

Abstract
The U2 auxiliary factor large subunit (U2AF65) is an essential pre-mRNA splicing factor for the initial stages of spliceosome assembly. Tandem RNA recognition motifs (RRM)s of U2AF65 recognize polypyrimidine tract signals adjacent to 3′ splice sites. Despite the central importance of U2AF65 for splice site recognition, the relative arrangement of the U2AF65 RRMs and the energetic forces driving polypyrimidine tract recognition remain unknown. Here, the solution conformation of the U2AF65 RNA binding domain determined using small angle x-ray scattering reveals a bilobal shape without apparent interdomain contacts. The proximity of the N and C termini within the inter-RRM configuration is sufficient to explain the action of U2AF65 on spliceosome components located both 5′ and 3′ to its binding site. Isothermal titration calorimetry further demonstrates that an unusually large enthalpy-entropy compensation underlies U2AF65 recognition of an optimal polyuridine tract. Qualitative similarities were observed between the pairwise distance distribution functions of the U2AF65 RNA binding domain and those either previously observed for N-terminal RRMs of Py tract-binding protein that lack interdomain contacts or calculated from the high resolution coordinates of a U2AF65 deletion variant bound to RNA. To further test this model, the shapes and RNA interactions of the wild-type U2AF65 RNA binding domain were compared with those of U2AF65 variants containing either Py tract-binding protein linker sequences or a deletion within the inter-RRM linker. Results of these studies suggest inter-RRM conformational plasticity as a possible means for U2AF65 to universally identify diverse pre-mRNA splice sites.
Pre-mRNA splicing is an essential source of transcript diversity in multicellular eukaryotes (reviewed in Refs. 1 and 2), as reflected by the significant number of cancers and hereditary diseases associated with mutations in pre-mRNA splice site signals or splicing factors (3-5). The splicing machinery (spliceosome) is faced with the task of recognizing relatively short exons (∼150 nucleotides on average in the human genome) located within vast stretches of intron RNA (>1500 nucleotides) (6). Although consensus sequences mark the 5′ and 3′ splice sites of the pre-mRNA, these sequences are relatively short and degenerate so that cryptic splice sites outnumber bona fide splice sites by an order of magnitude (7). Furthermore, to be distinguished and regulated in a specific manner, “weak” alternative splice sites may deviate substantially from the optimal consensus of “strong,” constitutive splice sites (8). The consensus sequences of 3′ splice sites recognized by the major spliceosome are more extensive than those of 5′ splice sites (9) and include a branch point sequence closely followed by a polypyrimidine (Py)4 tract, which is primarily composed of uridines and cytidines. How the spliceosome accurately identifies and pairs the splice sites remains an outstanding question.
Assembly of the core spliceosome (U1, U2, U4, U5, and U6 small nuclear ribonucleoprotein particles) first requires splice site identification by the U2 auxiliary factor large subunit (U2AF65) (10). The U2AF65 subunit serves as an extensive molecular surface, with domains responsible for critical functions (Fig. 1A). (i) As addressed here, two RNA recognition motifs (RRMs) recognize the Py tract splice site signal (11, 12); (ii) a region near the RRMs recruits the ATPase UAP56 to the assembling spliceosome (13); (iii) a C-terminal U2AF homology motif (UHM) domain organizes the SF1 (14) and SF3b155 (15) splicing factors at the branch point sequence; (iv) a tryptophan-containing UHM ligand motif (16) positions the U2AF35 small subunit at the 3′ splice site (17); and (v) an N-terminal arginine-serine-rich (RS) domain promotes branch point sequence/U2 small nuclear RNA annealing (18). To accomplish these tasks, both the C-terminal UHM of U2AF65 and the N-terminal RS domain are required to interact with splicing factors and RNA sites located upstream (5′), respectively, of the Py tract binding site. Simultaneously, the UHM ligand motif near the U2AF65 N terminus interacts with U2AF35 bound to the 3′ splice site. Directed hydroxyl radical footprinting shows that the U2AF65 N terminus contacts pre-mRNA sequences located both preceding and following the Py tract (19). To account for these distant interaction sites, the C- and N-terminal domains of U2AF65 must be brought in proximity of one another in the context of the folded three-dimensional structure, possibly by a bent configuration of the central RRMs.
FIGURE 1.

Design of constructs used for analysis. A, alignment of inter-RRM linker regions below the schematic diagram of U2AF65 domains. RS, RS domain; ULM, U2AF ligand motif. B, a ribbon diagram of a U2AF65 variant (dU2AF65R12) composed of core RNA binding domains (RRM1 and RRM2) connected by a shortened interdomain linker region (PDB ID 2G4B). Each RRM interacts with a distinct RNA oligonucleotide. An arrow indicates the position of the interdomain deletion. The C-terminal β-strand is colored red, and the N-terminal β-strand, which is also involved in RNA binding, is blue. Other β-strands that serve as the principle RNA contact sites are yellow. C, in vitro splicing of the AdML pre-mRNA substrate shows that activity of the ptbU2AF65 variant is comparable with wtU2AF65. The concentrations of U2AF65 proteins added to oligo(dT)-depleted nuclear extract were 0, 0.125, 0.25, 0.50, and 1.0 μm, increasing left to right. The products of the splicing reaction are shown schematically to the right.
