

Abstract
HNF4α (hepatocyte nuclear factor 4α) plays an essential role in the development and function of vertebrate organs, including hepatocytes and pancreatic β-cells by regulating expression of multiple genes involved in organ development, nutrient transport, and diverse metabolic pathways. As such, HNF4α is a culprit gene product for a monogenic and dominantly inherited form of diabetes, known as maturity onset diabetes of the young (MODY). As a unique member of the nuclear receptor superfamily, HNF4α recognizes target genes containing two hexanucleotide direct repeat DNA-response elements separated by one base pair (DR1) by exclusively forming a cooperative homodimer. We describe here the 2.0 Å crystal structure of human HNF4α DNA binding domain in complex with a high affinity promoter element of another MODY gene, HNF1α, which reveals the molecular basis of unique target gene selection/recognition, DNA binding cooperativity, and dysfunction caused by diabetes-causing mutations. The predicted effects of MODY mutations have been tested by a set of biochemical and functional studies, which show that, in contrast to other MODY gene products, the subtle disruption of HNF4α molecular function can cause significant effects in afflicted MODY patients.
HNF4α is a novel member (NR2A1) of the nuclear receptor (NR)2 family (1) and plays a crucial role in the development and function of vital organs. For example, HNF4α is essential for development of the liver (2), colon (3), and pancreas (4), and deletion of the HNF4α gene from the mouse genome results in embryonic lethality due to the failure to undergo normal gastrulation (5, 6). Conditional targeted gene disruption of HFN4α results in marked metabolic disregulation and increased mortality (7-9). In addition, HNF4α is known to activate a wide variety of genes involved in glucose, fatty acid, cholesterol, and amino acid metabolism in the liver, kidney, intestine, and pancreas (10-12), including counterpart transcription factors, such as HNF1α (13), HNF6 (14), and pregnane X receptor (15), that in turn control additional liver and β-cell-specific target genes. Further underscoring the importance of HNF4α in pancreatic β-cells, mutations on HNF4α in humans are directly linked to the onset of MODY1 (maturity onset diabetes of the young 1) (Fig. 1A), one of the most common monogenic causes of diabetes, mainly characterized by impairment of glucose-stimulated insulin secretion from the β-cells (16). A direct link between HNF4α and glucose-stimulated insulin secretion by pancreatic β-cells has been proven by cellular studies (17, 18) and pancreatic β-cell-specific HNF4α knock-out mice (17).
FIGURE 1.

Schematic diagrams of HNF4α and the sequence alignment with related NRs. A, HNF4α domain structure and MODY1 point mutations. B, secondary structures and sequence alignment of HNF4α-DBD with the related proteins. The related human NRs were extracted from Protein Data Bank entries whose DNA complex structures are available. Their Protein Data Bank accession codes and the starting residue numbers are shown on the left in parenthesis, and the oligomeric state of their DNA complexes and the arrangement of two hexanucleotide half-sites are indicated on the right. Residues that are identical or similar among the family members are shaded in blue and yellow, respectively. Hydrophobic core residues are indicated by green circles, and MODY1 point mutation residues are indicated by red stars above the sequences. Two post-translational modification sites (phosphorylation on Ser78 and methylation on Arg91) are also indicated. EcR, ecdsyone receptor; VDR, vitamin D receptor; TR, thyroid hormone receptor; LRH1, liver receptor homologue-1; ER, estrogen receptor; ERR2, estrogen-related receptor 2; REV-ERB, orphan nuclear receptor Rev-Erb; PR, progesterone receptor; DR1, direct repeat motif spaced by one nucleotide; IR1, inverted repeat motif spaced by one nucleotide, etc.
A recent genome-wide expression profiling study revealed that both HNF1α and HNF4α are the master regulators of the β-cells directly affecting their physiology (19, 20). HNF4α is functionally very closely related to HNF1α, since they cross-regulate each other and form a common network of transcription factors that controls the development and function of hepatocyte and pancreatic islets (21, 22). As a result, the diabetic phenotype in MODY1 due to HNF4α mutations is virtually indistinguishable from that due to HNF1α mutations (23). There is even an additional MODY mutation within the HNF4α recognition site of human HNF1α promoter (13), underscoring the interplay between HNF1α and HNF4α.
HNF4α belongs to the NR superfamily, and a typical NR comprises the all-cysteine zinc finger DNA binding domain (DBD), lipophilic ligand binding domain (LBD), and additional domains with activation function (24) (Fig. 1A). HNF4α displays a novel mode of ligand-dependent (or -independent) transactivation (11) and still remains as an orphan NR although recent crystal structures of HNF4α LBD alone (25) and in complex with the co-activator SRC-1 peptide (26) revealed fatty acids as structural ligands for HNF4α. These findings have been contested by the earlier findings (27) and the subsequent recent studies (28-31) that suggest that the true physiological ligands for HNF4α are fatty acyl-CoAs and that binding of these exchangeable ligands requires a longer construct that contains, in addition to the LBD, the regulatory F-domain toward the C-terminal end (Fig. 1A). These discrepancies await further investigations.
The DBD is the most highly conserved domain shared by NRs and consists of two all-cysteine zinc finger motifs. The crystal structures of several NR-DBDs revealed that the protein structures formed by zinc fingers facilitate sequence specific interactions with the major groove of the DNA double helix containing its response elements (32). These response elements contain hexameric sequences that can be arranged in various configurations (direct or inverted orientation). Orphan NRs can bind either one hexanucleotide (half-site or subrecognition site) or two direct hexanucleotides spaced by 1-5 nucleotides in between (DR1-DR5) with high selectivity (1). HNF4α predominantly recognizes target genes, including HNF1α, that contain the DR1 recognition sites although it can also bind a nonnatural DR2 with a much weaker affinity (33).
The NR-DBD also contains the C-terminal extension that facilitates dimerization through the T-box element and, in some cases, additional half-site recognition by the C-terminal A-helix (Fig. 1B). When the dimeric recognition occurs, the target DNA leads to the formation of a highly specific dimer interface, which places the DBDs in register with the half-sites of their respective response elements. Many of the nonsteroid hormone NRs form heterodimers with retinoid x receptor (RXR). However, HNF4α represents a nonsteroid receptor that exclusively functions as a homodimer despite its similar DNA binding specificity and amino acid sequence homologous to that of RXR (33). Although the major determinant for dimerization stability and selectivity appears to reside within the LDB, which spontaneously forms a dimer in solution (25), there is an additional dimerization determinant within the DBD that allows the formation of a dimer only in the presence of DNA and provides the cooperativity and additional selectivity when it binds DNA (33, 34).
To understand the molecular principles underlying the recognition of a naturally occurring DR1 target DNA sequence and the molecular basis of MODY mutations, we have solved the crystal structure of the homodimeric human HNF4α-DBDs in complex with a high affinity DNA containing the HNF1α (MODY3 gene) promoter sequence composed of a tandem direct repeat of half-sites separated by one base pair (DR1) and harboring an additional MODY single nucleotide mutation site. This structure, refined at 2.0 Å, shows the critical features of the protein/DNA and protein/protein interactions. The predicted effects of the MODY mutations were further tested by a set of biochemical and functional studies, and the regulatory post-translational modification sites are discussed.
EXPERIMENTAL PROCEDURES
Construction of Overexpression Vectors for Wild Type and Mutants and Protein Purification—Two different versions of the recombinant proteins were used in our studies: tag-free wild type for crystallization and maltose-binding protein (MBP) fusion proteins for biochemical characterizations of wild type and MODY mutants due to low yields or low solubility of some of the tag-free mutant proteins. However, the same vector constructs were used, either performing or skipping a tobacco etch virus (TEV) protease digestion to remove or retain the MBP tag. The cDNA harboring the full-length human HNF-4αB splice variant (35) was a kind gift from Dr. Steve Shoelson from Joslin Diabetes Center. A modified MBP fusion expression vector, pET41a MBP, was used for our studies. In this modified vector, the MBP fragment of pMAL-c2X (New England Biolabs) was amplified by PCR and cloned into pET41a vector (GE Healthcare) by inserting the fragment between the NdeI and HindIII sites to replace the GST tag, and a thrombin cleavage site was replaced by a TEV protease cleavage site for higher specificity. A fragment of human HNF4α cDNA (amino acids 46-126) was subcloned by standard PCR into pET41a MBP vector to produce the proteins fused with MBP at the N-terminal end with the TEV cleavage site.
