

Abstract
The small regulatory protein Crl binds to σS, the RNA polymerase stationary phase σ factor. Crl facilitates the formation of the σS-associated holoenzyme (EσS) and thereby activates σS-dependent genes. Using a real time surface plasmon resonance biosensor, we characterized in greater detail the specificity and mode of action of Crl. Crl specifically forms a 1:1 complex with σS, which results in an increase of the association rate of σS to core RNA polymerase without any effect on the dissociation rate of EσS. Crl is also able to associate with preformed EσS with a higher affinity than with σS alone. Furthermore, even at saturating σS concentrations, Crl significantly increases EσS association with the katN promoter and the productive isomerization of the EσS-katN complex, supporting a direct role of Crl in transcription initiation. Finally, we show that Crl does not bind to σ70 itself but is able at high concentrations to form a weak and transient 1:1 complex with both core RNA polymerase and the σ70-associated holoenzyme, leaving open the possibility that Crl might also exert a side regulatory role in the transcriptional activity of additional non-σS holoenzymes.
In Enterobacteria, σS, encoded by rpoS, is the master regulator of the general stress response and is also responsible for the transcription of stationary phase-specific genes. σS accumulates at the onset of the stationary phase and in response to harsh environmental conditions, including carbon and phosphate starvation and acidic and osmotic stress (1, 2). When associated with the RNA polymerase core enzyme (E),3 the σS-associated holoenzyme (EσS) transcribes rpoS-dependent genes and endows the cells with the ability to endure stationary phase and tolerate a multitude of stress conditions (3–5). The acquisition of this multiple stress resistance status, which is dependent on σS, has an energetic cost and decreases bacterial fitness in environments containing nutrients at low concentrations (6). Therefore, σS abundance and activity are tightly controlled by the interplay of a complex set of regulators that affect transcription, translation, and the stability of the protein. Indeed, the levels of σS are controlled primarily by the ClpXP protease, which, together with the catalytic adaptor protein RssB, targets free σS for degradation during exponential growth in the absence of stress (7, 8). σS concentration is not the sole parameter controlling the expression of rpoS-dependent genes. Another important checkpoint is the formation of EσS, which in Escherichia coli is restricted by the competition of σS with six other σ factors for binding to a limited amount of E (9–11). Even in stationary phase, when σS is most abundant, the concentration of the primary σ factor, σ70, remains 3-fold higher (12). Furthermore, of all of the E. coli σ factors, σS exhibits the lowest affinity for E (13). E. coli has developed different strategies that enable σS to capture enough E to transcribe its target genes, and several factors favor the formation of EσS during stationary phase or in response to toxic insult. First, by preventing ribosomal gene transcription, the alarmone ppGpp, together with DksA, dissociates the σ70-associated holoenzyme (Eσ70) from rRNA loci releasing E for binding to other σ factors (14–16). Second, by directly binding to σ70, Rsd, the level of which increases 2-fold in stationary phase, captures and inhibits free σ70 (17–20). Finally, in the late stationary phase, the small 6S RNA sequesters a large fraction of Eσ70, thereby restricting σ factor competition to the remaining six σ factors (21, 22). These mechanisms offer a partial solution, and clearly other elements are needed to assist σS in its competition with other σ factors and to directly facilitate the binding of σS to E. One such factor is Crl, a protein that was initially described as an activator of curli formation (23). Crl binds to σS (24) and positively regulates σS-dependent genes, especially when σS levels are low (25–29). Biochemical analyses confirm the direct effect of Crl on σS-dependent transcription (26, 27, 29, 30) and furthermore demonstrate that Crl favors EσS-dependent transcription over Eσ70-dependent transcription (29). Gel filtration experiments with crude E. coli cell extracts indicate that Crl enhances the binding of σS to E (29). Intriguingly, a recent biochemical study demonstrated that Crl could also enhance the transcriptional activity of Eσ70 and Eσ32, albeit significantly less than that of EσS (30). Two alternative, but not exclusive, models have been proposed to describe the action of Crl in activating transcription. First, Crl may promote the unfurling of free σ, allowing it to adopt the appropriate conformation for efficient binding to E, and second, Crl may function by stabilizing the Eσ once formed.
Eσ formation is a multistep process that involves major rearrangements in both E and σ (31). Both σ70 and σS are members of the σ70 family of σ factors, which are composed of four highly conserved domains that can adopt different conformations relative to one another (32–34). The minimal structural scaffold of all σ70 family members is composed of two large globular domains: σ domain 2 (σ2) and σ domain 4 (σ4). Domains σ2 and σ4, which are likely to interact with each other in the context of free σ (34), possess the main determinants for binding both E and promoter DNA. σ2 binds the β′ subunit coiled-coil and promoter -10 element, and σ4 binds the β subunit flap and the promoter -35 element (35–38). These interactions are crucial for orientating the σ2 and σ4 DNA-binding determinants with the correct spacing for binding to the promoter -10 and -35 elements, respectively (31). High resolution structural studies of thermophilic bacterial Eσ reveal that σ is spread out along one face of E, forming an extensive protein-protein interface with the β and β′ subunits (36–38). Both free σ70 and σS are unable to recognize promoter DNA (39, 40),4 most likely a result of the intramolecular interactions between σ2 and σ4 (34). However, variants of σ70 and several alternative σ factors with the N-terminal domain 1.1 (σ1.1) deleted exhibit poor but detectable promoter-specific binding (41). These results suggest that σ1.1, which is not resolved in any structure, inhibits the DNA-binding properties of σ70 and several alternative σ factors, including σS, by locking them in an inactive closed conformation. Upon binding to E, dramatic conformational changes in free σ ensue, resulting in a 15-Å increase in the distance between σ2 and σ4 and the unmasking of the DNA-binding determinants, as well as the displacement of σ1.1 by 20 Å (42).
To date, the binding properties of Crl to the components of the transcription machinery have not been characterized in a quantitative manner; nor has the mechanism by which Crl increases transcriptional activity been fully understood. In this study, we have used a real time surface plasmon resonance (SPR) microfluidic biosensor to decipher the specificity and mechanism of Crl-dependent activation. First, we investigated the binding of Crl to the components of the transcription machinery (σS, σ70, E, EσS, and Eσ70), and second, we examined the promoter binding properties of EσS and Eσ70, formed in the presence or absence of Crl. Our studies indicate that Crl binds to σS and not to σ70 and that Crl acts most likely by promoting a conformational change in σS that enhances its association rate to E. Furthermore, Crl is also able to bind to preformed EσS, resulting in a stable ternary complex. Finally, we find that Crl is able to bind directly to E, forming a transient complex that might be related to the reported Crl-dependent activation of transcription by other Eσ species (30).