We previously investigated the source of the Py tract specificity of U2AF65 by determining the atomic resolution structure of a modified U2AF65 RNA binding domain in complex with an optimal Py tract composed of uridines (20) (Fig. 1B). Despite revealing specific RNA interactions, this high resolution structure was insufficient to test models of a bent inter-RRM arrangement of U2AF65 because co-crystallization required a 20-residue deletion within the inter-RRM linker (21). Structures for two other Py tract binding factors composed of tandem RRMs currently are available for comparison with U2AF65, including Py tract-binding protein (PTB) (22) and FUSE-interacting repressor (FIR) (23). In PTB, an extended linker allows the N-terminal RRM1 and RRM2 to tumble independently, whereas the C-terminal RRM3 and RRM4 assemble into an integrated unit (22, 24, 25). In contrast, the FIR structure demonstrates a compact arrangement of its tandem RRM1 and RRM2 domains (23), with a detailed inter-RRM configuration distinct from that of PTB RRM3/RRM4. Either the PTB-type or the FIR-type models would be compatible with a bent configuration of the U2AF65 RRM surfaces; however, the RRM1 and RRM2 registers would be flexible in the model represented by the N-terminal PTB RRMs, whereas the FIR-type model would require a relatively rigid inter-RRM arrangement. Therefore, additional experimental information is required to address the conformation of U2AF65.
Here, we present the overall molecular shape of the U2AF65 RNA binding domain composed of RRM1 and RRM2, using small angle x-ray scattering (SAXS). A bilobal molecular envelope without apparent interdomain contacts between rigid body models of the known RRM coordinates was observed. Structural information was complemented by thermodynamic characterization, which revealed a significant enthalpy and entropy compensation drives association of the U2AF65 RNA binding domain with an optimal polyuridine RNA. To further explore a possible analogy between the bilobal shapes of U2AF65 and PTB RRM1-RRM2, a U2AF65 variant containing an inter-RRM linker region from PTB was investigated. Separately, a U2AF65 variant with a shortened inter-RRM linker was characterized for comparison with the crystal structure (PDB 2G4B) (20). Solution conformations, thermodynamic contributions to Py tract recognition, and in vitro pre-mRNA splicing activities of the U2AF65 variants were similar to those of the unmodified U2AF65 domain. Overall, these results support a model whereby the U2AF65 RRM1 and RRM2 domains act with relative independence, which has significant ramifications for current paradigms of splice site recognition.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification—The preparation of wild-type (wt)U2AF65R12 (residues 148-336, where R12 indicates RRM1-RRM2), dU2AF65 (residues 1-237 and 258-475), and dU2AF65R12 (residues 148-237 and 258-336) from pGEX-6p were described previously (21) and were derived from the full-length human wtU2AF65 pGEX-4T plasmid (11). Constructs containing the C-terminal UHM, as well as RRM1 and RRM2, wtU2AF65R123 (residues 148-475, where R123 indicates RRM1-RRM2-RRM3), and dU2AF65R123 (residues 148-237 and 258-475), were prepared in a manner similar to the R12 counterparts. The ptbU2AF65 construct was generated by three-way ligation of synthetic oligonucleotides encoding residues (148-167) of human PTB isoform a into the StuI-digested wtU2AF65 plasmid. From this, the ptbU2AF65R12 (U2AF65 residues 148-237 and 258-336, separated by PTB residues 148-167) and ptbU2AF65R123 constructs were subcloned to pGEX-6p and expressed and purified as described for other U2AF65 fragments (21).
RNA Preparation—RNA oligonucleotides composed of 20 uridines (U20) were synthesized by Dharmacon Research, Inc. (Lafayette, Colorado) and deprotected by the manufacturer's protocol for use in calorimetry experiments. RNA concentrations were estimated using calculated molar extinction coefficients as described (26).
Pre-mRNA Splicing Assay—U2AF65 was depleted from HeLa nuclear extract by chromatography through oligo(dT)-cellulose in the presence of 1 m KCl, and splicing reactions were carried out as described (20, 27). Spliced products from the adenovirus major late (AdML) substrate (28) were resolved on 10% denaturing polyacrylamide gels (8 m urea) in Tris-borate-EDTA buffer (Fig. 1C).
Small Angle X-ray Scattering—SAXS data were collected at the SIBYLS Beamline 12.3.1 of the Advanced Light Source (Lawrence Berkeley National Laboratory) using a MARCCD x-ray detector system located 1.6 m from the sample chamber to collect data in the q-spacing 0.01-0.32 Å-1, where q = 4πsinθ/λ (2θ is the scattering angle and λ = 1.03 Å is the wave-length). U2AF65R12 variants were exchanged into 100 mm NaCl, 15 mm HEPES, pH 7.4 by size exclusion chromatography (Superdex-75, GE Healthcare), and the scattering of this buffer was collected before or after each protein sample was subtracted to correct the scattering data. Monodispersity of the samples was further checked prior to SAXS data collection by dynamic light scattering. SAXS data were collected at concentrations of 2.5, 5.0, and 10.0 mg/ml for 6- and 60-s exposures followed by a 6-s exposure to check for radiation damage. Low and high resolution data were scaled and merged from the short and long exposures, respectively. The radii of gyration were analyzed using the Guinier approximation (RgG) (29) with low angle data (q < 1.3/Rg) to evaluate possible interparticle effects and showed little or no variation with concentration (ΔRg < 0.3 Å). Accordingly, scattering profiles at all three concentrations of each variant superimposed within the errors of the experiments after scaling and were merged using PRIMUS (30). The Rg values (RgP) and maximum dimensions (Dmax) were also computed from the entire scattering profiles of the merged files using the program GNOM (31), as given in Fig. 2B and Table 1.
FIGURE 2.