For tag-free protein purification, HNF4α-DBD was overexpressed in Escherichia coli BL21-Gold (Novagen) with induction of 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at an A600 of 0.8-1.0 at 37 °C and harvested after culturing for an additional 4-6 h. The cells were lysed by sonication, and the expressed MBP-fusion proteins were isolated in the presence of 0.6 m NaCl to prevent nonspecific binding to bacterial DNA. HNF4α-DBD was released by TEV digestion from amylose magnetic beads (New England Biolabs) after overnight incubation at 4 °C and further purified by ion exchange chromatography (Mono-S FPLC). The purified protein was estimated to be at least 98% pure as judged by staining with Coomassie Brilliant Blue on an 8-25% gradient SDS-polyacrylamide gel (36). Fractions were pooled; concentrations were measured by UV absorption and stored at -80 °C as a 10% (v/v) glycerol stock.
For MBP fusion protein purification for in vitro biochemical studies with wild type and MODY mutants, the TEV digestion step was skipped due to insolubility of the cleavage products (e.g. the G115S mutant heavily precipitated after digestion). Instead, the MBP fusion proteins were directly eluted from the beads with 10 mm maltose in 50 mm Tris-Cl, pH 8.0, and subsequently purified by ion exchange chromatography (Mono-S FPLC). Fractions were pooled and stored at -80 °C as a 10% (v/v) glycerol stock.
Site-directed Mutagenesis Analysis—QuikChange multisite-directed mutagenesis kit (Stratagene) was used to generate constructs with each point mutation of HNF4α-DBD according to the manufacturer's instructions. The plasmid templates used in the mutation were pET41a MBP-HNF4α DBD (amino acids 46-126) and pcDNA3 HNF4α. All of the generated constructs with the mutated sequences were verified with DNA sequencing.
Preparation of DNA Oligonucleotides—Tritylated oligonucleotides were purchased from the Midland Certified Reagent Company (Midland, TX) and further purified by reverse phase HPLC. Excess mobile phase-containing acetonitrile was removed using HiTrapQ (GE Healthcare), and the trityl groups were removed with 80% acetic acid. The deprotected oligonucleotides were precipitated with 75% ethanol, dissolved in water for concentration measurement, and lyophilized before storage at -80 °C. When needed, double-stranded DNAs were generated by heating equimolar amounts of complementary oligonucleotides to 95 °C for 10 min and slowly cooling to 4 °C.
Crystallization and Data Collection—Crystallization of the complex has been reported previously (36). Briefly, protein-DNA complexes were made by dialyzing them with a molar ratio of 2 HNF4α and 1.2 double-stranded DNA in 20 mm Tris, pH 7.5, 75 mm NaCl, and 1 mm dithiothreitol at 4 °C for 2.5 h and concentrated to at least 10 mg ml-1. The crystals were grown at 22 °C using the hanging drop vapor diffusion method with an overhang 21-mer duplex DNA. The optimized crystals were grown with the well solution containing 26% (v/v) polyethylene glycol 4000, 80 mm magnesium acetate, and 50 mm sodium citrate, pH 4.8, and transferred into the mother liquor containing an additional 15% (v/v) glycerol as cryoprotectant before being directly plunged into liquid nitrogen and stored for data collection. The native data were collected at 100 K at APS (SER-CAT 22BM) using a MAR-225 CCD detector and processed using HKL2000 (37).
Structure Determination and Refinement—The crystals belong to the space group C2 with unit cell dimensions a = 121.628 Å, b = 35.425 Å, c = 70.985 Å, and β = 119.364° and diffraction to 2.0 Å resolution. There is one complex in the asymmetric unit (41.5% solvent content). The structure was solved by the molecular replacement method by the use of MOLREP (38). As a search model, we used the previous structure of RXR·RAR·DNA complex (Protein Data Bank accession code 1DSZ) (39). The best solution had a correlation coefficient of 38.5%, 11.4% above the second best solution. The Rcryst value after molecular replacement was 0.514. After one round of rigid body refinement, Rcryst and Rfree dropped to 0.502 and 0.498, respectively. Protein residues were mutated in order to match the sequence of HNF4α, and the model was refined by simulated annealing with CNS (40), initially with tight restraints for tetrahedral zinc coordination and Watson-Crick base pairing and global restraints placed on bond lengths, bond angles, non-bonded contacts, and temperature factors of neighboring atoms. The σA-weighted 2Fo - Fc maps as well as omit maps were calculated at regular intervals to allow manual rebuilding. Tight restraints on the zinc-sulfur coordination and base pairing were released, and solvent water molecules based on higher than 3σ peaks in Fo - Fc σA-weighted maps were added conservatively at appropriate sites. Inclusion of individual atomic temperature factors during the final stages of refinement was accompanied by a substantial decrease in Rfree values (Table 1). Iterative building and refinement were performed using the program O (41) and CNS (40).
TABLE 1.
Data and refinement statistics
| Parameter | Total | Outer shell |
|---|---|---|
| Native diffraction data | ||
| Resolution (Å) | 30.3-2.0 | 2.07-2.00 |
| Rmergea (%) | 6.4 | 30.4 |
| I/σ(I) | 20.7 | 3.8 |
| Redundancy | 5.6 | 3.6 |
| Refinement | ||
| No. of reflections | (20.0-2.0 Å) | |
| Work | 16,841 | |
| Free FLAGb | 811 | |
| Modeled regions | Amino acids 49-124 in molecule A | |
| Amino acids 49-126 in molecule B | ||
| DNA 1-21 in chain C and 1-21 in chain D 4 zinc atoms | ||
| 157 solvent atoms | ||
| R factor (%) | 21.3 | |
| Rfree (%) | 25.1 | |
| <B> for all atoms (Å2) | 29.37 | |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.005 | |
| Bond angles (degrees) | 1.031 |
Rmerge = ∑h ∑i|I(h)i - <I(h)>|/∑h ∑i I(h)i, where I(h) is the intensity of reflection h, ∑h is the sum over all reflections, and ∑i is the sum over i measurements of reflection h.
5% of the reflection data excluded from refinement.
HNF4α/DNA Binding Titration through Electromobility Shift Assay (EMSA)—Single-stranded oligonucleotides 1 and 2 (Fig. 2B) were dissolved in 10 mm Tris (pH 7.5 at 20 °C) and 1 mm EDTA. Oligonucleotide 1 was 5′-end-labeled with 32P as described (42). Labeled oligonucleotide 1 was mixed with a 1.1-fold molar excess of oligonucleotide 2, and the samples were heated to 90 °C and cooled slowly to 20 °C. DNA was transferred by dialysis into binding buffer (10 mm Tris (pH 8.0), 1 mm EDTA, 100 mm NaCl, 1 mm MgCl2, 1 mm dithiothreitol, 4% (v/v) glycerol). DNA concentrations were measured by absorbance, using a molar extinction coefficient (ε1cm260 = 2.57 × 105). Pure recombinant wild type HNF4α-DBD-(46-126) was prepared as described previously (36). Samples were stored at -20 °C until use. EMSAs were carried out as described (43), using 10% (w/v) polyacrylamide gels cast and run in 45 mm Tris-borate, 2.5 mm EDTA (pH 8.3 at 20 °C). Autoradiographic images were captured on storage phosphor screens (type GP; GE Healthcare) detected with a Typhoon PhosphorImager. Band intensities were determined using ImageQuant software (GE Healthcare).
FIGURE 2.

Overall structure of the HNF4α-DBD·DNA complex and schematic diagrams of the protein/DNA interactions. A and B, schematic diagrams of the protein construct sequences (A) and natural response element (B) constructs used in crystallization and biochemical studies. The amino acid and nucleotide sequence numbers are shown in parenthesis at both ends. The HNF1α promoter sequence numbers were extracted from Ref. 13. C and D, schematic diagrams of protein-DNA contacts at the upstream consensus sequence half-site (C) and at the downstream half-site (D). Sequences and interactions (key is provided within the figure) are shown together for each monomer of HNF4α. Sequence-specific protein/DNA interactions and nonspecific DNA backbone interactions are indicated by brown arrows and black arrows, respectively. Water molecules are depicted as blue circles. MODY1 mutation (Arg125) and MODY3 mutation within the HNF4α recognition sequence are shaded in light blue.
Binding affinity (K) and cooperativity (ω) were evaluated
using the McGhee-von Hippel isotherm
(44), as modified by Record
and co-workers (45) to account
for finite lattice size (Equations 1 and
2).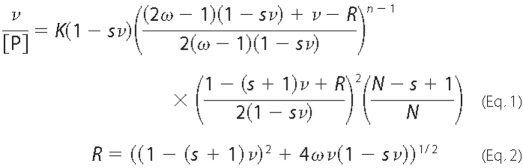
Here ν is the binding density (protein molecules/base pair), K is the equilibrium association constant for binding a single site, ω is the cooperativity parameter, N is the length of the DNA in nucleotides, and s is the size of the site (in base pairs) that a protein molecule occupies to the exclusion of others. Because the total concentration of protein binding sites on DNA was always much less than that of the protein, the approximation [P] = [P]total was used.