EXPERIMENTAL PROCEDURES
Cloning Procedures—The DNA encoding a His12 tag was appended to the 5′ end of both rpoD, encoding σ70, and rpoS, encoding σS, in a multistep procedure. First, the XbaI-HindIII fragment of pGEMHisrpoD (43) was cloned between the XbaI and HindIII sites of pET21a, generating pET21His6rpoD and the internal rpoD NdeI site was removed by site-directed mutagenesis. Second, oligonucleotides, 5′-T ATG CAT CAC CAT CAC CAC CA-3′ (K81) and 5′-T ATG GTG GTG ATG GTG ATG CA-3′ (K82), encoding an NsiI site and a His6 tag, were annealed and cloned into the unique NdeI site of pET21His6rpoD, creating pET21His12rpoD. Finally, the BamHI-HindIII fragment from pET21His12rpoD was replaced with the wild type rpoD BamHI-HindIII fragment. Similarly, the K81/K82 fragment was inserted into the unique NdeI site of pFC0, that encodes σS (44), resulting in pFC0His6rpoS, after which the vector-encoded NsiI-HindIII fragment was replaced by the NsiI-HindIII vector-encoded fragment of pET21His12rpoD, generating pET21His12rpoS.
The DNA encoding Salmonella enterica Crl was amplified by PCR using primers L73 5′-GTT GCT TCA TTA AAG GAG ATC CAT ATG ACG TTA CCG AGT GGA CAC C-3′ and L74 5′-GGC ATG GCA GAA TTC TTA TGC CGA CAG TTT TAC CGG C-3′ using S. enterica genomic DNA as a template. The resulting fragment was cleaved with NdeI and EcoRI and cloned between the NdeI and EcoRI sites of a pET28a-based plasmid (20), creating pSKB2ppXcrl.
Derivatives of pUT18 encoding N-terminal Crl-T18 fusion proteins used in the bacterial adenylate cyclase two-hybrid (BACTH) assays were constructed by cloning PCR-amplified DNA fragments encoding Crl between the XbaI and KpnI sites of pUT18. The DNA encoding E. coli Crl was amplified using primers M75 (5′-CG ACT CTA GAG ATG ACG TTA CCG AGT GGA CAC CCG-3′) and M76 (5′-GCT CGG TAC CCG TTA TTA TGC CGA CAG TTT TAC CGG CTC GTC G-3′) and plasmid pQEcrl as the template (26). The DNA encoding S. enterica Crl was amplified by PCR using primers M82 (5′-C CCT CTA GAA ATG ACG TTA CCG AGT GGA CAC CCG-3′) and M83 (5′-C TCG GTA CCC GCG CCG TTA ACT TCA CCG G-3′) and plasmid pSKB2ppXcrl as the template. The resultant PCR products were cloned between the XbaI and KpnI sites of pUT18. All plasmids were confirmed to be correct by DNA sequencing.
Purification of Proteins—Expression plasmids were transformed into BL21 (DE3) E. coli cells, and the transformants were selected in the presence of the appropriate antibiotic. Cultures were grown at 37 °C to an A600 ∼0.6 and induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 3 h at 30°C. Cells containing overexpressed proteins were harvested by centrifugation and stored at -80 °C.
His12-σS and His12-σ70 were prepared using an adapted protocol (45). The untagged σ70 and σS factors were purified from the M5219/pMRG8 and BL21(DE3)/pLysS/pFC0 strains, respectively, according to previously described protocols (46, 47). E was prepared according to Ref. 48.
The His6-ppXCrl protein, containing a vector-encoded N-terminal His6 tag and a PreScission protease cleavage site, was purified by HiTrap Ni2+-charged affinity chromatography (GE Healthcare). The N-terminal His6 tag was removed using the PreScission protease (GE Healthcare). Crl produced in this manner contains three additional N-terminal amino acids, followed by the methionine residue of the wild type 133-amino acid protein. The sample was further purified by a second subtractive HiTrap Ni2+-charged affinity chromatography step to remove uncleaved His6-ppXCrl protein and the His6 tag, followed by an ion exchange chromatography step (HiTrap Q Sepharose; GE Healthcare) and a gel filtration chromatography step (Superdex 75; GE Healthcare). The peak fractions were dialyzed against storage buffer (10 mm Tris-HCl (pH 8.0), 10 mm MgCl2, 200 mm KCl and 50% (v/v) glycerol) and stored at -20 °C. His6-Crl was purified as previously described (26) and used in place of untagged Crl with identical results. Aliquots were passed over a Microcon YM-3 to exchange the storage buffer for buffer A (40 mm Hepes (pH 8.0), 10 mm MgCl2 and 100 mm potassium glutamate) prior to SPR analysis.
BACTH Assays—The E. coli cya strain BTH101 was transformed with derivatives of plasmids pKT25 and pUT18 encoding the T25 and T18 fragments of Bordetella pertussis adenyl cyclase. Plasmids pKT25-σ70, pKT25-σS, and pUT18-Rsd have been described previously (19). Co-transformants were plated onto MacConkey maltose plates supplemented with 100 μg/ml ampicillin, 50 μg/ml kanamycin, and 0.5 mm isopropyl 1-thio-β-d-galactopyranoside. After incubating the plates at 30 °C for 3 days, colonies were collected and lysed with chloroform and 0.05% (w/v) SDS, and their β-galactosidase activities were measured as described by Miller (49). Each transformation was performed twice, and the S.E. values on the β-galactosidase activities were below 15%.
SPR Experiments—All SPR binding assays were conducted on a Biacore 2000 instrument (GE Healthcare), equilibrated at 25 °C in buffer A supplemented with 0.034% (v/v) Tween 20.
Binding of Crl to σ—5000 RU of penta-His monoclonal antibody (Qiagen) were covalently immobilized through their solvent-accessible primary amine groups to the carboxymethylated dextran matrix of a CM5 sensor chip, using the Amine Coupling Kit (GE Healthcare), according to the manufacturer's instructions. Briefly, each flow cell, equilibrated at a flow rate of 5 μl/min in phosphate-buffered saline (pH 7.4), supplemented with 0.005% (v/v) Tween 20, was activated for 12 min with a solution of 50 mm N-hydroxysuccinimide and 200 mm N-ethyl-N′-(3 dimethylaminopropyl)carbodiimide, followed by an injection of the antibody (5 μg/ml) in 10 mm sodium acetate (pH 4.5). The surface was finally deactivated for 12 min with 1 m ethanolamine (pH 8.5).