Small angle x-ray scattering analysis of U2AF65 R12 variants. Color schemes are consistent throughout: U2AF65 sequences, blue; PTB variants, maroon. A, experimental x-ray scattering profiles as compared with data calculated from the most typical BUNCH model (solid lines). Scattering intensities from the low q-region for short exposures and high q-region for long exposures were integrated and merged to achieve the experimental scattering profiles shown. The relative scattering intensities are arbitrarily displaced along a logarithmic y axis for clarity. B, comparison of P(r) functions for wtU2AF65R12, ptbU2AF65R12, and dU2AF65R12 calculated from the experimental scattering profiles using the program GNOM (31). The functions are presented in arbitrary units. The radius of gyration (Rg) and maximum intraparticle size (Dmax) of the variants are in the inset. C, the P(r) functions calculated from the experimental dU2AF65R12 or wtU2AF65R12 scattering data, respectively, as compared with data calculated from the protein coordinates of the dU2AF65R12 (PDB ID 2G4B) or FIR R12 structures (PDB ID 2QFJ) using the program CRYSOL (46). D, envelope restorations of wtU2AF65R12 (colored as in Fig. 1B), ptbU2AF65R12, and dU2AF65R12. Mean ab initio shapes resulting from the program GASBOR (32) are superimposed with the most typical model built by the program BUNCH (33). For the BUNCH models, the ab initio models of the inter-RRM linker regions are shown as spheres, and the rigid body models of the individual RRMs are depicted by ribbon diagrams. The mean χ2 value for the GASBOR models and the NSD value of the most typical BUNCH model are given. E, for comparison, the solvent accessible surfaces and ribbon diagrams of the dU2AF65R12 and FIR R12 coordinates are shown following removal of nucleotides. The locations of RRM1 and RRM2 are indicated for wtU2AF65R12 and FIR R12, and remaining models are oriented similarly. Panels D and E were drawn using PyMOL.
TABLE 1.
Overall parameters and quality indicators derived from scattering data for U2AF65R12 variants
RgG, radius of gyration value from the Guinier analysis; RgP, radius of gyration value from the P(r) analysis; Dmax, maximum size; χ2 values are the discrepancies between the experimental data and scattering from the kth model as indicated by the subscript: χ2ab, ab initio GASBOR model; χ2RB, rigid body/ab initio BUNCH model; χ2dU2AF, dU2AF65R12 crystal structure (PDB ID 2G4B with RNA coordinates removed); χ2FIR, FIR R12 crystal structure (PDB ID 2QFJ, chain b with nucleotide removed). CRYSOL was used to calculate scattering curves to q = 0.2 Å−1 from PDB coordinates with a single hydration layer of density 0.38 e/Å3 added to the molecular surface. NSD is the average normalized spatial discrepancy among: NSDab, 10 ab initio envelopes; NSDRB, 10 BUNCH models for PTB U2AF65R12 and dU2AF65R12, 15 BUNCH models for wtU2AF65R12; NSDSUP, between the average ab initio envelope and the most typical BUNCH model superimposed using SUPCOMB.
| Sample | RgG | RgP | Dmax | χ2ab | NSDab | χ2RB | NSDRB | NSDSUP | χ2dU2AF | χ2FIR |
|---|---|---|---|---|---|---|---|---|---|---|
| Å | Å | Å | ||||||||
| wtU2AF65R12 | 24.6 ± 0.2 | 24.5 ± 0.04 | 79 ± 5 | 1.99 | 0.85 | 1.84 | 0.82 | 0.82 | 13.00 | |
| ptbU2AF65R12 | 25.1 ± 0.3 | 25.6 ± 0.05 | 83 ± 5 | 1.74 | 0.84 | 2.10 | 0.79 | 0.90 | ||
| dU2AF65R12 | 20.7 ± 0.2 | 20.5 ± 0.02 | 63 ± 5 | 1.33 | 0.79 | 2.14 | 0.81 | 0.84 | 2.74 |
Molecular Modeling—GASBOR (32) was used for ab initio modeling with the default settings, assuming a starting particle with no symmetry and of unknown shape. The program BUNCH (33) combined rigid body modeling of the known RRM1 and RRM2 structures from PDB 2G4B with ab initio modeling of the inter-RRM linker regions (corresponding to residues 230-257 of human U2AF65). For each modeling method, 10 independent models were aligned and averaged to determine common structural features using SUPCOMB (34) and DAMAVER (35), with the exception of the wtU2AF65R12 BUNCH data, for which 15 independent models were analyzed. Each set of models agreed well with one another, as indicated by normalized spatial discrepancies (NSD < 1.0), as detailed in Table 1.
Fluorescence Anisotropy—The apparent equilibrium
dissociation constants (KD) for association of the
wild-type and variant U2AF65R12 domains with U20 RNA
were determined using fluorescence anisotropy. Anisotropy changes were
measured after the addition of U2AF65 protein to a solution of 30
nm 5′-fluorescein-labeled U20 in 100 mm
NaCl, 25 mm Hepes, pH 6.8. The average KD and
standard deviation of more than two independent experiments are indicated on
representative fitted curves in Fig.
3. Data were fit by non-linear regression assuming single site
binding to obtain the apparent KD using the following
equation, where x is the total protein concentration, [RNA] is the
total RNA concentration, r is the observed anisotropy at the
ith titration, rB is the anisotropy at
zero protein concentration, and rF is the anisotropy at
saturating protein concentration (floated in
fit).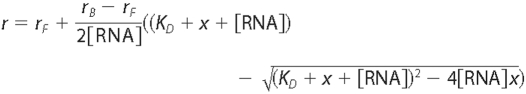
FIGURE 3.

Representative fluorescence anisotropy curves for binding of U2AF65 variants to the polyuridine fluorescein-U20 RNA. Solid lines correspond to the fit of the data using the model described under “Results” with the program Prism4 (Graphpad, Inc.). The average KD and standard deviation of two experiments are given. A, wtU2AF65R12; B, wtU2AF65R123; C, ptbU2AF65R12; D, dU2AF65R12. a.u., arbitrary units.
Isothermal Titration Calorimetry—The heats generated by the addition of U20 RNA to U2A)F65R123 variants were measured at 30 °C using a VP-ITC calorimeter (MicroCal, LLC). Proteins were dialyzed extensively against buffer containing 25 mm Hepes, pH 7.4, 100 mm NaCl, and 0.2 mm tris(2-carboxyethyl)phosphine, and concentrated RNA stock solutions were diluted >40-fold into this dialysis buffer. Samples were filtered and degassed before loading the calorimeter. RNAs at 200-250 μm concentrations were titrated into 1.4 ml of 12-17 μm U2AF65 fragments over 28 injections of 10 μl each, with constant stirring at 307 rpm and 2-min injection spacings. Data were corrected for dilution and buffer effects by subtracting the average of 3-5 terminal injection points from the saturated tail of the binding curve. A control experiment titrating U20 RNA into buffer showed that the heats of U20 dilution were insignificant. Data were analyzed using the least-squares fitting routines available in the Origin v7.0 software (MicroCal, LLC). Values shown in Fig. 4D and Table 2 are the averages of two experiments.