Cell Cultures—The HeLa cell line was cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Invitrogen), 50 units/ml penicillin, and 50 μg/ml streptomycin and 0.1 mm nonessential amino acids (Invitrogen). MIN6 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 15% fetal bovine serum (Invitrogen), penicillin and streptomycin, and 10 μm β-mercaptoethanol (Sigma), and MIN6 cells of passages 26-28 were used in this study.
Transient Transfection and Luciferase Assay—The full-length cDNA of human HNF4α wild type or mutant was subcloned into the pcDNA3(+)/Neo vector (Invitrogen), and the reporter vector pGL3-(BA)3 containing three copies of the HNF4α response element (-77 to -65) within the promoter of human apolipoprotein B was constructed by subcloning this promoter fragment from p(BA1)5CAT (kindly provided by Dr. Margarita Hadzopoulou-Cladaras (Aristotle University of Thessaloniki, Greece)) into the firefly luciferase reporter vector pGL3-Basic (Promega). Similarly, the reporter vector pGL3-HNF1α-298 containing one copy of the HNF4α response element (-64 to -52) within the promoter of human HNF1α (-298 to the first AGT) was constructed and used for Fig. 4B. HeLa cells or MIN6 cells were transfected using Opti-MEM and Lipofectamine 2000 reagent (Invitrogen) or Nucleofactor reagent V (Amaxa), respectively, according to the manufacturer's recommendations. Briefly, a total of 50 ng of pcDNA3-HNF4α and 50 ng of pGL3-(BA)3 or pGL3-HNF1α-298 and 10 ng of pRL-TK (control Renilla luciferase vector) were used for transfection of 1 × 105 HeLa cells seeded on 24-well plate 1 day before transfection. For MIN6 cells, a total of 500 ng of pcDNA3-HNF4α and 500 ng of pGL3-(BA)3 and 100 ng of pRL-TK were used for transfection of 2 × 106 MIN6 cells. Forty-eight hours after transfection, cells were washed with 1× phosphate-buffered saline and lysed with luciferase lysis buffer supplied with the Luciferase assay kit (Promega). Luciferase activity was measured using the dual luciferase assay system (Promega) and a luminometer. All values were normalized in a relative ratio of firefly luciferase activity and Renilla luciferase activity. At least three independent transfections were performed in quadruplicate.
FIGURE 4.

Diabetes mutations on HNF4α-DBD and their functional studies. A, mapping of MODY1 mutations on the HNF4α-DBD structure. Side chains of residues affected by MODY point mutations are displayed by a ball-and-stick model. Mutations are found toward the C-terminal end of HNF4α-DBD, including the T-box, and no mutations are found at the main HNF4α/DNA binding interface or the hydrophobic core of HNF4α. B, overall transcriptional activity by the MODY mutants compared with wild type with different cell types or on different promoters. Top, HeLa cells transfected with wild type or mutated pcDNA3-HNF4α and the luciferase reporter construct pGL3-(BA)3; middle, HeLa cells transfected with wild type or mutated pcDNA3-HNF4α and the luciferase reporter construct pGL3-HNF1α-298; bottom, MIN6 cells transfected with wild type or mutated pcDNA3-HNF4α and the luciferase reporter construct pGL3-(BA)3. Standard luciferase-based transcriptional reporter assays were conducted using HeLa cells or MIN6 cells transfected with wild type or mutated pcDNA3-HNF4α and the luciferase reporter construct containing three copies of the HNF4α response element (-77 to -65) within the promoter of human apolipoprotein B (pGL3-(BA)3) or one copy of the HNF4α response element (-64 to -52) within the promoter of human HNF1α (pGL3-HNF1α-298). CTL in the first lane refers to an empty vector, and all data have been normalized against the firefly Renilla luciferase activity. C, EMSA DNA binding assay. The first two lanes (from the left) were used as controls (DNA probe only and nuclear extract from the transfection with the empty vectors), and the third lane confirmed the identity of the binding proteins. Wild type (WT) is shown in lane 4, and the mutants are shown in the subsequent set of lanes (lanes 5-9). The lower bands correspond to free DNA, whereas the upper bands represent the shifted HNF4α·DNA complex or the supershifted HNF4α·antibody·DNA complex. The Western blot (top) compares the amount of wild type and mutated forms of HNF4α in the loaded nuclear extracts. D, lifetime of the HNF4α mutants compared with wild type. Left, raw data. HeLa cells were labeled with [35S]methionine/cysteine for 30 min (0 h) and pulse-chased for various lengths of time (3-18 h) in the presence of excess nonradioactive methionine/cysteine. Samples were immunoprecipitated under normal stringency conditions using polyclonal anti-HNF4α antibody and subjected to SDS-PAGE followed by autoradiography. Right, fraction of initial protein. The percentage of the remaining protein (relative value) is calculated by the relative amount of the protein at a given postchase time to the initial amount at 0 has determined by the intensity of each band using the Image-Quanta software. E and F, overall secondary structure contents and thermal denaturation analysis of the wild type and the G115S mutant. For these experiments, HNF4α-(46-126)-MBP fusion proteins were used. E, far-UV CD spectrum of the G115S mutant measured from 260 and 198 nm along with the wild type and D126H mutants as control. The amounts of secondary structures were calculated by CDSSTR deconvolution software and are given in the inserted table. NRMSD, normalized standard deviation. F, temperature-induced denaturation profile of the wild type and the G115S and D126H mutants. The melting temperatures (TM) were taken as the midpoint of each unfolding transition by fitting to Gaussian curves of the first derivative function.
Protein Stability Assays (Pulse-Chase Experiment)—Stability of the mutants in cultured cells was measured by means of pulse-chase experiments. Twenty-four hours after transfection, HeLa cells were serum-starved for 1 h before being incubated in Dulbecco's modified Eagle's medium minus Met/Cys for 30 min and then labeled with 100 μCi/ml Trans35S labeling mix (MP Biomedicals) for 30 min at 37 °C. Labeling medium was removed thereafter, and the 35S-labeled cells were incubated in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum for the indicated periods of time (0-18 h) and lysed. The nuclear extracts were prepared using the Nuc-Buster protein extraction kit (Novagen). Proteins were immunoprecipitated with anti-HNF4α antibody (Santa Cruz Biotechnology) and Protein A-Sepharose beads (Amersham Biosciences) and resolved by SDS-PAGE, and incorporated radioactivity was analyzed by autoradiography using BioMax film (Eastman Kodak Co.) or storage phosphor screens (type GP; GE Healthcare) for quantification with a Typhoon PhosphorImager equipped with Image-Quanta software (GE Healthcare).
Circular Dichroism—The secondary structure content and the denaturation profile of HNF4α-DBD in solution were determined using far-UV CD. CD spectra were collected on a Jasco J-810 spectropolarimeter using a 1-mm path length quartz cuvette with solutions containing 1-5 μm protein in 30 mm MES buffer, pH 6.0, 30 mm NaCl, 1 mm dithiothreitol, and 1% glycerol. The temperature was controlled via a Peltier block. For the regular scan, spectra were collected at 20 °C with a 0.5 nm resolution and a scan rate of 200 nm min-1. Reported spectra were measured only once. Observed ellipticities were converted to mean residue ellipticity, [θ] = degrees cm2 dmol-1 (molar ellipticity). Reported molar ellipticities are estimated to have errors of ±3%. Secondary structure analysis was performed using the secondary structure estimation algorithm CDSSTR (46), provided by the CDPro software package (47). The quality of the fit between experimental and back-calculated spectrum corresponding to the derived secondary structure element fractions was assessed from the normalized root mean square deviation, with a value of <0.1 considered as a good fit (48).
For thermal denaturation profile acquisition, samples containing 1-5 μm protein were heated from 0 to 100 °C at a rate of 1.0 °C/min, and ellipticity was monitored at 222 nm; melting temperatures (Tm) were taken as the midpoint of each unfolding transition by fitting to Gaussian curves of the first derivative function (49).
Preparation of the Nuclear Extracts and the Subsequent Comparative EMSA Analysis for the Wild Type and the MODY Mutants—The HeLa cells were harvested 10 h after transfection, and nuclear extracts were prepared using the Nucbuster protein extraction kit (Novagen) according to the manufacturer's protocol. The loading amount of HNF4α wild type and mutants were adjusted to an equal amount after quantification by an enzyme-linked immunosorbent assay and visualization by a Western blot.
