25.Pramanik J, Keasling J D. Biotechnol Bioeng. 1997;56:398–421. doi: 10.1002/(SICI)1097-0290(19971120)56:4<398::AID-BIT6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 26.Bonarius H P J, Hatzimanikatis V, Meesters K P H, DeGooijer C D, Schmid G, Tramper J. Biotechnol Bioeng. 1996;50:299–318. doi: 10.1002/(SICI)1097-0290(19960505)50:3<299::AID-BIT9>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 27.Varma A, Palsson B O. J Theor Biol. 1993;165:503–522. doi: 10.1006/jtbi.1993.1202. [DOI] [PubMed] [Google Scholar]
- 28.Neidhardt F C, Umbarger H E. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 13–16. [Google Scholar]
- 29.Hanson R L, Rose C. J Bacteriol. 1980;141:401–404. doi: 10.1128/jb.141.1.401-404.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraenkel D G. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 189–198. [Google Scholar]
- 31.Hutchison C A, Peterson S N, Gill S R, Cline R T, White O, Fraser C M, Smith H O, Venter J C. Science. 1999;286:2165–2169. doi: 10.1126/science.286.5447.2165. [DOI] [PubMed] [Google Scholar]
- 32.Winzeler E A, Shoemaker D D, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke J D, Bussey H, et al. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 33.Edwards J S. Ph.D thesis. La Jolla: Univ. of California-San Diego; 1999. [Google Scholar]
- 34.Varma A, Palsson B O. Appl Environ Microbiol. 1994;60:3724–3731. doi: 10.1128/aem.60.10.3724-3731.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanbogelen R A, Abshire K Z, Moldover B, Olson E R, Neidhardt F C. Electrophoresis. 1997;18:1243–1251. doi: 10.1002/elps.1150180805. [DOI] [PubMed] [Google Scholar]
- 36.Link A J, Robison K, Church G M. Electrophoresis. 1997;18:1259–1313. doi: 10.1002/elps.1150180807. [DOI] [PubMed] [Google Scholar]
- 37.Richmond C S, Glasner J D, Mau R, Jin H, Blattner F R. Nucleic Acids Res. 1999;27:3821–3835. doi: 10.1093/nar/27.19.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown P O, Botstein D. Nat Genet. 1999;21:33–37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- 39.Link A J, Phillips D, Church G M. J Bacteriol. 1997;179:6228–6237. doi: 10.1128/jb.179.20.6228-6237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reizer J, Reizer A, Saier M H., Jr Microbiology. 1997;143:2519–2520. doi: 10.1099/00221287-143-8-2519. [DOI] [PubMed] [Google Scholar]
- 41.Jensen P R, Michelsen O. J Bacteriol. 1992;174:7635–7641. doi: 10.1128/jb.174.23.7635-7641.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reitzer L J. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 391–407. [Google Scholar]
- 43.Greene R C. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 542–560. [Google Scholar]
- 44.McFall E, Newman E B. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 358–379. [Google Scholar]
- 45.Berlyn M K B, Low K B, Rudd K E, Singer M. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 2. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 1715–1902. [Google Scholar]
- 46.Glansdorff N. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 408–433. [Google Scholar]
- 47.Matthews R G. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 600–611. [Google Scholar]
- 48.Neuhard J, Kelln R A. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 580–599. [Google Scholar]
- 49.Penfound T, Foster J W. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 721–730. [Google Scholar]
- 50.Jackowski S. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 687–694. [Google Scholar]
- 51.White R L, Spenser I D. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 680–686. [Google Scholar]
- 52.Meganathan R. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 642–656. [Google Scholar]
- 53.Funk C R, Zimniak L, Dowhan W. J Bacteriol. 1992;174:205–213. doi: 10.1128/jb.174.1.205-213.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mengin-Lecreulx D, van Heijenoort J. J Biol Chem. 1996;271:32–39. doi: 10.1074/jbc.271.1.32. [DOI] [PubMed] [Google Scholar]
- 55.Raetz C R H. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 1035–1063. [Google Scholar]
- 56.Postma P W, Lengeler J W, Jacobson G R. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 1149–1174. [Google Scholar]
- 57.Lin E C C. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 307–342. [Google Scholar]
- 58.Cronan J E, Jr, Laporte D. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 189–198. [Google Scholar]
- 59.Tran Q H, Bongaerts J, Vlad D, Unden G. Eur J Biochem. 1997;244:155–160. doi: 10.1111/j.1432-1033.1997.00155.x. [DOI] [PubMed] [Google Scholar]
- 60.Creaghan I T, Guest J R. J Gen Microbiol. 1978;107:1–13. doi: 10.1099/00221287-107-1-1. [DOI] [PubMed] [Google Scholar]
- 61.Kumari S, Tishel R, Eisenbach M, Wolfe A J. J Bacteriol. 1995;177:2878–2886. doi: 10.1128/jb.177.10.2878-2886.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calhoun M W, Oden K L, Gennis R B, de Mattos M J, Neijssel O M. J Bacteriol. 1993;175:3020–3025. doi: 10.1128/jb.175.10.3020-3025.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Courtright J B, Henning U. J Bacteriol. 1970;102:722–728. doi: 10.1128/jb.102.3.722-728.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vinopal R T, Fraenkel D G. J Bacteriol. 1974;118:1090–1100. doi: 10.1128/jb.118.3.1090-1100.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Singer M, Walter W A, Cali B M, Rouviere P, Liebke H H, Gourse R L, Gross C A. J Bacteriol. 1991;173:6249–6257. doi: 10.1128/jb.173.19.6249-6257.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harold F M, Maloney P C. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 283–306. [Google Scholar]
- 67.von Meyenburg K, Jørgensen B B, Nielsen J, Hansen F G. Mol Gen Genet. 1982;188:240–248. doi: 10.1007/BF00332682. [DOI] [PubMed] [Google Scholar]
- 68.Boogerd F C, Boe L, Michelsen O, Jensen P R. J Bacteriol. 1998;180:5855–5859. doi: 10.1128/jb.180.22.5855-5859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karp P D, Krummenacker M, Paley S, Wagg J. Trends Biotechnol. 1999;17:275–281. doi: 10.1016/s0167-7799(99)01316-5. [DOI] [PubMed] [Google Scholar]