FIGURE 4.

Thermodynamic comparison of polyuridine 20-mer RNA binding by U2AF65 variants. A-C, representative isotherms for U20 titration into wtU2AF65R123 (A), ptbU2AF65R123 (B), and dU2AF65R123 (C). Apparent dissociation constants (KD) for a single binding site are given. D, comparison of the magnitudes of the free energy, enthalpy, and entropy changes for U20 RNA binding determined using ITC.
TABLE 2.
Thermodynamic parameters derived from isothermal titration calorimetry experiments
Averages of two experimental titrations for each protein are given with standard deviations. ΔG calculated using ΔG° = −RT In (KD −1). TΔS° calculated using ΔG° = ΔH° - TΔS°, where T = 303 K.
| Protein/U20 | KD | ΔG° | ΔH° | −TΔS° |
|---|---|---|---|---|
| nm | kcal mol−1 | kcal mol−1 | kcal mol−1 | |
| wtU2AF65R123 | 820 ± 9 | −8.45 ± 0.02 | −69.5 ± 0.3 | 61.0 ± 0.3 |
| ptbU2AF65R123 | 975 ± 45 | −8.34 ± 0.02 | −52.8 ± 5.0 | 44.4 ± 5.0 |
| dU2AF65R123 | 697 ± 128 | −8.55 ± 0.11 | −67.6 ± 6.1 | 59.0 ± 6.1 |
RESULTS
Overall Shape of U2AF65 RNA Binding Domain—A major goal of this investigation was to determine the solution conformation of the Py tract recognition domain of U2AF65 using SAXS. The solution x-ray scattering profile of the wtU2AF65 domain composed of RRM1 and RRM2 (R12, residues 148-336) is shown in Fig. 2A. The average dimensions (radius of gyration, Rg) and maximum size (Dmax) of the wtU2AF65R12 molecule were 24.5 and 79 Å, respectively, as estimated from the paired-distance distribution function (Fig. 2B and Table 1). Similar values of Rg were obtained from the Guinier plot. The Dmax of wtU2AF65 R12 is ∼26 Å less than that of the analogous R12 domain of PTB (Rg = 29 Å, Dmax = 105 Å (25)). In light of the 23-residue greater length of the PTB inter-R12 linkers, the solution dimensions of wtU2AF65R12 are consistent with comparable or weaker interactions between the U2AF65 RRM1 and RRM2 domains as compared with those of PTB.
The Dmax from the paired-distance distribution function of wtU2AF65R12 was used as a starting point for ab initio shape restorations using the program GASBOR (32), which represents the protein structure as a chain-like ensemble of dummy residues. Ten independent restorations gave reproducible results (mean NSD 0.85). The average most populated envelope demonstrates a distinctly bilobal shape consistent with two loosely associated RRMs (Fig. 2D). Next, the rigid body modeling program BUNCH (33) was used to position the high resolution structures of the separated RRM1 (residues 148-228) and RRM2 (residues 260-334) from PDB ID 2G4B and to connect these independent domains via ab initio modeling of the inter-RRM linker region (residues 229-259). Fifteen independent BUNCH models are compatible with the overall shape of the reconstructions from GASBOR, as reflected by the overlay shown in Fig. 2D (NSD = 0.82 between the typical BUNCH and mean GASBOR models shown, calculated using the program SUPCOMB (34)). Like the bilobal ab initio models, the rigid body arrangements of the RRM1 and RRM2 domains remain beyond distances compatible with direct contacts between the domains (>14 Å closest separation). To further illustrate this point, the wtU2AF65R12 solution data are a better match for the relatively extended RRMs of the crystal structure of a deletion mutant (d)U2AF65 (PDB 2G4B) (20) that lacks a portion of the inter-RRM linker (Rg 22.7 Å, discrepancy value χ2 = 2.1) than for the compact fold of FIR R12 (PDB 2QFJ) (23) (Rg 18.9 Å, χ2 = 13.0) (Fig. 2E). Thus, SAXS data are inconsistent with a closely packed arrangement for U2AF65 RRM1 and RRM2 in the context of the core R12 domain studied here.
Affinity of Polyuridine Tract Binding—As a prelude to rigorously investigating the enthalpy and entropy changes responsible for Py tract recognition by U2AF65, the apparent equilibrium dissociation constants (KD) were determined using fluorescence anisotropy assays. The interactions with an RNA site composed of 20 uridines (U20) were studied for several reasons. (i) A homogeneous sequence avoided complicated isotherms due to sites with different affinities; (ii) the in vitro selected site of U2AF65 contains 20 tandem uridines (36, 37); and (iii) uridine is the nucleotide most frequently observed in natural Py tracts (9). Anisotropy changes were monitored as fluorescein-labeled fluorescein-U20 solutions were titrated with U2AF65 proteins (Fig. 3).
The domain necessary and sufficient for Py tract binding, the wtU2AF65R12-bound fluorescein-U20, revealed an apparent affinity (KD 2.35 ± 0.30 μm) comparable with that previously measured for an immobilized biotin-labeled U20 RNA using surface plasmon resonance (KD 3.4 ± 1.8 μm) (20). This value was compared for a larger construct including all three RRM-like motifs (RRM1-RRM2-UHM, R123). Despite the absence of detectable RNA cross-linking to the U2AF65UHM or chemical shift changes in the presence of RNA (12, 38, 39), the wtU2AF65R123 bound U20 with ∼7-fold higher affinity (KD 0.35 ± 0.07 μm) than the minimal R12 construct, perhaps due to RNA interactions by residues following RRM2. Because greater amounts of material are required for studying lower affinity interactions by calorimetry, the R123 variants were used for thermodynamic characterization.