For EMSA, synthetic 15-base pair oligonucleotides containing the HNF4α response element within the HNF1α promoter were labeled with an infrared dye (IRDye 700 phosphoramidite) (LI-COR Biosciences). EMSAs were performed in a similar way described above on a 6% native polyacrylamide gel using a Tris-glycine-EDTA buffer and analyzed with the Odyssey infrared imaging system (LI-COR Biosciences). Initially, a 21-mer DNA with the 13-mer HNF4α recognition site in the middle (the same DNA construct we used for crystallization and structure determination) was used for EMSA with the nuclear extracts; however, it showed one major extra band throughout the lanes due to the binding of an additional nuclear protein (which later turned out to be a heat shock protein, HSF-90, by mass spectrometry analysis) to the DNA probe. Thus, subsequently, a 15-mer DNA was used, and the binding of an additional nuclear protein was eliminated.
RESULTS AND DISCUSSION
Overall Structure of HNF4α-DBD and Comparison with the Related Nuclear Receptors on Various DNA Recognition Elements—The crystals of HNF4α-(46-126), containing two zinc finger motifs and the C-terminal extension “T-box” in complex with DNA were prepared as previously reported (36), and the structure was determined by conventional methods as described under “Experimental Procedures.” Fig. 2, A and B, shows the protein and DNA constructs used in co-crystallization. HNF4α and the homologous NRs, such as RXR and RAR, do not appear to have an “A-helix” at the C-terminal end (Fig. 1B), which has been shown to make additional contact with DNA through the minor groove (50, 51). The secondary structure predictions for HNF4α, RXR, and RAR do not indicate the presence of an α-helix in this region, whereas they do for TR and VDR (see Fig. S1B for superposition of their crystal structures).
In our studies, we used the HFN4α recognition DNA sequence element from the human HNF1α promoter, another MODY gene (MODY3). The best crystal diffracted to 2.0 Å with the synchrotron x-ray source, the data collection and refinement statistics are provided in Table 1, and the representative electron density map is shown in Fig. S1A. In the asymmetric unit, there was one HNF4α·HNF1α promoter element complex, which consists of two HNF4α monomers in a head-to-tail orientation (Fig. 3A), each monomer with two zinc finger motifs (Fig. 2A). The two monomers bind to a 21-bp duplex with a 1-bp overhang at the end (Fig. 2B), which mediates an A-T Watson-Crick base pair and stacking interactions with the symmetry-related DNA to form a pseudocontinuous DNA helix in the crystal. The geometry of DNA duplex is canonical B-form DNA with 29.2° of overall bending (Fig. 3A and Fig. S2) when calculated with CURVES (52) and with an axis kink of 8° occurring at the C-G pair right after the 3′-end of the downstream half-site (Fig. 2B).
FIGURE 3.

Functional features of the HNF4α-DBD·DNA complex. A, surface and ribbon representation of the HNF4α·DNA complex structure. Two proteins (red, upstream; blue, downstream) are bound to double-stranded DNA, and the hexanucleotide direct repeat DNA-response elements separated by one base pair (DR1) on the HNF1α promoter are shown in green. Protein residues at the interface participating in dimeric interactions are highlighted by a ball-and-stick model. B, DNA binding properties of HNF4α-(46-126) measured by forward titration. Top, titration of the 21-mer duplex (0.45 nm) with protein (0 μm ≤ [P] ≤ 20.7 μm), detected by EMSA. Bottom, Scatchard plot summarizing results of three independent experiments. The smooth curve is a fit of the data with Equations 1 and 2 returning association constant K = 1.36 ± 0.26 × 104 m-1, cooperativity factor ω = 67 ± 14, and stoichiometry 2.04 ± 0.20 protein/DNA. C, stereo view of the detailed contacts of the HNF4α-DBD/DNA interface through the recognition helix (helix I) at the major groove. This figure was made based on the protein/DNA interactions at the downstream half-site. D, hydrophobic core of a monomeric HNF4α-DBD made of two unrelated zinc fingers. The core residues, including a stretch of aromatic residues, are shown as a ball-and-stick model.
Superimposition of the HNF4α-DBD·DNA complex with the previously reported NR-DBD·DNA complexes shows a high degree of structural similarity in the core zinc finger region (Fig. S1B), including the linker between helices I and II, despite their sequence variations in the region (Fig. 1B). The most structurally divergent region of these NR-DBD·DNA complex structures is the C-terminal extension of the zinc finger region where the T-box and A-helix are located. Sequence variations are also extremely high in this region (Fig. 1B), reflecting the necessary elements required for specificity and plasticity in various target recognitions in terms of orientation of two half-sites and the number of base pairs in between. For direct repeat recognition, dimeric interactions are made by the residues in the T-box of the downstream monomer and the residues in the zinc finger II of the upstream monomer (Figs. 2A and 3A), whereas the residues in the zinc finger II from both monomers contribute dimeric interactions for inverted repeat recognition sites (53-55). The beginning of the T-box of the majority in NRs, including HNF4α, forms a single-turn 310-helix, whereas the corresponding segments of some other NRs adopt coiled structures. Some NRs, such as TR and VDR, possess the distinctive A-helix, which makes additional DNA contacts through the minor groove (Fig. S1B), whereas HNF4α is believed to lack this secondary structure element.
Dimeric Protein Interactions and Cooperative DNA Binding—The correct NR dimerization on a target DNA facilitates target selection from a pool of genomic sequences that contain consensus hexameric half-sites and is essential for programmed recruitment of co-activators or co-repressors to the transcription complex (56, 57). Typical orphan NRs can either heterodimerize with RXR or can bind as monomers at the responsive elements to carry out their function (32). HNF4α and RXR share 61% amino acid sequence identity within the DBD and over 35% identity within the LBD (Fig. 1B). However, HNF4α does not form a heterodimer with RXR, and it exclusively binds its target genes as a homodimer independent of putative ligand binding (33). Although the main binding interface for HNF4α homodimeric assembly is provided by the LBD (25, 58, 59), there are additional protein/protein interactions between the two HNF4α-DBD monomers in the presence of DNA that provides a high degree of cooperativity in DNA binding.
In the absence of DNA, the HNF4α-DBD is monomeric in solution; however, quantitative analysis of gel mobility shift assays returns a binding density equivalent to 2.02 ± 0.2 molecules of protein per DNA (Fig. 3B), consistent with the stoichiometric ratio of 2:1 found previously (36). When examined by an EMSA titration experiment, binding is significantly cooperative (ω = 67 ± 14) despite the small dimer interface between the proteins. This interface is formed by the T-box of the monomer associated with the downstream half-site and the zinc finger II region of the monomer associated with the upstream half-site (Fig. 2A), which covers the majority of the minor groove of the spacing base pair (Fig. 3A). These interactions bury 179 Å2 of solvent-accessible surface from both monomers upon binding, which is substantially smaller than other dimeric interactions observed in the crystal structures of related NR-DBD·DNA complexes (39, 54), yet induce considerable cooperativity in DNA binding. There could be additional dimeric interactions made by the residues beyond Asp126 (the last residue in our construct), and it is worth pointing out that another MODY1 point mutation is found at the Arg127 residue (R127W). However, among the various constructs of HNF4α we have tried for crystallization with DNA, only the 46-126 construct produced diffracting quality crystals. The key dimeric interaction in our structure is the salt bridge between the T-box residue Asp126 of the downstream monomer recognizing the downstream half-site and the zinc finger II region residue Arg88 from the upstream monomer (Fig. 3A). The inter-half-site spacing of the direct repeat provides the geometry needed for two subunits to interact effectively at their interface, and the spacing of one base pair appears to be optimal for the salt bridge between Asp126 and Arg88 to be formed. This critical dimeric interaction also ensures the head-to-tail orientation of two monomers in recognition of target genes containing a direct repeat. Thus, these dimeric interactions appear to serve as a molecular “ruler” that helps HNF4α effectively select the intended target genes against the genes containing non-HNF4α NR response elements.
Two of the MODY1 point mutations, D126H and D126Y, are found at this residue, and the carriers of these mutations develop severe diabetes with defective metabolic pathways (60). The significance of this interaction for dimer formation and cooperativity in target gene recognition is underscored by a previous finding in which a similar construct of HNF4α-DBD-(1-125), but critically lacking the Asp126 residue, was unable to form a homodimer upon binding to DNA (33). Another notable dimeric interaction is the hydrogen bond between the Nε of Gln102 from the upstream monomer and the carbonyl oxygen of Glu124 from the downstream monomer. These interactions are similar in nature but distinct from the dimeric interactions found in the RXR·RXR homodimer or the RXR·RAR heterodimer bound to the DR1 elements (39, 61), since there is little structural or sequence similarity in the C-terminal extensions of the DBD, including the T-box (Fig. 1B).