330–350 RU of either His12-σS or His12-σ70 were noncovalently captured on the penta-His antibody surface. Another surface was left unliganded and used as a reference flow cell. Crl (50 nm to 15 μm) was then injected for 1 min at a flow rate of 100 μl/min, and the dissociation was followed for 3 min. Regeneration of the surfaces was performed by successive injections of 10 mm glycine-HCl (pH 2) and 0.05% (w/v) SDS.
Binding of σ to Core RNA Polymerase—10,000 RU of 4RA2 monoclonal antibody (Neoclone), purified on a Protein A-Sepharose column (GE Healthcare), were covalently immobilized on the surface of a CM5 sensor chip, as described above. 2000–2500 RU of E were captured on a 4RA2 antibody surface. Another surface was left unliganded and used as a reference flow cell.
σS (0.4–250 nm) or σ70 (0.25–15 nm), in the presence or absence of Crl, was then injected for 7 min at 20 μl/min, and the dissociation was followed for 5 min. Control experiments were performed by injecting Crl alone. Regeneration of the surfaces was performed by successive injections of 10 mm glycine-HCl (pH 1.5) and 0.1% (w/v) SDS.
Binding of Crl to Core RNA Polymerase or Holoenzymes—1500–2000 RU of E were captured on a 4RA2 antibody surface, whereas a reference flow cell was left unliganded, followed by 130–150 RU of σS or σ70 (or running buffer for the study of direct E-Crl interactions). Crl (0.1–25 μm) was then injected for 5 min at 20 μl/min, and the dissociation was followed for 10 min. Regeneration of the surfaces was performed by successive injections of 10 mm glycine-HCl (pH 1.5) and 0.1% (w/v) SDS.
SPR Data Analysis—All of the association and dissociation profiles were double-referenced using the Scrubber 2.0 software (BioLogic Software) (i.e. both the signals from the reference surfaces and from blank experiments using running buffer instead of Crl or σ factors were subtracted). The binding curves were globally analyzed with a nonlinear least squares algorithm implemented in the BIAevaluation 4.1 software (Biacore), using single exponential (Langmuir model) or coupled double-exponential functions of time (“conformation change” model). Kinetic parameters (kon and koff), half-lives (t½), equilibrium dissociation constants (Kd), and maximal binding capacities (Rmax) were determined based on at least two experiments. Steady-state signals (Req; measured or extrapolated) were plotted against the Crl or σS concentration (C). Fitting was performed with the following equations,
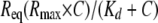 |
(Eq. 1) |
(single class of binding sites) or
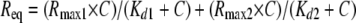 |
(Eq. 2) |
(two independent classes of binding sites). The stoichiometry of binding of Crl or σS was determined with the equation,
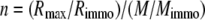 |
(Eq. 3) |
where M and Mimmo represent the molecular weight of the injected molecule (Crl or σS) and of the tethered molecule (E or σS), respectively, and Rimmo is the density of tethered molecules on the sensor chip surface.
Gel Retardation Assays—The 123-bp labeled wild-type katN fragment was generated by PCR using primers 5′-[32P]CGA GCT CGT GTT CCT CGT TGC TTG C-3′ and 5′-biotin-TTA CGC GGT AAA TCA CAA CTA TTT CCG-3′, and pJCDkatN (27) as a template. A variant of the katN promoter with substitutions in the -35 element (TTGA to CCAG) and -10 region (CTAATTTTA to GAGCTCGGC) was synthesized using the QuikChange multisite-directed mutagenesis kit (Stratagene). The fragments were purified on a 7.5% polyacrylamide gel run in Tris-borate-EDTA buffer. E (16 nm) was preincubated in buffer A supplemented with 0.034% Tween 20 with σS (0, 16, 80, or 2000 nm). 5 μl of the protein complexes were then added to 5 μl of 32P-labeled katN fragment in buffer A supplemented with 0.034% Tween 20 and 0.5 μg/ml of heparin and incubated at 25 °C for 20 min. After the addition of 2 μl of loading buffer (buffer A containing 50% sucrose, 0.025% xylene cyanol blue), the mixture was loaded onto a 6% native polyacrylamide gel run in TG buffer (25 mm Tris, 192 mm glycine, pH 8.5) at 10 V/cm. The gel was dried before being autoradiographed and quantified using a PhosphorImager (GE Healthcare).
Potassium Permanganate Reactivity—Complex formation was initiated by adding a mixture of EσS with or without Crl to the labeled wild-type katN fragment at 37 °C or at 20 °C in buffer A containing 500 μg/ml bovine serum albumin. At various times, 10-μl aliquots were withdrawn and allowed to react with 3 μl of 18 mm potassium permanganate solution. The reaction was stopped after 15 s by adding 6 μl of 200 mm dithiothreitol, and the samples were phenol-extracted, precipitated with ethanol, and rinsed with 70% (v/v) ethanol. The ethanol precipitates were resuspended in 100 μl of piperidine (1 m), heated at 90 °C for 30 min, and evaporated until dry. Then 20 μl of water was added and evaporated (twice). The samples were resuspended in 5 μl of 20 mm EDTA in formamide containing xylene cyanol and bromphenol blue and loaded onto a 9% (w/v) denaturing polyacrylamide gel.
RESULTS
SPR instruments sense small refractive index changes, which are proportional to local concentration variations in the vicinity of a solid surface. SPR can be used to monitor protein-protein and protein-DNA interactions in real time and measure association and dissociation rates (50–54). SPR is especially appropriate for analyzing the properties of Crl, which in contrast to many transcription activators, exhibits no DNA binding activity but has been shown to bind directly to σS and affect the rate of open complex formation.
Crl Binds to σS but Not to σ70—Real time SPR assays were first conducted to investigate the direct binding of Crl to either σS or σ70. Variants of σS with the N-terminal portion of the protein deleted are responsive to Crl (24); hence, to immobilize σS and σ70 to the solid surface (sensor chip), we used a noncovalent orientated strategy, relying on the stable interaction between the His12 moiety appended to the N termini of both σS and σ70, and an anti-His5 monoclonal antibody attached to the dextran surface of the sensor chip. We observed that Crl readily bound to σS, at concentrations as low as 60 nm, and reached a steady state within a few seconds (Fig. 1A). The analysis of the concentration dependence of the steady-state responses allowed us to determine a dissociation equilibrium constant (Kd) of 2.46 ± 0.13 μm and a 1:1 stoichiometry for the Crl-σS complex (Fig. 1A, inset). The dissociation rate (koff) of the complex was ∼0.3 s-1 (corresponding to a t½ of ∼3 s), and the association rate (kon) was 1.5 × 105 m-1 s-1 (calculated from the koff/Kd ratio). Under the same conditions, no binding of Crl to σ70 could be detected, even at the highest Crl concentration used (15 μm; Fig. 1B), indicating a minimal Kd threshold of 750 μm for a hypothetical Crl-σ70 interaction. The inability of Crl to bind σ70 was not due to the quality of the σ70 preparation used, since it was at least 50% active in abortive and run-off transcription assays using the lacUV5 promoter, and furthermore, when immobilized as above, it was able to interact tightly with its specific ligand, Rsd (regulator of sigma D; data not shown).