Thermodynamic Characteristics of Polyuridine Tract Binding—Isothermal titration calorimetry (ITC) was used to fully analyze the thermodynamic basis for U2AF65 interactions with a representative Py tract (Fig. 4 and Table 2). For the wild-type wtU2AF65R123 protein, the U20 binding enthalpy (ΔH°) of -69 ± 0.3 kcal mol-1 was nearly offset by a corresponding change in binding entropy (-TΔS°) of 61 ± 0.3 kcal mol-1, demonstrating that recognition of the uridine tract is enthalpically driven. The enthalpy-entropy compensation is unusually large as compared with the typical thermodynamic signatures for protein-protein or protein-double-stranded DNA interactions (for example, ΔH° = -10 kcal mol-1 and -TΔS° = 6 kcal mol-1 for U2AF65UHM binding to an SF3b155 fragment (40)). The one other available example of ITC characterization of single-stranded RNA binding (by the bacterial RNA chaperone, Hfq) also demonstrates a large enthalpy-entropy compensation (for example, ΔH° = -41 kcal mol-1 and -TΔS° = 30 kcal mol-1 for Hfq binding an 18-mer polyadenosine) (41). Although further experiments are needed to test the generality and source of such effects, these results serve as a preliminary indication that remarkably large enthalpy and entropy changes may be a general characteristic of single-stranded RNA recognition.
Design of Interdomain Linker Variants—The elongated bilobal shape of the U2AF65R12 fragment implied that the relative RRM1 and RRM2 arrangement lacked significant interdomain constraints, consistent with the previous observation that both U2AF65 RRM1 and RRM2 slide with overlapping cross-linking patterns across Py tracts (12). The U2AF65R12 pairwise distance distribution function was qualitatively similar to that of the corresponding PTB R12 domain (25). The independent action of PTB RRM1 and RRM2 has been established by the absence of interdomain nuclear Overhauser effects, rotational correlation times, and elongated average shape determined using SAXS (22, 24, 25). Thus, to further test to the possibility that the U2AF65 RRM1 and RRM2 act independently of a tightly packed, intramolecular conformation, we tested the ability of the PTB RRM1-RRM2 interdomain linker to functionally substitute for that of U2AF65.
A hybrid ptbU2AF65 construct replaced 20 residues (residues 238-257) of the U2AF65 inter-RRM linker with sequences from the PTB linker between RRM1 and RRM2 (Fig. 1A). Given that this RRM1-RRM2 linker of PTB has greater length than that of U2AF65, the central region of the PTB linker (residues 148-167 of human isoform a) was chosen for substitution. The low sequence identity of these PTB and U2AF65 regions (two identical out of 20 residues) allows the majority of the region to be replaced with unrelated amino acids. Importantly, these PTB residues lack detectable contacts with the flanking RRMs or RNA in the context of their native protein (22).
Separately, we constructed a deletion variant of these residues in the U2AF65 inter-RRM linker region (Fig. 1A). These residues were absent from the atomic resolution dU2AF65R12 structure because the wtU2AF65 RNA binding domain eluded co-crystallization in the absence of the internal linker deletion (21). Here, we expanded previous work demonstrating that these residues were dispensable for polyuridine binding affinity and in vitro splicing of the AdML substrate (20) by further analyzing their contribution to the molecular shape of the domain and thermodynamic forces underlying Py tract recognition by ITC. The possible effects of these linker modifications on the RNA binding characteristics and nanostructures of U2AF65 are described below.
PtbU2AF65 Linker Variant Supports Pre-mRNA Splicing—The ability of the PTB linker sequences to function in place of the natural U2AF65 sequences was investigated by pre-mRNA splicing assays with the ptbU2AF65 variant (Fig. 1C). Deletion of the corresponding inter-RRM linker region in dU2AF65 had been shown previously to lack detectable effects on in vitro pre-mRNA splicing assays with the prototypical AdML pre-mRNA substrate (20). In an analogous experiment, the ability of ptbU2AF65 to restore splicing activity was tested by the addition of the variant protein to nuclear extracts depleted of wtU2AF65. The recombinant ptbU2AF65 restored pre-mRNA splicing to levels indistinguishable from wtU2AF65 when similar amounts were added to the splicing reaction (Fig. 1C). Thus, the sequence composition of the PTB RRM1-RRM2 linker supports the fundamental ability of U2AF65 to promote splicing of an optimal pre-mRNA substrate.
Comparison of Apparent Polyuridine Tract Affinities—Fluorescence anisotropy assays were used to compare the RNA affinities of the ptbU2AF65R12 and dU2AF65R12 variants with wtU2AF65R12 (Fig. 3, C and D). All three R12 proteins bound the U20 RNA binding site with comparable affinities. Given that sequences within the PTB RRM1-RRM2 linker lack detectable interactions with RNA in the context of the native protein (22, 24), this result suggests that residues 238-257 of U2AF65 likewise do not substantially contribute to Py tract affinity.
Thermodynamic Characteristics of RNA Binding are Comparable for Wt-, Ptb-, and dU2AF65R123 Variants—ITC characterization allowed the detailed thermodynamic similarities or differences to be compared among the inter-RRM variants of U2AF65. Representative isotherms for the titration of U20 RNA into the wtU2AF65R123, ptbU2AF65R123, or dU2AF65R123 are shown in Fig. 4, A-C. Consistent with the results of fluorescence anisotropy assays, the free energy changes for uridine tract binding by all three variants are the same within error. The enthalpy and entropy changes are also very similar (Fig. 4D and Table 2), with the qualification that ∼20% decreases in their magnitudes are conferred by the PTB-linker substitution. Overall, residues 238-257 of the inter-RRM linker do not contribute significantly to the thermodynamic basis for polyuridine binding by U2AF65.