HNF1α Promoter Recognition by HNF4α—The DBD is the most conserved structural and functional element of NRs and consists of two nonequivalent zinc finger motifs, each containing four highly conserved cysteine residues coordinating the binding of a zinc ion. This results in the formation of a tertiary structure containing three main helices (Fig. S1B), among which the N-terminal helix (helix I) directly and specifically interacts with the major groove of each DNA half-site sequence element, whereas the C-terminal helix (helix III) overlays the N-terminal helix in a perpendicular fashion and contributes to stabilization of the overall protein structure by forming the base of a hydrophobic core. The hydrophobic core of HNF4α is made of a stretch of aromatic groups, such as Tyr63, Phe74, Phe75, Tyr85, and Phe112, which are either strictly or highly conserved throughout the members of the NR superfamily (Figs. 1B and 3D). Two other residues, Val79 and Leu108, complete the core structure. The resulting extensive hydrophobic core provides the integrity of the HNF4α-DBD and fixes the relative orientation of the two helical substructures for optimal DNA interactions.
Most nonsteroid NRs recognize one or two of the consensus hexameric sequence 5′-AGGTCA-3′ or its slightly degenerate variations (32). HNF4α recognizes target genes with two direct repeats, the DR1 element, as a homodimer (33). The synthetic double-stranded DNA used in our study corresponds to the HNF4α recognition site within the HNF1α promoter with optimal binding (13, 62, 63) and contains the upstream “AGTTCA” and downstream “AGTCCA” half-sites (Fig. 2B) as well as natural flanking sequences. This crystal structure of the complex allowed us to elucidate high resolution atomic details of intimate protein/DNA interactions at 2.0 Å resolution (Fig. S1A), and schematic representations of the intimate protein/DNA interactions at both half-sites are shown in Fig. 2, A-D.
The two HNF4α-DBD molecules bind to DNA in a similar manner. Briefly, each DNA-binding domain of HNF4α is arranged in two compact zinc finger motifs consisting of an N-terminal β-hairpin, three main α-helices followed by a single helical turn (310-helix), and a C-terminal extension (the remainder of T-box) (Figs. 1B and 3A). DNA contacts are made in both the major groove and minor groove, and schematic details of the interactions between HNF4α and each half-site are shown in Fig. 2, C and D. The recognition helix (helix I) docks into the major groove roughly perpendicular to the DNA axis (Figs. 3A and 4A) and forms extensive interactions with the HNF4α consensus binding sequence (Fig. 3C). The key residues making specific DNA interactions, such as Asp69, Lys72, Arg76, and Arg77 on helix I, are highly conserved throughout the NR superfamily (Figs. 1B and 2, C and D). Among these, Arg76 displays the plasticity through its ability to bind DNA in alternative ways to recognize differential sequences between the two half-sites of our DNA construct and might play a role in selecting specific targets. Additional residues making specific DNA interactions outside helix I, such as His62 and Tyr63 protruding from the loop proceeding helix I, are also highly conserved. These sequence-specific DNA contacts, either direct or water-mediated, are made by five of the six base pairs of each half-site (Figs. 2, C and D). This is in contrast to RXR, in which no more than three conserved base pairs are recognized in each half-site (61), reflecting its promiscuity in DNA binding as a heterodimer for multiple nonsteroid NRs. HNF4α appears to have more stringent target selectivity. Intriguingly, a MODY mutation is found on the only base pair that does not participate in the sequence-specific interactions (Fig. 2C) and is therefore believed to play a minor role in protein/DNA interactions (see “Diabetes Mutations” for further discussion). Numerous protein-phosphate backbone interactions occur with only three phosphate backbone groups within the 5′-half of each recognition site, as shown in Fig. 2, C and D, and this feature appears to be a common mode of DNA recognition among NRs (39, 64, 65).
The HNF4α-DBD contains a short C-terminal extension, known as the T-box (Figs. 1B and 2A), that provides protein dimer interactions and additional DNA interactions in the minor groove upon dimer formation (Fig. 3A). In this minor groove, typically no DNA base-specific contacts are found, and only the phosphate backbone interactions are made by the residues within the T-box. In our structure, Gln122 forms a pair of hydrogen bonds to DNA backbone phosphates, and Arg125 makes an additional ion pair with a DNA backbone phosphate only at the downstream half-site, where it becomes ordered upon dimeric interactions. Since these interactions are all with the DNA backbone atoms, they may not greatly influence the target sequence specificity.
To analyze structural deviations, two HNF4α-DBD monomers present in the asymmetric unit were superimposed. The 0.46 Å root mean square deviation for the entire 76 Cα atoms of each monomer reflects the lack of gross structural disparity (Fig. S2). The only notable differences between the two half-site protein/DNA interactions are the alternative interactions needed for the recognition of differential sequences between the two half-sites (AGTTCA versus AGTCCA), displaying its plasticity in target recognition, and the additional DNA contact in the downstream half-site made by the C-terminal end of HNF4α, such as an ion pair between Arg125 and a DNA backbone phosphate in the minor groove, when the C-terminal end becomes ordered when the two monomers meet (Figs. 2, C and D).
Diabetes Mutations—Missense point mutations and the encoded single amino acid substitutions can be instructive site-specific measures of protein function and structure. Within the region of our construct, there are five MODY1 missense mutations that are listed in the Human Gene Mutation Data base at the Institute of Medical Genetics (available on the World Wide Web). These mutations were mapped to our structure, and they were found in the peripheral regions away from both the protein/DNA binding interface and the hydrophobic core (Fig. 4A). None of the mutations are found at the residues that make important DNA contacts or comprise the core of the zinc finger motifs. Only 1 of 17 DNA contacting residues (Arg125 at the downstream half-site but not at the upstream half-site) is mutated in MODY1 patients (Figs. 2, C and D), which is in contrast to related MODY gene products, such as HNF1α (MODY3) and HNF1β (MODY5), in which numerous loss-of-function mutations are found on residues involved in DNA binding or protein core packing (66, 67). Consistent with these findings, all of the known mutations cause only modest reductions in overall transcriptional activity, with the greatest reduction being slightly over 50% as a result of the G115S mutation (Fig. 4B). These modest effects are consistent across different cell types and on different promoters (Fig. 4B). It seems likely, given the absence of mutations that profoundly affect function, that drastic disruption of HNF4α cannot be tolerated. This notion is consistent with embryonic lethality observed with the embryonic targeted disruption of HNF4α (5, 6). Our data suggest that a nearly 50% reduction in activity can be tolerated, albeit with significant effects on the organism. DBD mutants would probably have a dominant negative effect, since they dimerize with the wild type protein present (68). In that regard, no homozygous mutants of HNF4α have been found in humans, and MODY1 has a much lower prevalence compared with MODY3 and MODY5 (69).
The patients with the G115S mutation develop severe diabetes with very low insulin secretion (70), and this mutation was proven to result in strong impairment of HNF4α transcriptional activity and creation of a repressive phosphorylation site (71). Gly115 and the adjacent residue Met116 are strictly conserved in the related NR superfamily (Fig. 1B), implying a strict requirement for a glycine residue at this position. They are located in the solvent-exposed loop at the C-terminal end of helix III and appear to play no direct role in DNA binding (Fig. 4A). However, Gly115 is essential for forming a sharp turn in the loop, adopting unusual backbone torsion angles of ϕ = 118° and ψ = -174° for both monomers. Substitution by a nonglycine residue is expected to perturb the local conformation and potentially protein stability. We therefore measured the secondary structure content and protein stability of the G115S mutant in vitro as well as in vivo. To characterize the secondary structure contents of the HNF4α wild type and the G115S mutant, we used the CD technique. Fig. 4E shows the CD spectra and the estimates of the secondary structure element fractions for each of the fusion protein. The secondary structure content of the wild type HNF4α-DBD-MBP fusion protein was estimated as 64% in α-helix, 2% in β-strands, 4% in turns, and 16% in coils. These values are in good agreement with the actual secondary structure content for this presenting structure and the previously determined MBP structure (Protein Data Bank code 1N3X). However, the α-helical content for the G115S mutant was estimated as 36% (44% reduction compared with the wild type), whereas the content of the unordered regions (coils) went up to 35% (an over 200% increase). These changes were markedly greater than the control mutant (D126H) and could be responsible for its reduced protein stability, which was measured by both in vitro and in vivo assays. In the in vitro protein unfolding studies, G115S yielded a thermal denaturation midpoint (Tm) value of 53.5°, corresponding to TM shift of -3.0° compared with the wild type and the D126H mutant as controls (Fig. 4F). The in vivo pulse-and-chase experiments also showed similar results (Fig. 4D). Although the differences were not large, the G115S mutant along with the V121I mutant showed more rapid degradation in mammalian cell lines. As a result, there was very little DNA binding activity of the G115S mutant by EMSA analysis with nuclear extract from the transfected cells overexpressing the full-length proteins (Fig. 4C, lane 5). It is worth pointing out that the in vivo transcription assay with the G115S mutant showed some activity (but considerably less than the activities by other MODY1 mutants) (Fig. 4B), whereas it showed no binding in in vitro EMSA assays. These differences between the in vitro and in vivo studies could be due to the presence of cellular factors, such as co-activators, that may help stabilize DNA binding in vivo. The reduced transcriptional activity of the G115S mutant was not due to lower protein expression levels, based on our observations and the previous report (71).