FIGURE 1.

Binding of Crl to immobilized His12-tagged σ factors. Association and dissociation real time profiles are shown for the following Crl concentrations: 50 nm, 110 nm, 225 nm, 450 nm, 890 nm, 1.79 μm, 3.58 μm, 7.15 μm, and 14.3 μm. A, immobilized σS surface. The inset shows the steady state response as a function of Crl concentration. B, immobilized σ70 surface. No binding is observed.
In all SPR experiments, we used σS and σ70 from E. coli and the Crl protein from S. enterica. Crl proteins from both species share 95% similarity and 85% identity, and the S. enterica protein supports transcription activation of E. coli EσS at each promoter tested (26, 27). In order to probe the interplay of these proteins with one another in vivo, we utilized the BACTH system. The BACTH system is dependent upon the functional reconstitution of the B. pertussis adenyl cyclase T18 and T25 subdomains by two interacting partners (55, 56). The resultant cAMP binds to and activates the transcription activator CRP, a positive regulator of β-galactosidase expression. C-terminal fusions of E. coli σ70 and E. coli σS to T25 and N-terminal fusions of E. coli Crl and S. enterica Crl to T18 were constructed. Table 1 shows the different fusion proteins, which were overproduced in the E. coli cya lac+ strain BTH101, and the resultant β-galactosidase activities. As shown previously (19) and confirmed by our experiments, the T25-σ70 fusion interacted with the Rsd-T18 fusion protein (Table 1, row 4). However, T25-σ70 did not interact with either E. coli or S. enterica Crl fused to T18 (Table 1, rows 2 and 3). In contrast, both the E. coli and S. enterica Crl-T18 fusion proteins interacted with T25-σS (rows 6 and 7), showing that Crl from both species was functionally selective for σS but unable to recognize σ70. Taken together, our SPR and BACTH data clearly show that Crl has exquisite binding specificity for free σS but does not bind to σ70.
TABLE 1.
BACTH analysis of Crl-σ interactions The table lists measured β-galactosidase activities in BTH101 cya cells carrying pKT25 derivatives encoding a T25-σ70 or a T25-σs fusion protein and pUT18 derivatives encoding Crl-T18 and Rsd-T18 fusions. Cells were grown on MacConkey maltose plates containing 100 μg/ml ampicillin, 50 μg/ml kanamycin, and 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at 30°C before measuring β-galactosidase activities. No difference in activity was detected between E. coli Crl-T18 and S. enterica Crl-T18.
| Protein fusion 1 | Protein fusion 2 | β-Galactosidase activity | |
|---|---|---|---|
| Miller units | |||
| 1 | T25-σ70 | T18 | 63 |
| 2 | T25-σ70 | E. coli Crl-T18 | 60 |
| 3 | T25-σ70 | S. enterica Crl-T18 | 62 |
| 4 | T25-σ70 | E. coli Rsd-T18 | 1022 |
| 5 | T25-σs | T18 | 60 |
| 6 | T25-σs | E. coli Crl-T18 | 696 |
| 7 | T25-σs | S. enterica Crl-T18 | 698 |
| 8 | T25-σs | E. coli Rsd-T18 | 72 |
Crl Specifically Facilitates the Formation of the σS-associated Holoenzyme—We reasoned that the N-terminally oriented immobilization of σ might not be optimal for the study of the interactions of σ with E, since it might restrict the mobility of σ1.1. Therefore, to monitor the effect of Crl on σ-E interactions, we resorted to an alternative strategy that utilized a monoclonal antibody specific for the C terminus of the RNA polymerase α subunit (α-CTD; which plays no role in the association of σ with E) to noncovalently tether E on the surface of the chip. In the absence of Crl, σS bound to E with a stoichiometry of 1:1 and a Kd value of 68.2 ± 8.4 nm (Fig. 2A). The association process followed a complex mechanism best described by an initial encounter step between σS and E, followed by subsequent isomerizations of the EσS complex, as previously reported for the formation of the Eσ70 holoenzyme (57). The dissociation curves were biphasic, corresponding to two different populations of σS-E complexes dissociating with markedly different koff rates of 2.5 ± 0.2 × 10-2 s-1 and 2.3 ± 0.3 × 10-3 s-1. The experiment was repeated in the presence of a range of Crl (40 nm to 5 μm), showing that the Kd of the σS-E interaction steadily decreased as a function of Crl concentration, reaching an optimum for 5 μm Crl, with a Kd of 9.41 ± 1.86 nm, more than 7-fold lower than in the absence of Crl. Interestingly, the presence of Crl did not affect the biphasic nature of the σS-E dissociation curves or the corresponding koff rates, indicating that the difference in Kd was primarily due to a 7-fold increase in the association rate (kon) of σS with E (Fig. 2B). A concomitant increase in the maximum steady-state response (Rmax) could be observed, reaching 285 ± 13 RU at [Crl] = 5 μm, against 205 ± 9 RU in the absence of Crl (Fig. 2C). This 1.4-fold increase can be directly correlated to the fact that, at 5 μm Crl, σS should essentially be present in the form of a σS-Crl complex (1.4 times heavier than σS).
FIGURE 2.

Binding of σS to immobilized E. Association and dissociation real time profiles are shown for the following σS concentrations: 0.4 nm (black), 2 nm (red), 10 nm (blue), 50 nm (green), and 250 nm (cyan). A, in the absence of Crl. B, in the presence of 5 μm Crl. C, plot representing the steady-state responses as a function of σS concentration in the presence of five different Crl concentrations.
In contrast, no effect of Crl on the binding of σ70 to E could be detected, regardless of the σ70 concentrations tested (0.25–15 nm; we determined a Kd of 0.3 nm for the σ70-E interaction). The total absence of effect of Crl on σ70 incorporation (Fig. 3B, [σ70] = 0.25 nm) can be compared with its large effect on the formation of EσS (Fig. 3A, [σS] = 5 nm).
FIGURE 3.

Effect of Crl on the binding of either σS or σ70 to E. The sensorgrams shown in black, red, blue, green, and cyan correspond to Crl concentrations of 0, 0.2, 1, 5, and 25 μm. A, σS, 5 nm; B, σ70, 0.25 nm.