U2AF65 Variants Exhibit Bilobal Shapes—To determine how substitution with PTB sequences or reduction in length influences the overall arrangements of the U2AF65 RRMs, the dU2AF65R12 and ptbU2AF65R12 variants were characterized using SAXS (Fig. 2 and Table 1). An experimental distance distribution plot of the dU2AF65R12 variant as compared with the calculated profile of the high resolution, RNA-bound dU2AF65R12 coordinates (PDB 2G4B) (χ2 = 2.75) shows that the Rg and Dmax dimensions decreased by 2 and 6 Å, respectively, in solution. These differences reflect a somewhat more collapsed average conformation in solution than in the RNA-bound crystal structure of the deletion variant, although in both cases, a bilobal shape is observed. As compared with the unmodified wtU2AF65R12 protein, the dU2AF65R12 variant demonstrates a decreased average size Rg (4 Å difference) and maximum length Dmax (16-20 Å difference), consistent with the 20-residue deletion within the dU2AF65R12 interdomain linker. Substitution of the U2AF65 linker residues with the PTB sequences in ptbU2AF65R12 results in ∼5% increases in Rg and Dmax, which could reflect partial structural differences between the PTB and the U2AF65 linker regions and/or a greater proclivity for the natural linker to associate with its cognate RRMs. Both U2AF65 variants display extended, bilobal ab initio molecular envelopes qualitatively similar to wtU2AF65R12, without apparent interdomain contacts between the rigid body models of the RRM1 and RRM2 coordinates (Fig. 2D). These qualitatively similar shapes indicate that residues (238-257) are unlikely to directly determine the relative RRM arrangement of U2AF65.
The respective low resolution shape restorations of the U2AF65 variants are consistent with bilobal mass distributions separated by a flexible linker (Fig. 2B). This observation is also supported by P(r) functions, which display bimodal distributions. However, the low resolution shapes of U2AF65 variants are unable to elucidate the structural elements within the natural inter-RRM linker. As such, it is worth considering experimental measurements of the average per-residue dimensions for sequences of well characterized structural composition. A short polymer of glycines, the residue with the fewest backbone restrictions, adopts a more extended conformation than an α-helical peptide (Rg of ∼1.5 Å and Dmax of ∼5.7 Å per glycine residue (42) as compared with an average Rg of ∼0.4 Å per α-helical residue (43)). However, chimeric multidomain proteins with α-helical linkers are more elongated than counterparts containing flexible linkers, as reflected by increases in the Rg and Dmax values derived from SAXS analysis (44). This difference is thought to arise from rearrangements of the flexible linker to accommodate molecular attraction between the protein domains, whereas sequences with α-helical propensity confer rigidity. The similar bilobal shapes of wtU2AF65R12, dU2AF65R12, and ptbU2AF65R12 support the view that the U2AF65 RRM1 and RRM2 domains are relatively separated in solution, although additional linker variants would need to be analyzed to fully evaluate the role of interdomain sequences. These observations are relevant to our consideration of weak versus strong Py tract recognition.
DISCUSSION
The overall structures for U2AF65 and its 3′ splice site assemblies are currently unknown, which presents a major obstacle to understanding their critical role during initiation of pre-mRNA splicing. Previously, we determined the detailed interactions of U2AF65 with an optimal polyuridine binding site from the high resolution structure of a variant lacking a portion of the interdomain linker (20). Here, we reveal key features of the intact U2AF65 RNA binding domain and further investigate how modification of the interdomain linker influences these features. First, the intact RNA binding domain of U2AF65 possesses a bilobal shape consistent with the physical separation of the tandem RRMs observed in the crystal structure (PDB 2G4B). Second, unusually large enthalpy and entropy changes serve as the energetic basis for U2AF65 association with an optimal Py tract. Third, a region from the well characterized, flexible inter-RRM linker of PTB is capable of substituting for native U2AF65 sequences to support in vitro splicing, thermodynamic characteristics of uridine tract recognition, and the bilobal shape of the RNA binding domain. Although the exact structural features of the linker region remain to be elucidated, the separation of the U2AF65 RRM1 and RRM2 domains, coupled with the functionally neutral interchange of the PTB and U2AF65 linker sequences, supports a model in which the U2AF65 RRM1 and RRM2 act independently in a manner comparable with the N-terminal RRM1 and RRM2 of PTB, rather than the tightly coupled domain architecture observed for the RRMs of FIR or the C-terminal RRM3 and RRM4 of PTB.
Our structural and biochemical results have important implications for the mode of U2AF65 recognition of the Py tract consensus sequences of the pre-mRNA substrates (Fig. 5). As shown in Fig. 5A, a map of the known interactions with U2AF65 requires a spatial organization of protein domains and RNA sites beyond the simple linearity of the primary sequences. Because the U2AF65 RRMs responsible for identifying the Py tract consensus sequences are centrally located (11, 12), a hinge motion between the RRMs is one means for bringing the N- and C-terminal domains of U2AF65 into proximity. Our SAXS analysis demonstrates that the average organization of the U2AF65 RRMs is relatively straight in the absence of RNA or other factors, illustrating that a bent RRM1-RRM2 conformation is not prearranged. Closer inspection reveals that given the well established topology of the RRMs, there is no need to invoke an acute inter-RRM angle as an explanation for the proximity of the flanking N- and C-terminal U2AF65 domains. The inter-RRM linker by definition connects the C terminus of RRM1 with the N terminus of the RRM2. Concurrently, the topology of the RRM fold constrains the C terminus to protrude adjacent the N terminus of the same domain (Fig. 5B). When connected by a linker of finite length, the relative RRM rotations are constrained so that the U2AF65 sequences directly preceding or following the tandem RRMs are naturally positioned close to one another in three-dimensional space. Accordingly, the N and C termini of the RRM1 and RRM2 domains are oriented toward one another in all rigid body models docked within the U2AF65 molecular envelope. This orientation is an outcome of the simple requirement for the ab initio linker to connect the termini of the two RRMs (Fig. 5B).