The V121I mutation was identified from MODY patients who have abnormal glucose tolerance along with other typical diabetic symptoms (72, 73); however, the functional and biochemical studies of this mutant have not been conducted. The Val121 residue is located at the fringe of the hydrophobic core but optimally situated at the top of the DNA recognition helix (helix I) and helps to anchor the helix I in a position for DNA binding (Fig. 4A). Substitution with the bulkier isoleucine residue may shift helix I sufficiently to affect its interactions with DNA. Thus, the V121I mutation is predicted to cause disease by a mechanism wherein it alters DNA binding by preventing optimal positioning of the recognition helix (helix I). Our DNA binding assay with the full-length mutant (Fig. 4C, lane 6) showed a modest yet clearly noticeable reduction in DNA binding activity. Since slightly reduced protein stability of the V121I mutant was also observed in the pulse-and-chase experiment (Fig. 4D), the decrease in in vitro transcription (Fig. 4B) could result from a combination of decrease in both DNA binding activity and protein stability.
The R125W mutation was identified from the MODY patients in an isolated ethnic group and causes relatively mild diabetic symptoms (73), and no functional studies have been done. Arg125 is at the C-terminal end of the T-box, and it makes DNA backbone interactions in the minor groove through the guanidium group as well as the backbone amide nitrogen (Fig. 4A). However, these interactions were only seen at the downstream half-site, where the C-terminal end of HNF4α T-box becomes ordered through dimeric interactions. Mutation to a tryptophan will probably disrupt the organization of the protein/DNA interface in afflicted individuals through steric repulsion and the loss of the salt bridge interaction. This structural finding was tested by a set of biochemical studies in which the R125W mutant showed markedly reduced DNA binding activities (Fig. 4C, lane 7) and over 50% reduction in transactivation potential (Fig. 4B). Since this residue was found outside the HNF4α-DBD hydrophobic core, this substitution appears to have no effect on protein stability (Fig. 4D).
Mutations on the Asp126 residue (D126H and D126Y) were proven to cause severe pancreatic β-cell dysfunction and mild liver abnormalities (60). Preliminary functional studies on these mutations indicated loss-of-function primarily due to their suboptimal binding to target genes, especially with HNF1α (60). Our crystal structure now provides the molecular basis of functional disruption by these mutations. The Asp126 residue is located at the dimeric interface, where it forms an ion-pair with Arg88 of the other monomer and provides DNA binding cooperativity and target gene selectivity (Figs. 3A and 4A). This crucial interaction by Asp126 occurs only for the downstream monomer where two monomers meet. The upstream Asp126 residue is disordered in the crystal structure. Substitutions at this site seen in the D126H and D126Y mutants displayed only marginal reduction in their overall transcriptional activities (Fig. 4B), although they, especially the D126H mutant, lost most of their DNA binding activities (Fig. 4C, lanes 8 and 9), which suggests that their in vitro DNA binding activities did not directly correlate with the final levels of transactivation and may have been affected by other cellular factors. These mutations are expected to cause the loss of the salt bridge formation between the monomers and the subsequent DNA binding cooperativity seen with the wild type (Figs. 3A and 4A). Similar findings were also made with the nuclear extracts containing the full-length version of the mutants (60). These mutations appeared to retain the wild type-like protein stabilities (Fig. 4D).
Finally, an additional MODY mutation has been identified through the linkage studies on the HNF4α response element within the HNF1α promoter (A → C substitution at nucleotide -58; Fig. 2, B and C), reinforcing the HNF1α gene as a downstream target for HNF4α both in the β-cell and hepatocyte (13). The A-T base pair at the 3′-end of the upstream half-site does not participate in any base-specific interactions with HNF4α (Fig. 2, B and C). Only water-mediated and direct hydrogen bonds with the backbone phosphate atoms of one of the A-T base pair nucleotides (thymidine) is observed. Thus, a change at this promoter position would not be likely to cause major functional disruption, consistent with the idea that substantial defects in HNF4α function can be lethal to the survival of the embryos. This mutation might cause altered “indirect readout” by the protein, resulting in a disease state, or affect the local DNA conformation within the chromatin assembly, altering its accessibility to other transcription combinatorial elements, such as co-activators and mediators (74, 75).
Additional Regulatory Residues within the DNA Binding Domain of HNF4α—The activity of HNF4α is known to be regulated by several post-translational modifications. For example, HNF4α DNA binding activity and transactivation potential are tightly regulated by its state of phosphorylation induced by various signal-dependent kinases (76-79). Among the positively identified phosphorylation sites (76-78), only Ser78, a protein kinase C phosphorylation site, lies within the DBD (Fig. 1B) (79). In our structure, Ser78 is at the back side of the DNA binding surface with its side chain facing the core (Fig. 5). Although Ser78 forms a hydrogen bond with the neighboring residue Tyr85, it is near the surface of the protein and should be accessible for an acting kinase (protein kinase C) in the absence of DNA. Phosphorylation of Ser78 would probably disturb a hydrogen bond with Tyr85 and destabilize the core electrostatically, reducing protein stability. The effect of this modification has been recently tested with the mutants that either block or mimic this specific modification, and the data showed that this modification indeed reduced the protein stability and the endogenous HNF4α protein amount (79). Reduced DNA binding and impaired nuclear localization also have been reported for the phosphomimetic mutant at this position (S78D) by the same group. This can be explained by our structure that indicates bringing in a highly negatively charged group near both the DNA backbones and a set of positively charged residues serving as a nuclear localization signal would alter the local charge distributions, which could prevent the optimal DNA binding and nuclear localization, thereby acting as a repressive regulatory modification.
FIGURE 5.

Mapping of regulatory post-translational modification sites on the HNF4α-DBD structure. Phosphorylation and methylation sites are indicated by PO-23 (red) and CH3 (purple), respectively, among which the Ser78 residue makes a hydrogen bond with the neighboring Tyr85 residue through their hydroxyl groups.
Methylation of Arg91 by PRMT1 (protein arginine methyltransferase 1) has also been recently reported, and it was shown that this modification enhances DNA binding and facilitates the protein assembly needed for transcription preinitiation complex formation (80). Thus, this modification functions as an activator, as opposed to the repressive role played by phosphorylation. As shown in Fig. 5, Arg91 is fully exposed to solvent and should be readily available for the catalysis by PRMT1. However, Arg91 is far away from the DNA binding sites; thus, it does not appear to have a direct role in DNA binding. Although the modifications at remote sites can have an indirect effect on DNA binding, it is worth pointing out that in these studies (80), DNA binding was measured by chromatin immunoprecipitation or EMSA studies with the chromatinized DNA; thus, the indirect effects through additional protein factors cannot be ruled out. It is very clear from our structure that since Arg91 is fully exposed, this modification could alter the protein-protein interaction pattern, leading to differential recruitment of additional factors needed for a transcription preinitiation complex assembly, which in turn could affect the stability of the premade HNF4α/DNA interactions.
Conclusions—NRs have evolved to be able to recognize a variety of specific target genes with high specificity from a pool of genomic sequences within a regulated chromatin structure. HNF4α, as a member of the orphan NR family, binds target DNAs exclusively as a homodimer and mainly recognizes the DR1 elements. Our structural results indicate that faithful target selection and recognition by HNF4α result from DNA sequence-specific interactions as well as unique dimeric interactions made by the T-box residues, which also provide high cooperativity in DNA binding. MODY mutations on the HNF4α gene product and the HNF1α promoter were found to be peripheral residues or a nucleotide that do not directly participate in major protein/DNA interactions or protein core formation. In contrast to other MODY mutations, all of these mutations on HNF4α cause only a modest loss of function, and none appear to affect nuclear localization or are found on the protein surface for potential protein-protein interactions. Nevertheless, these substitutions result in sufficient reduction in overall transcriptional activity to cause severe forms of diabetes. On the other hand, two known post-translational modifications within the HNF4α-DBD appear to regulate activity mainly by protein core destabilization or modification of protein-protein interactions. These findings should help in the targeting of selective residues for correcting mutational defects or modulating the overall activity of HNF4α as a means of therapeutic intervention.