Crl Is Able to Bind Directly to the Core Enzyme and to the σS-associated Holoenzyme—Interestingly, we demonstrated that Crl could bind directly to E with a 1:1 stoichiometry and a low affinity (Kd = 13.8 ± 1.8 μm; Fig. 4A). The dissociation of the Crl-E complex was too fast (half-life < 1 s) to determine reliably its dissociation rate. Crl also bound transiently to preformed Eσ70 (Fig. 4B) with a 1:1 stoichiometry and an affinity 2-fold lower than that for E (Kd = 27.5 ± 1.8 μm). In contrast, the interaction between Crl and preformed EσS exhibited several interesting characteristics (Fig. 4C). First, Crl bound to EσS with a stoichiometry of 2:1, with two clearly distinct affinities (Kd1 = 227 ± 27 nm, and Kd2 = 40 ± 9 μm), which can be interpreted as two independent binding events per EσS holoenzyme. Kd1 is 10-fold lower than the Kd of the interaction between Crl and free σS, and could correspond to the binding of Crl to σS incorporated into EσS. The close match of Kd2 with the Kd values determined for the Crl-E and Crl-Eσ70 interactions suggests that it could correspond to the direct binding of Crl to E within EσS. The sensorgrams (Fig. 4C) were best fitted assuming that an isomerization of the ternary Crl-EσS complex occurred after the initial encounter between Crl and EσS. Accordingly, the dissociation curves were biphasic, with two distinct koff rates, 4.1 ± 1.0 × 10-2 s-1 and 4.2 ± 1.3 × 10-3 s-1. The half-life of the ternary Crl-EσS was ∼50 s, more than 15-fold higher than the half-life of the Crl-σS binary complex. Altogether, these data suggest that Crl not only favors the incorporation of σS within EσS but that it could bind to and persist within the EσS holoenzyme long enough to play an active role during the transcription initiation process.
FIGURE 4.

Binding of Crl to E, EσS, and Eσ70. Injection of Crl (1.5–25 μm) over an E (A) or an Eσ70 (B) surface. C, interaction of Crl (0.1–25 μm) with immobilized EσS. D, steady state responses measured on the E, Eσ70, and EσS surfaces as a function of Crl concentration.
Crl Increases the DNA Binding Ability of EσS—We therefore investigated by electrophoretic mobility shift assays whether Crl had an effect on E and EσS binding to a katN promoter fragment, which is transcribed by EσS exclusively in vivo and preferentially in vitro (27, 58). To limit the nonspecific binding of E to the DNA fragment, heparin was added to the Eσ and E solutions, at a very low concentration (0.25 μg/ml), which does not affect the σ-E interactions. In all cases, we used a Crl concentration of 1 μm, at which direct binding of Crl to E is virtually negligible. Interestingly, although we could not detect any effect of Crl on the binding of E, a 2–3-fold increase was observed for EσS binding, even at concentrations of σS (1 μm), at which E should be fully saturated (Fig. 5). This strongly suggests that Crl may selectively promote DNA binding by EσS. To probe the specificity of this effect, we constructed a variant of the katN fragment where both the -10 and the -35 regions were severely mutated; the -35 element was changed from TTGACT to CCAGCT, and the -10 region was changed from CTAATTTTA to GAGCTCGGC. The mutant fragment bound EσS 4–5-fold less than the wild-type fragment, and the positive effect of Crl on this residual binding was lower than that on the specific binding to wild-type katN (Fig. 5).
FIGURE 5.

Band shift analysis of E and EσS binding to wild type (left) or mutant (right) katN-labeled fragments in the presence (lanes 6–9) or absence of Crl (lanes 2–5) at 8 nmE and [σS] = 0(lanes 2 and 6), 8 nm (lanes 3 and 7), 40 nm (lanes 4 and 8), and 1 μm (lanes 5 and 9); lane 1, no protein. A typical autoradiogram is shown. The bands corresponding to free and bound DNAs are indicated, and the percentage of bound DNA is quantified in the histogram below each lane.
Crl Stimulates Isomerization of the EσS-katN Promoter Complex en Route to Open Complex Formation—To investigate EσS open complex formation at the katN promoter, potassium permanganate reactivity experiments (59) were performed at both 20 and 37 °C (Fig. 6, lanes 2–5). At both temperatures, thymines at positions -10 and -11 on the template strand were reactive, indicative of the nucleation of the transcription bubble. At 20 °C, in the presence of Crl (lanes 8 and 10), but not in its absence (lanes 7 and 9), the transcription bubble extended downstream to thymines at positions -5, -4, and -2. This experiment was repeated after incubating the katN promoter and EσS for long periods of time; even after 30 h of incubation at 20 °C, no reactivity of T -5, -4, or -2 could be detected in the absence of Crl. In contrast, after a prolonged incubation time at 37 °C, the patterns of permanganate reactivity were identical, irrespective of the presence or absence of Crl (Fig. 6, lanes 4 and 5). After only 10 min of incubation, a difference between the complexes formed with and without Crl could be noticed, especially in the intensity of the reactive bands (Fig. 6, lanes 2 and 3), suggesting that Crl at 37 °C enhanced the rate of open complex formation but did not affect the extent of the transcription bubble. Altogether, these data demonstrate that at 20 °C, the katN-EσS complex is trapped in a nonfunctional intermediate and that Crl is required to overcome the energetic barrier necessary to facilitate the transcription bubble formation around the transcription start site. Other steps subsequent to the opening of the transcription bubble to position -2 might be required to obtain the fully competent transcription complex. In support of this notion, we observed that the katN-EσS complex formed at 20 °C in the presence of Crl led to only a few run-off transcripts (less than 5% of the amount synthesized at 37 °C).
FIGURE 6.

Potassium permanganate reactivity of the EσS-katN promoter complexes formed at 37 °C (lanes 2–5) and 20 °C (lanes 7–10) with or without Crl (1 μm). Lanes 1 and 6, controls without EσS. Lanes 2, 4, 6, 7, and 9, without Crl. Lanes 1, 3, 5, 8, and 10, with Crl. T, thymine.
DISCUSSION
This study aimed to elucidate the mechanism by which Crl promotes E-σ association and involved the two principal σ factors of Enterobacteria: σ70, the σ factor required for vegetative growth, and σS, the σ factor required for stationary phase survival. Both σ factors exhibit a high degree of sequence similarity and bind to almost identical -35 and -10 promoter elements in vitro (60). Both σ70 and σS consist of four domains, which all have a role in mediating the binding of σ to both E and promoter DNA. However, σ70 is larger than σS and contains an extended σ1.1 and a 265-residue insertion between σ1.1 and region 1.2. These structural differences might explain why σ70 has a higher affinity for E than σS (12, 13, 61).