FIGURE 5.

Models for Py tract recognition by U2AF65. A, model of initial 3′ splice site complex derived from biochemical experiments and known interacting domains. BPS, branch point sequence; RS, RS domain. B, most typical (lowest NSD) BUNCH model of wtU2AF65R12, colored in a blue-to-red gradient from N terminus (N) to C terminus (C), overlaid with a representation of the solvent-accessible surface. Asterisks mark the junctions of the RRMs with the ab initio linker (green). The N and C termini are restricted to extend from adjacent regions of the modeled polypeptide by the physical limitations of the intermediary linker. C, a model for adaptable recognition of diverse natural Py tracts by adjustment of the U2AF65 inter-RRM configuration.
The Py tracts of multicellular organisms are often marked by interspersed rather than contiguous uridine tracts, in some cases as markers for selective regulation of alternative splice sites (8). For example, in the α-tropomyosin transcript, a continuous, uridine-rich Py tract directs inclusion of a default exon, whereas an alternatively spliced exon is preceded by several short uridine tracts interrupted by guanosines (45). Although the default Py tract is considerably stronger in its ability to direct splicing, the constitutive splicing factor U2AF65 manages to recognize the weaker splice site, albeit with lower affinity (11). Based on our previous high resolution structure, we suggested that U2AF65 could adjust to recognize weak Py tracts such as that found in α-tropomyosin by rearranging flexible side chains or intermediary water molecules. The separation of the U2AF65 RRM1 and RRM2 observed here, coupled with the ability of flexible PTB inter-RRM linker to support U2AF65 activities, opens a new avenue for diverse splice site recognition; a malleable conformation of inter-RRM linker sequences could serve as a potential means for U2AF65 to adapt to diverse Py tract sequences (Fig. 5C). Accordingly, multiple binding registers are observed in cross-linking experiments between U2AF65 and Py tracts of different lengths and sequences (12). We have shown that linker residues 238-257 are dispensable for U2AF65 to recognize polyuridine sequences and to promote splicing of an optimal substrate marked by eight consecutive uridines. Nevertheless, a larger number of constructs needs to be examined to unambiguously relate U2AF65 functions to its inter-RRM sequence composition and length. In particular, shortening of this linker may interfere with the ability of U2AF65 to recognize divergent splice sites by limiting the ability of the inter-RRM register to adjust. This potential means for U2AF65 action, along with the conformation of the overall U2AF65-splice site complex, are thus highlighted as significant areas for future investigation as we progress toward a more sophisticated three-dimensional view of pre-mRNA splice site identification.
Acknowledgments
We thank K. E. Frato and Dr. S. R. Paranawithana for construction of the PTB-linker variants, Dr. D. Braddock for sharing FIR coordinates prior to publication, Dr. J. E. Wedekind for sharing experiences with SAXS data analysis and for comments on the manuscript, and Dr. G. L. Hura for indispensable training and guidance with SAXS data collection and for constructive criticism of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM070503 (to C. L. K.) and R01 GM035490 (to M. R. G.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: Py, polypyrimidine; PTB, Py tract-binding protein; U2AF, U2 auxiliary factor; UHM, U2AF homology motif; RRM, RNA recognition motif; RS, arginine-serine-rich; wt, wild-type; d, deletion; FIR, FUSE-interacting repressor; FUSE, far upstream element; AdML, adenovirus major late; SAXS, small angle X-ray scattering; PDB, Protein Data Bank; ITC, isothermal titration calorimetry; NSD, normalized spatial discrepancies.
References
- 1.Stamm, S., Ben-Ari, S., Rafalska, I., Tang, Y., Zhang, Z., Toiber, D., Thanaraj, T. A., and Soreq, H. (2005) Gene (Amst.) 344 1-20 [DOI] [PubMed] [Google Scholar]
- 2.Maniatis, T., and Tasic, B. (2002) Nature 418 236-243 [DOI] [PubMed] [Google Scholar]
- 3.Lopez-Bigas, N., Audit, B., Ouzounis, C., Parra, G., and Guigo, R. (2005) FEBS Lett. 579 1900-1903 [DOI] [PubMed] [Google Scholar]
- 4.Liu, H. X., Cartegni, L., Zhang, M. Q., and Krainer, A. R. (2001) Nat. Genet. 27 55-58 [DOI] [PubMed] [Google Scholar]
- 5.Garcia-Blanco, M. A., Baraniak, A. P., and Lasda, E. L. (2004) Nat. Biotechnol. 22 535-546 [DOI] [PubMed] [Google Scholar]
- 6.Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., Funke, R., Gage, D., Harris, K., Heaford, A., Howland, J., Kann, L., Lehoczky, J., LeVine, R., et al. (2001) Nature 409 860-921 [DOI] [PubMed] [Google Scholar]
- 7.Sun, H., and Chasin, L. A. (2000) Mol. Cell. Biol. 20 6414-6425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itoh, H., Washio, T., and Tomita, M. (2004) RNA (Cold Spring Harbor) 10 1005-1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senapathy, P., Shapiro, M. B., and Harris, N. L. (1990) Methods Enzymol. 183 252-278 [DOI] [PubMed] [Google Scholar]
- 10.Ruskin, B., Zamore, P. D., and Green, M. R. (1988) Cell 52 207-219 [DOI] [PubMed] [Google Scholar]