Supplementary Material
Acknowledgments
We thank the staff at SER-CAT beamlines 22-ID and 22-BM for data collection. We also thank Veronique Chellgren and Claire Adams for assistance in the CD experiments and data analysis, Dan Noonan for assistance in the transcriptional reporter assays, Becky Dutch for assistance in the pulse-chase experiments, and David Rodgers for critical review of the manuscript.
The atomic coordinates and structure factors (code 3CBB) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grant P20RR20171 from the COBRE program of the National Center for Research Resources (to Y. I. C.) and National Institutes of Health Grant GM070662 (to M. G. F.). This work was also supported by Juvenile Diabetes Research Foundation Grant 1-2004-506 (to Y. I. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: NR, nuclear receptor; HNF, hepatocyte nuclear factor; RXR, retinoid X receptor; RAR, retinoic acid receptor; DBD, DNA binding domain; LBD, lipophilic ligand binding domain; MODY, maturity onset diabetes of the young; MBP, maltose-binding protein; TEV, tobacco etch virus; EMSA, electrophoretic mobility shift assay; MES, 4-morpholineethanesulfonic acid.
References
- 1.Benoit, G., Cooney, A., Giguere, V., Ingraham, H., Lazar, M., Muscat, G., Perlmann, T., Renaud, J. P., Schwabe, J., Sladek, F., Tsai, M. J., and Laudet, V. (2006) Pharmacol. Rev. 58 798-836 [DOI] [PubMed] [Google Scholar]
- 2.Parviz, F., Matullo, C., Garrison, W. D., Savatski, L., Adamson, J. W., Ning, G., Kaestner, K. H., Rossi, J. M., Zaret, K. S., and Duncan, S. A. (2003) Nat. Genet. 34 292-296 [DOI] [PubMed] [Google Scholar]
- 3.Garrison, W. D., Battle, M. A., Yang, C., Kaestner, K. H., Sladek, F. M., and Duncan, S. A. (2006) Gastroenterology 130 1207-1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gupta, R. K., and Kaestner, K. H. (2004) Trends Mol. Med. 10 521-524 [DOI] [PubMed] [Google Scholar]
- 5.Duncan, S. A., Nagy, A., and Chan, W. (1997) Development 124 279-287 [DOI] [PubMed] [Google Scholar]
- 6.Chen, W. S., Manova, K., Weinstein, D. C., Duncan, S. A., Plump, A. S., Prezioso, V. R., Bachvarova, R. F., and Darnell, J. E., Jr. (1994) Genes Dev. 8 2466-2477 [DOI] [PubMed] [Google Scholar]
- 7.Hayhurst, G. P., Lee, Y. H., Lambert, G., Ward, J. M., and Gonzalez, F. J. (2001) Mol. Cell Biol. 21 1393-1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inoue, Y., Yu, A. M., Inoue, J., and Gonzalez, F. J. (2004) J. Biol. Chem. 279 2480-2489 [DOI] [PubMed] [Google Scholar]
- 9.Inoue, Y., Yu, A. M., Yim, S. H., Ma, X., Krausz, K. W., Inoue, J., Xiang, C. C., Brownstein, M. J., Eggertsen, G., Bjorkhem, I., and Gonzalez, F. J. (2006) J. Lipid Res. 47 215-227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watt, A. J., Garrison, W. D., and Duncan, S. A. (2003) Hepatology 37 1249-1253 [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez, F. J. (2008) Drug Metab. Pharmacokinet. 23 2-7 [DOI] [PubMed] [Google Scholar]
- 12.Kamiyama, Y., Matsubara, T., Yoshinari, K., Nagata, K., Kamimura, H., and Yamazoe, Y. (2007) Drug Metab. Pharmacokinet. 22 287-298 [DOI] [PubMed] [Google Scholar]
- 13.Gragnoli, C., Lindner, T., Cockburn, B. N., Kaisaki, P. J., Gragnoli, F., Marozzi, G., and Bell, G. I. (1997) Diabetes 46 1648-1651 [DOI] [PubMed] [Google Scholar]
- 14.Lahuna, O., Rastegar, M., Maiter, D., Thissen, J. P., Lemaigre, F. P., and Rousseau, G. G. (2000) Mol. Endocrinol. 14 285-294 [DOI] [PubMed] [Google Scholar]
- 15.Kamiya, A., Inoue, Y., and Gonzalez, F. J. (2003) Hepatology 37 1375-1384 [DOI] [PubMed] [Google Scholar]
- 16.Yamagata, K., Furuta, H., Oda, N., Kaisaki, P. J., Menzel, S., Cox, N. J., Fajans, S. S., Signorini, S., Stoffel, M., and Bell, G. I. (1996) Nature 384 458-460 [DOI] [PubMed] [Google Scholar]
- 17.Miura, A., Yamagata, K., Kakei, M., Hatakeyama, H., Takahashi, N., Fukui, K., Nammo, T., Yoneda, K., Inoue, Y., Sladek, F. M., Magnuson, M. A., Kasai, H., Miyagawa, J., Gonzalez, F. J., and Shimomura, I. (2006) J. Biol. Chem. 281 5246-5257 [DOI] [PubMed] [Google Scholar]
- 18.Gupta, R. K., Vatamaniuk, M. Z., Lee, C. S., Flaschen, R. C., Fulmer, J. T., Matschinsky, F. M., Duncan, S. A., and Kaestner, K. H. (2005) J. Clin. Invest. 115 1006-1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kulkarni, R. N., and Kahn, C. R. (2004) Science 303 1311-1312 [DOI] [PubMed] [Google Scholar]
- 20.Odom, D. T., Zizlsperger, N., Gordon, D. B., Bell, G. W., Rinaldi, N. J., Murray, H. L., Volkert, T. L., Schreiber, J., Rolfe, P. A., Gifford, D. K., Fraenkel, E., Bell, G. I., and Young, R. A. (2004) Science 303 1378-1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boj, S. F., Parrizas, M., Maestro, M. A., and Ferrer, J. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 14481-14486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaxillaire, M., and Froguel, P. (2008) Endocr. Rev. 29 254-264 [DOI] [PubMed] [Google Scholar]
- 23.Pearson, E. R., Pruhova, S., Tack, C. J., Johansen, A., Castleden, H. A., Lumb, P. J., Wierzbicki, A. S., Clark, P. M., Lebl, J., Pedersen, O., Ellard, S., Hansen, T., and Hattersley, A. T. (2005) Diabetologia 48 878-885 [DOI] [PubMed] [Google Scholar]
- 24.Bain, D. L., Heneghan, A. F., Connaghan-Jones, K. D., and Miura, M. T. (2007) Annu. Rev. Physiol. 69 201-220 [DOI] [PubMed] [Google Scholar]
- 25.Dhe-Paganon, S., Duda, K., Iwamoto, M., Chi, Y. I., and Shoelson, S. E. (2002) J. Biol. Chem. 277 37973-37976 [DOI] [PubMed] [Google Scholar]
- 26.Duda, K., Chi, Y.-I., and Shoelson, S. E. (2004) J. Biol. Chem. 279 23311-23316 [DOI] [PubMed] [Google Scholar]
- 27.Hertz, R., Magenheim, J., Berman, I., and Bar-Tana, J. (1998) Nature 392 512-516 [DOI] [PubMed] [Google Scholar]
- 28.Petrescu, A. D., Hertz, R., Bar-Tana, J., Schroeder, F., and Kier, A. B. (2005) J. Biol. Chem. 280 16714-16727 [DOI] [PubMed] [Google Scholar]
- 29.Petrescu, A. D., Hertz, R., Bar-Tana, J., Schroeder, F., and Kier, A. B. (2002) J. Biol. Chem. 277 23988-23999 [DOI] [PubMed] [Google Scholar]
- 30.Schroeder, F., Huang, H., Hostetler, H. A., Petrescu, A. D., Hertz, R., Bar-Tana, J., and Kier, A. B. (2005) Lipids 40 559-568 [DOI] [PubMed] [Google Scholar]
- 31.Hertz, R., Kalderon, B., Byk, T., Berman, I., Za'tara, G., Mayer, R., and Bar-Tana, J. (2005) J. Biol. Chem. 280 24451-24461 [DOI] [PubMed] [Google Scholar]
- 32.Khorasanizadeh, S., and Rastinejad, F. (2001) Trends Biochem. Sci. 26 384-390 [DOI] [PubMed] [Google Scholar]