We used SPR to monitor the binding of Crl to either His12-σ70 or His12-σS tethered on a sensor chip through their His12 moiety. A 1:1 Crl-σS complex formed readily and the equilibrium was characterized by a Kd of ∼2 μm. On the other hand, no Crl-σ70 complex could be observed, setting a minimal threshold of 750 μm for the Kd of a putative Crl-σ70 interaction, a value almost 50-fold higher than the intracellular concentration of σ70 (62). These data clearly show that Crl binds to σS but not to σ70. The inability of Crl to bind to σ70 was unexpected, since previous biochemical studies demonstrated that Crl could activate Eσ70-dependent transcription (30). However, our observation was confirmed by two further experiments. First, σS, but not σ70, bound to His6-Crl tethered to a nickel-nitrilotriacetic acid sensor chip (data not shown); second, Crl interacted with σS, but not with σ70, in a bacterial two-hybrid assay. Furthermore, our results are also fully consistent with the transcriptomic analysis of the crl regulon, where the only genes positively regulated by Crl are all dependent on σS for expression (29). Although we have not excluded that Crl might bind to σ factors other than σ70, the specificity of Crl for σS is reminiscent of that exhibited by the RssB adaptor protein, which specifically binds and targets σS for proteolysis via the ClpXP proteolytic machine (63).
E-σ association involves major rearrangements in both E and σ (42), the most dramatic of which is the unmasking of the σ promoter DNA-binding determinants, which are occluded by extensive interdomain contacts within σ (64). Previous results on the formation of Eσ70 showed that the association mechanism involves a rapid bimolecular encounter step followed by isomerization(s) of the initial complex (57). Our data support a similar mechanism for EσS formation in the presence and absence of Crl. In our study, we showed that Crl induced a 7-fold increase in the affinity of σS for E but had no effect on the affinity of σ70 for E. The effect of Crl originated solely from an increase in the rate of association (kon) of σS, which, at saturating Crl concentrations (5 μm), appeared to bind to E almost exclusively as a σS-Crl complex. No significant effect of Crl could be detected on the rate of dissociation (koff) of the EσS complex. Crl therefore appears to function as a σS-specific chaperone, which most likely favors an “open” conformation of σS with a high E-binding propensity.
At high concentrations ([Crl] > 5 μm), we however observed that Crl could also bind directly and transiently to E or Eσ70 with a 1:1 stoichiometry. At similar concentrations, we consistently showed that the stoichiometry of binding of Crl to the preformed EσS was 2:1 and that two distinct binding phases could be distinguished, which could correspond to the binding of Crl to two independent sites. The low affinity Crl-binding site (Kd2 > 10 μm) is present on E, Eσ70, and EσS and is therefore situated on E itself, whereas the high affinity one (Kd1 ≈ 0.2 μm) is specific to the σS molecule incorporated within EσS. Interestingly, Kd1 is 10-fold higher than the affinity of Crl for free σS. Two explanations can be provided; either the conformation of σS in EσS is more favorable for Crl binding than that of free σS, or the first Crl binding site on EσS involves contacts with both σS and E. If Crl bound simultaneously to both σS and E, one would expect a stabilization of the E-σS-Crl ternary complex, contrary to our observations (Fig. 2). Therefore, our results rather indicate that the isomerization process that σS undergoes after its encounter with E leads to a conformation that is able to form a stable complex with Crl (half-life t½ ≈ 50 s), unlike that with free σS (t½ ≈ 3 s). This long persistence within the EσS holoenzyme opens up the possibility that Crl, beyond its chaperone-like role on the process of incorporation of σS, could also directly influence transcription initiation.
We indeed found that Crl increased 2–3-fold EσS association with the katN promoter fragment at saturating (1 μm) concentrations of σS (Fig. 5). However, a lower Crl-induced increase could also be observed for a katN mutant fragment, suggesting that the stimulatory effect of Crl was not entirely promoter-specific at least under our electrophoretic mobility shift assay conditions. The EσS residual binding to the mutant fragment might be attributed to nonspecific binding (65) or binding to fragment ends (66), as suggested by the faster mobility of the mutant complexes (compare lanes 3–5 and 6–9 in Fig. 5 for wild type and mutant DNAs). Furthermore, all the early stages preceding closed complex formation at the promoter involve nonspecific binding events mediated by electrostatic interactions. Although Crl is not a DNA-binding protein, it could assist EσS DNA binding by masking the negatively charged regions of the holoenzyme.
We also showed that, beyond its partially nonspecific impact in the early steps of promoter search, Crl also exhibits a direct effect on katN promoter melting. Permanganate footprinting experiments revealed that in the absence of Crl, the EσS-katN complexes at 20 °C are locked in an inactive state with only thymines at positions -11 and -12 single-stranded. In the presence of Crl, both the size of the transcription bubble increased (up to thymine at position -2) and productive transcription occurred, suggesting that Crl is also able to directly facilitate transcription bubble formation. This isomerization step, strictly independent of RNA polymerase concentrations, argues for a direct role of Crl in the process of transcription initiation.
Our results show that Crl exerts two widely different positive roles on σS-dependent transcription. Initially, at the onset of stationary phase, the fast binding of Crl to σS may alter the dynamic equilibrium between σS conformations, thereby facilitating the association of σS with E. This response is the most likely to be physiologically relevant when σS levels are low. It ensures significant EσS levels under conditions when σS has to compete with the major σ factor, σ70, for binding to E. Asstationary phase proceeds and EσS levels become more abundant, the ternary EσS-Crl complex could become predominant, and Crl may play a direct role in the pathway from promoter recognition to transcription initiation at some promoters. Different reports have already suggested the existence and functionality of a ternary EσS-Crl complex; Crl is indeed specifically co-isolated with E or EσS in extracts from cells grown in Dulbecco's modified Eagle's medium (67), and cell extracts containing Crl increase the recruitment of EσS at the csgBA promoter (24).
In vivo, Crl levels exceed σS levels throughout the growth phase (29), and in stationary phase, the intracellular concentration of Crl is at least 2–3-fold higher than that of σS (68), implying that it could reach up to 10–15 μm. Thus, although Crl predominantly associates to σS and EσS, significant amounts of excess Crl remain available for binding to other molecules. In this study, we could not detect any binding of Crl to σ70, but we were able to show a transient binding of Crl to E and to Eσ70 at Crl concentrations higher than 1 μm. The nature of the Crl binding site on E and the physiological significance of these results are still unclear. Indeed, we were unable to detect any positive effect of Crl on in vitro σ70-dependent transcription at the katN and lacUV5 promoters, irrespective of temperature, buffer conditions, and the order of the addition of proteins. Besides the stimulatory role of Crl on a fraction of σS-dependent genes (27–29), Crl is known to inhibit the expression of some members of the σ70- and σ54-regulons in the presence of σS (25, 27, 28, 69). This phenomenon most likely results from the Crl-dependent increase in EσS formation. Interestingly, Crl also appears able to regulate a small number of genes in the absence of σS (70, 71). These regulatory effects might result from the interactions of Crl with E (this study) and/or other proteins (70, 72). Whatever the significance of these side effects, the major regulatory functions of Crl are to facilitate E-σS association and to promote the expression of σS-dependent genes.