- 11.Zamore, P. D., Patton, J. G., and Green, M. R. (1992) Nature 355 609-614 [DOI] [PubMed] [Google Scholar]
- 12.Banerjee, H., Rahn, A., Davis, W., and Singh, R. (2003) RNA (Cold Spring Harbor) 9 88-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fleckner, J., Zhang, M., Valcarcel, J., and Green, M. R. (1997) Genes Dev. 11 1864-1872 [DOI] [PubMed] [Google Scholar]
- 14.Rain, J. C., Rafi, Z., Rhani, Z., Legrain, P., and Kramer, A. (1998) RNA (Cold Spring Harbor) 4 551-565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gozani, O., Potashkin, J., and Reed, R. (1998) Mol. Cell. Biol. 18 4752-4760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kielkopf, C. L., Rodionova, N. A., Green, M. R., and Burley, S. K. (2001) Cell 106 595-605 [DOI] [PubMed] [Google Scholar]
- 17.Wu, S., Romfo, C. M., Nilsen, T. W., and Green, M. R. (1999) Nature 402 832-835 [DOI] [PubMed] [Google Scholar]
- 18.Valcarcel, J., Gaur, R. K., Singh, R., and Green, M. R. (1996) Science 273 1706-1709 [DOI] [PubMed] [Google Scholar]
- 19.Kent, O. A., Reayi, A., Foong, L., Chilibeck, K. A., and MacMillan, A. M. (2003) J. Biol. Chem. 278 50572-50577 [DOI] [PubMed] [Google Scholar]
- 20.Sickmier, E. A., Frato, K. E., Shen, H., Paranawithana, S. R., Green, M. R., and Kielkopf, C. L. (2006) Mol. Cell 23 49-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sickmier, E. A., Frato, K. E., and Kielkopf, C. L. (2006) Acta Crystallogr. F Struct. Biol. Crystalliz. Comm. 62 457-459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oberstrass, F. C., Auweter, S. D., Erat, M., Hargous, Y., Henning, A., Wenter, P., Reymond, L., Amir-Ahmady, B., Pitsch, S., Black, D. L., and Allain, F. H. (2005) Science 309 2054-2057 [DOI] [PubMed] [Google Scholar]
- 23.Crichlow, G. V., Zhou, H., Hsiao, H. H., Frederick, K. B., Debrosse, M., Yang, Y., Folta-Stogniew, E. J., Chung, H. J., Fan, C., De la Cruz, E. M., Levens, D., Lolis, E., and Braddock, D. (2008) EMBO J. 27 277-289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simpson, P. J., Monie, T. P., Szendröi, A., Davydova, N., Tyzack, J. K., Conte, M. R., Read, C. M., Cary, P. D., Svergun, D. I., Konarev, P. V., Curry, S., and Matthews, S. (2004) Structure (Camb.) 12 1631-1643 [DOI] [PubMed] [Google Scholar]
- 25.Petoukhov, M. V., Monie, T. P., Allain, F. H., Matthews, S., Curry, S., and Svergun, D. I. (2006) Structure (Camb.) 14 1021-1027 [DOI] [PubMed] [Google Scholar]
- 26.Cavaluzzi, M. J., and Borer, P. N. (2004) Nucleic Acids Res. 32 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kan, J. L., and Green, M. R. (1999) Genes Dev. 13 462-471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia-Blanco, M. A., Jamison, S. F., and Sharp, P. A. (1989) Genes Dev. 3 1874-1886 [DOI] [PubMed] [Google Scholar]
- 29.Guinier, A., and Fournet, G. (1955) Small-angle Scattering of X-rays, pp. 167-170, Wiley Interscience, New York
- 30.Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., and Svergun, D. I. (2003) J. Appl. Crystallogr. 36 1277-1282 [Google Scholar]
- 31.Svergun, D. I. (1992) J. Appl. Crystallogr. 25 495-503 [Google Scholar]
- 32.Svergun, D. I., Petoukhov, M. V., and Koch, M. H. (2001) Biophys. J. 80 2946-2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petoukhov, M. V., and Svergun, D. I. (2005) Biophys. J. 89 1237-1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kozin, M. B., and Svergun, D. I. (2000) J. Appl. Crystallogr. 34 33-41 [Google Scholar]
- 35.Volkov, V., and Svergun, D. I. (2003) J. Appl. Crystallogr. 36 860-864 [Google Scholar]
- 36.Singh, R., Banerjee, H., and Green, M. R. (2000) RNA (Cold Spring Harbor) 6 901-911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh, R., Valcarcel, J., and Green, M. R. (1995) Science 268 1173-1176 [DOI] [PubMed] [Google Scholar]
- 38.Selenko, P., Gregorovic, G., Sprangers, R., Stier, G., Rhani, Z., Krämer, A., and Sattler, M. (2003) Mol. Cell 11 965-976 [DOI] [PubMed] [Google Scholar]
- 39.Banerjee, H., Rahn, A., Gawande, B., Guth, S., Valcarcel, J., and Singh, R. (2004) RNA (Cold Spring Harbor) 10 240-253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thickman, K. R., Swenson, M. C., Kabogo, J. M., Gryczynski, Z., and Kielkopf, C. L. (2006) J. Mol. Biol. 356 664-683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mikulecky, P. J., Kaw, M. K., Brescia, C. C., Takach, J. C., Sledjeski, D. D., and Feig, A. L. (2004) Nat. Struct. Mol. Biol. 11 1206-1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohnishi, S., Kamikubo, H., Onitsuka, M., Kataoka, M., and Shortle, D. (2006) J. Am. Chem. Soc. 128 16338-16344 [DOI] [PubMed] [Google Scholar]
- 43.Zagrovic, B., Jayachandran, G., Millett, I. S., Doniach, S., and Pande, V. S. (2005) J. Mol. Biol. 353 232-241 [DOI] [PubMed] [Google Scholar]
- 44.Arai, R., Wriggers, W., Nishikawa, Y., Nagamune, T., and Fujisawa, T. (2004) Proteins 57 829-838 [DOI] [PubMed] [Google Scholar]
- 45.Mullen, M. P., Smith, C. W., Patton, J. G., and Nadal-Ginard, B. (1991) Genes Dev. 5 642-655 [DOI] [PubMed] [Google Scholar]
- 46.Svergun, D. I., Barberato, C., and Koch, M. H. J. (1995) J. Appl. Crystallogr. 28 768-773 [Google Scholar]