- 33.Jiang, G., and Sladek, F. M. (1997) J. Biol. Chem. 272 1218-1225 [DOI] [PubMed] [Google Scholar]
- 34.Holmbeck, S. M., Dyson, H. J., and Wright, P. E. (1998) J. Mol. Biol. 284 533-539 [DOI] [PubMed] [Google Scholar]
- 35.Kritis, A. A., Argyrokastritis, A., Moschonas, N. K., Power, S., Katrakili, N., Zannis, V. I., Cereghini, S., and Talianidis, I. (1996) Gene (Amst.) 173 275-280 [DOI] [PubMed] [Google Scholar]
- 36.Lu, P., Liu, J., Melikishvili, M., Fried, M. G., and Chi, Y. I. (2008) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64 313-317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 38.Vagin, A., and Teplyakov, A. (1997) J. Appl. Crystallogr. 30 1002-1025 [Google Scholar]
- 39.Rastinejad, F., Wagner, T., Zhao, Q., and Khorasanizadeh, S. (2000) EMBO J. 19 1045-1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 41.Jones, T. A., Zou, J. Y., Cowan, S. W., and Kjeldgaard. (1991) Acta Crystallogr. Sect. A 47 110-119 [DOI] [PubMed] [Google Scholar]
- 42.Maxam, A. M., and Gilbert, W. (1977) Proc. Natl. Acad. Sci. U. S. A. 74 560-564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hellman, L. M., and Fried, M. G. (2007) Nat. Protoc. 2 1849-1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGhee, J. D., and von Hippel, P. H. (1974) J. Mol. Biol. 86 469-489 [DOI] [PubMed] [Google Scholar]
- 45.Tsodikov, O. V., Holbrook, J. A., Shkel, I. A., and Record, M. T., Jr. (2001) Biophys. J. 81 1960-1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson, W. C. (1999) Proteins 35 307-312 [PubMed] [Google Scholar]
- 47.Sreerama, N., and Woody, R. W. (2000) Anal. Biochem. 287 252-260 [DOI] [PubMed] [Google Scholar]
- 48.Mao, D., Wachter, E., and Wallace, B. A. (1982) Biochemistry 21 4960-4968 [DOI] [PubMed] [Google Scholar]
- 49.Borges, J. C., Pereira, J. H., Vasconcelos, I. B., dos Santos, G. C., Olivieri, J. R., Ramos, C. H., Palma, M. S., Basso, L. A., Santos, D. S., and de Azevedo, W. F., Jr. (2006) Arch. Biochem. Biophys. 452 156-164 [DOI] [PubMed] [Google Scholar]
- 50.Rastinejad, F., Perlmann, T., Evans, R. M., and Sigler, P. B. (1995) Nature 375 203-211 [DOI] [PubMed] [Google Scholar]
- 51.Shaffer, P. L., and Gewirth, D. T. (2002) EMBO J. 21 2242-2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lavery, R., and Sklenar, H. (1989) J. Biomol. Struct. Dyn. 6 655-667 [DOI] [PubMed] [Google Scholar]
- 53.Shaffer, P. L., Jivan, A., Dollins, D. E., Claessens, F., and Gewirth, D. T. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 4758-4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Devarakonda, S., Harp, J. M., Kim, Y., Ozyhar, A., and Rastinejad, F. (2003) EMBO J. 22 5827-5840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roemer, S. C., Donham, D. C., Sherman, L., Pon, V. H., Edwards, D. P., and Churchill, M. E. (2006) Mol. Endocrinol. 20 3042-3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glass, C. K., Rose, D. W., and Rosenfeld, M. G. (1997) Curr. Opin. Cell Biol. 9 222-232 [DOI] [PubMed] [Google Scholar]
- 57.Zamir, I., Zhang, J., and Lazar, M. A. (1997) Genes Dev. 11 835-846 [DOI] [PubMed] [Google Scholar]
- 58.Bogan, A. A., Dallas-Yang, Q., Ruse, M. D., Jr., Maeda, Y., Jiang, G., Nepomuceno, L., Scanlan, T. S., Cohen, F. E., and Sladek, F. M. (2000) J. Mol. Biol. 302 831-851 [DOI] [PubMed] [Google Scholar]
- 59.Aggelidou, E., Iordanidou, P., Demetriades, C., Piltsi, O., and Hadzopoulou-Cladaras, M. (2006) FEBS J. 273 1948-1958 [DOI] [PubMed] [Google Scholar]
- 60.Oxombre, B., Moerman, E., Eeckhoute, J., Formstecher, P., and Laine, B. (2002) J. Mol. Med. 80 423-430 [DOI] [PubMed] [Google Scholar]
- 61.Zhao, Q., Chasse, S. A., Devarakonda, S., Sierk, M. L., Ahvazi, B., and Rastinejad, F. (2000) J. Mol. Biol. 296 509-520 [DOI] [PubMed] [Google Scholar]
- 62.Miura, N., and Tanaka, K. (1993) Nucleic Acids Res. 21 3731-3736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tian, J. M., and Schibler, U. (1991) Genes Dev. 5 2225-2234 [DOI] [PubMed] [Google Scholar]
- 64.Jakob, M., Kolodziejczyk, R., Orlowski, M., Krzywda, S., Kowalska, A., Dutko-Gwozdz, J., Gwozdz, T., Kochman, M., Jaskolski, M., and Ozyhar, A. (2007) Nucleic Acids Res. 35 2705-2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao, Q., Khorasanizadeh, S., Miyoshi, Y., Lazar, M. A., and Rastinejad, F. (1998) Mol. Cell 1 849-861 [DOI] [PubMed] [Google Scholar]
- 66.Chi, Y. I., Frantz, J. D., Oh, B. C., Hansen, L., Dhe-Paganon, S., and Shoelson, S. E. (2002) Mol. Cell 10 1129-1137 [DOI] [PubMed] [Google Scholar]
- 67.Lu, P., Rha, G. B., and Chi, Y. I. (2007) Biochemistry 46 12071-12080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sladek, F. M., Dallas-Yang, Q., and Nepomuceno, L. (1998) Diabetes 47 985-990 [DOI] [PubMed] [Google Scholar]
- 69.Vaxillaire, M., and Froguel, P. (2006) Endocrinol. Metab. Clin. North Am. 35 371-384 [DOI] [PubMed] [Google Scholar]
- 70.Malecki, M. T., Yang, Y., Antonellis, A., Curtis, S., Warram, J. H., and Krolewski, A. S. (1999) Diabet. Med. 16 193-200 [DOI] [PubMed] [Google Scholar]
- 71.Oxombre, B., Kouach, M., Moerman, E., Formstecher, P., and Laine, B. (2004) Biochem. J. 383 573-580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Monney, C. T., Kaltenrieder, V., Cousin, P., Bonny, C., and Schorderet, D. F. (2002) Hum. Mutat. 20 230-231 [DOI] [PubMed] [Google Scholar]
- 73.Pruhova, S., Ek, J., Lebl, J., Sumnik, Z., Saudek, F., Andel, M., Pedersen, O., and Hansen, T. (2003) Diabetologia 46 291-295 [DOI] [PubMed] [Google Scholar]
- 74.Rochette-Egly, C. (2005) J. Biol. Chem. 280 32565-32568 [DOI] [PubMed] [Google Scholar]
- 75.Torchia, J., Glass, C., and Rosenfeld, M. G. (1998) Curr. Opin. Cell Biol. 10 373-383 [DOI] [PubMed] [Google Scholar]
- 76.Guo, H., Gao, C., Mi, Z., Wai, P. Y., and Kuo, P. C. (2006) Biochem. J. 394 379-387 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 77.Hong, Y. H., Varanasi, U. S., Yang, W., and Leff, T. (2003) J. Biol. Chem. 278 27495-27501 [DOI] [PubMed] [Google Scholar]
- 78.Viollet, B., Kahn, A., and Raymondjean, M. (1997) Mol. Cell Biol. 17 4208-4219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun, K., Montana, V., Chellappa, K., Brelivet, Y., Moras, D., Maeda, Y., Parpura, V., Paschal, B. M., and Sladek, F. M. (2007) Mol. Endocrinol. 21 1297-1311 [DOI] [PubMed] [Google Scholar]
- 80.Barrero, M. J., and Malik, S. (2006) Mol. Cell 24 233-243 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.