Acknowledgments
We thank Samita Sally Goyal for performing the β-galactosidase assays and Christelle Talhouarne and Sylviane Hoos for monitoring the first SPR experiments. We thank Henri Buc for helpful suggestions.
This work was supported, in whole or in part, by National Institutes of Health Grant GM53759 (in support of L. F. W.; awarded to Seth A. Darst). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: E, RNA polymerase core enzyme; EσS, σS-associated holoenzyme; SPR, surface plasmon resonance; BACTH, bacterial adenylate cyclase two-hybrid; RU, response units.
A. Kolb, unpublished data.
References
- 1.Hengge-Aronis, R. (2002) Microbiol. Mol. Biol. Rev. 66 373-395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klauck, E., Typas, A., and Hengge, R. (2007) Sci. Prog. 90 103-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lacour, S., and Landini, P. (2004) J. Bacteriol. 186 7186-7195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patten, C. L., Kirchhof, M. G., Schertzberg, M. R., Morton, R. A., and Schellhorn, H. E. (2004) Mol. Genet. Genomics 272 580-591 [DOI] [PubMed] [Google Scholar]
- 5.Weber, H., Polen, T., Heuveling, J., Wendisch, V. F., and Hengge, R. (2005) J. Bacteriol. 187 1591-1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nystrom, T. (2004) Mol. Microbiol. 54 855-862 [DOI] [PubMed] [Google Scholar]
- 7.Becker, G., Klauck, E., and Hengge-Aronis, R. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 6439-6444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou, Y., Gottesman, S., Hoskins, J. R., Maurizi, M. R., and Wickner, S. (2001) Genes Dev. 15 627-637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishihama, A. (2000) Annu. Rev. Microbiol. 54 499-518 [DOI] [PubMed] [Google Scholar]
- 10.Gruber, T. M., and Gross, C. A. (2003) Annu. Rev. Microbiol. 57 441-466 [DOI] [PubMed] [Google Scholar]
- 11.Grigorova, I. L., Phleger, N. J., Mutalik, V. K., and Gross, C. A. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 5332-5337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jishage, M., Iwata, A., Ueda, S., and Ishihama, A. (1996) J. Bacteriol. 178 5447-5451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeda, H., Fujita, N., and Ishihama, A. (2000) Nucleic Acids Res. 28 3497-3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jishage, M., Kvint, K., Shingler, V., and Nystrom, T. (2002) Genes Dev. 16 1260-1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paul, B. J., Ross, W., Gaal, T., and Gourse, R. L. (2004) Annu. Rev. Genet. 38 749-770 [DOI] [PubMed] [Google Scholar]
- 16.Gourse, R. L., Ross, W., and Rutherford, S. T. (2006) J. Bacteriol. 188 4589-4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jishage, M., and Ishihama, A. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 4953-4958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Westblade, L. F., Ilag, L. L., Powell, A. K., Kolb, A., Robinson, C. V., and Busby, S. J. (2004) J. Mol. Biol. 335 685-692 [DOI] [PubMed] [Google Scholar]
- 19.Mitchell, J. E., Oshima, T., Piper, S. E., Webster, C. L., Westblade, L. F., Karimova, G., Ladant, D., Kolb, A., Hobman, J. L., Busby, S. J., and Lee, D. J. (2007) J. Bacteriol. 189 3489-3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patikoglou, G. A., Westblade, L. F., Campbell, E. A., Lamour, V., Lane, W. J., and Darst, S. A. (2007) J. Mol. Biol. 372 649-659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wassarman, K. M. (2007) Mol. Microbiol. 65 1425-1431 [DOI] [PubMed] [Google Scholar]
- 22.Gildehaus, N., Neusser, T., Wurm, R., and Wagner, R. (2007) Nucleic Acids Res. 35 1885-1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnqvist, A., Olsen, A., Pfeifer, J., Russell, D. G., and Normark, S. (1992) Mol. Microbiol. 6 2443-2452 [DOI] [PubMed] [Google Scholar]
- 24.Bougdour, A., Lelong, C., and Geiselmann, J. (2004) J. Biol. Chem. 279 19540-19550 [DOI] [PubMed] [Google Scholar]
- 25.Pratt, L. A., and Silhavy, T. J. (1998) Mol. Microbiol. 29 1225-1236 [DOI] [PubMed] [Google Scholar]
- 26.Robbe-Saule, V., Jaumouille, V., Prevost, M. C., Guadagnini, S., Talhouarne, C., Mathout, H., Kolb, A., and Norel, F. (2006) J. Bacteriol. 188 3983-3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robbe-Saule, V., Lopes, M. D., Kolb, A., and Norel, F. (2007) J. Bacteriol. 189 2976-2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lelong, C., Aguiluz, K., Luche, S., Kuhn, L., Garin, J., Rabilloud, T., and Geiselmann, J. (2007) Mol. Cell Proteomics 6 648-659 [DOI] [PubMed] [Google Scholar]
- 29.Typas, A., Barembruch, C., Possling, A., and Hengge, R. (2007) EMBO J. 26 1569-1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaal, T., Mandel, M. J., Silhavy, T. J., and Gourse, R. L. (2006) J. Bacteriol. 188 7966-7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gruber, T. M., Markov, D., Sharp, M. M., Young, B. A., Lu, C. Z., Zhong, H. J., Artsimovitch, I., Geszvain, K. M., Arthur, T. M., Burgess, R. R., Landick, R., Severinov, K., and Gross, C. A. (2001) Mol. Cell 8 21-31 [DOI] [PubMed] [Google Scholar]
- 32.Lonetto, M., Gribskov, M., and Gross, C. A. (1992) J. Bacteriol. 174 3843-3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young, B. A., Anthony, L. C., Gruber, T. M., Arthur, T. M., Heyduk, E., Lu, C. Z., Sharp, M. M., Heyduk, T., Burgess, R. R., and Gross, C. A. (2001) Cell 29 935-944 [DOI] [PubMed] [Google Scholar]
- 34.Sorenson, M. K., and Darst, S. A. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 16722-16727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campbell, E. A., Muzzin, O., Chlenov, M., Sun, J. L., Olson, C. A., Weinman, O., Trester-Zedlitz, M. L., and Darst, S. A. (2002) Mol. Cell 9 527-539 [DOI] [PubMed] [Google Scholar]
- 36.Murakami, K. S., Masuda, S., Campbell, E. A., Muzzin, O., and Darst, S. A. (2002) Science 296 1285-1290 [DOI] [PubMed] [Google Scholar]
- 37.Murakami, K. S., Masuda, S., and Darst, S. A. (2002) Science 296 1280-1284 [DOI] [PubMed] [Google Scholar]
- 38.Vassylyev, D. G., Sekine, S., Laptenko, O., Lee, J., Vassylyeva, M. N., Borukhov, S., and Yokoyama, S. (2002) Nature 417 712-719 [DOI] [PubMed] [Google Scholar]
- 39.Dombroski, A. J., Walter, W. A., Record, M. T., Jr., Siegele, D. A., and Gross, C. A. (1992) Cell 70 501-512 [DOI] [PubMed] [Google Scholar]
- 40.Gowrishankar, J., Yamamoto, K., Subbarayan, P. R., and Ishihama, A. (2003) J. Bacteriol. 185 2673-2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dombroski, A. J., Walter, W. A., and Gross, C. A. (1993) Genes Dev. 7 2446-2455 [DOI] [PubMed] [Google Scholar]
- 42.Callaci, S., Heyduk, E., and Heyduk, T. (1999) Mol. Cell 3 229-238 [DOI] [PubMed] [Google Scholar]
- 43.Igarashi, K., and Ishihama, A. (1991) Cell 65 1015-1022 [DOI] [PubMed] [Google Scholar]
- 44.Colland, F., Fujita, N., Kotlarz, D., Bown, J. A., Meares, C. F., Ishihama, A., and Kolb, A. (1999) EMBO J. 18 4049-4059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhi, H., and Jin, D. J. (2003) Methods Enzymol. 370 174-180 [DOI] [PubMed] [Google Scholar]
- 46.Gribskov, M., and Burgess, R. R. (1983) Gene (Amst.) 26 109-118 [DOI] [PubMed] [Google Scholar]
- 47.Tanaka, K., Takayanagi, Y., Fujita, N., Ishihama, A., and Takahashi, H. (1993) Proc. Natl. Acad. Sci. U. S. A. 90 3511-3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lederer, H., Mortensen, K., May, R. P., Baer, G., Crespi, H. L., Dersch, D., and Heumann, H. (1991) J. Mol. Biol. 219 747-755 [DOI] [PubMed] [Google Scholar]
- 49.Miller, J. H. (1972) in Experiments in Molecular Genetics (Miller, J. H., ed) pp. 352-355, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
- 50.Fenton, M. S., Lee, S. J., and Gralla, J. D. (2000) EMBO J. 19 1130-1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferguson, A. L., Hughes, A. D., Tufail, U., Baumann, C. G., Scott, D. J., and Hoggett, J. G. (2000) FEBS Lett. 481 281-284 [DOI] [PubMed] [Google Scholar]
- 52.Greive, S. J., Weitzel, S. E., Goodarzi, J. P., Main, L. J., Pasman, Z., and von Hippel, P. H. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 3315-3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pemberton, I. K., and Buckle, M. (1999) J. Mol. Recognit. 12 322-327 [DOI] [PubMed] [Google Scholar]
- 54.Stockley, P. G., Baron, A. J., Wild, C. M., Parsons, I. D., Miller, C. M., Holtham, C. A., and Baumberg, S. (1998) Biosens. Bioelectron. 13 637-650 [DOI] [PubMed] [Google Scholar]
- 55.Karimova, G., Pidoux, J., Ullmann, A., and Ladant, D. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 5752-5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karimova, G., Ullmann, A., and Ladant, D. (2001) J. Mol. Microbiol. Biotechnol. 3 73-82 [PubMed] [Google Scholar]
- 57.Wu, F. Y.-H., Yarbrough, L. R., and Wu, C.-W. (1976) Biochemistry 18 3254-3258 [DOI] [PubMed] [Google Scholar]
- 58.Robbe-Saule, V., Coynault, C., Ibanez-Ruiz, M., Hermant, D., and Norel, F. (2001) Mol. Microbiol. 39 1533-1545 [DOI] [PubMed] [Google Scholar]
- 59.Sasse-Dwight, S., and Gralla, J. D. (1989) J. Biol. Chem. 264 8074-8081 [PubMed] [Google Scholar]
- 60.Gaal, T., Ross, W., Estrem, S. T., Nguyen, L. H., Burgess, R. R., and Gourse, R. L. (2001) Mol. Microbiol. 42 939-954 [DOI] [PubMed] [Google Scholar]
- 61.Colland, F., Fujita, N., Ishihama, A., and Kolb, A. (2002) Genes Cells 7 233-247 [DOI] [PubMed] [Google Scholar]
- 62.Mooney, R. A., and Landick, R. (2003) Genes Dev. 17 2839-2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Studemann, A., Noirclerc-Savoye, M., Klauck, E., Becker, G., Schneider, D., and Hengge, R. (2003) EMBO J. 22 4111-4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sorenson, M. K., Ray, S. S., and Darst, S. A. (2004) Mol. Cell 14 127-138 [DOI] [PubMed] [Google Scholar]
- 65.de Haseth, P. L., Lohman, T. M., Burgess, R. R., and Record, M. T., Jr. (1978) Biochemistry 17 1612-1622 [DOI] [PubMed] [Google Scholar]
- 66.Melancon, P., Burgess, R. R., and Record, M. T., Jr. (1983) Biochemistry 22 5169-5176 [DOI] [PubMed] [Google Scholar]
- 67.Lee, D. J., Busby, S. J., Westblade, L. F., and Chait, B. T. (2007) J. Bacteriol. 190 1284-1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robbe-Saule, V., Carreira, I., Kolb, A., and Norel, F. (2008) J. Bacteriol. 190 4453-4459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schnetz, K. (2002) Microbiology 148 2573-2578 [DOI] [PubMed] [Google Scholar]
- 70.Lelong, C., Rolland, M., Louwagie, M., Garin, J., and Geiselmann, J. (2007) Mol. Cell Proteomics 6 660-668 [DOI] [PubMed] [Google Scholar]
- 71.Dong, T., Kirchhof, M. G., and Schellhorn, H. E. (2008) Mol. Genet Genomics 1 267-277 [DOI] [PubMed] [Google Scholar]
- 72.Arifuzzaman, M., Maeda, M., Itoh, A., Nishikata, K., Takita, C., Saito, R., Ara, T., Nakahigashi, K., Huang, H. C., Hirai, A., Tsuzuki, K., Nakamura, S., Altaf-Ul-Amin, M., Oshima, T., Baba, T., Yamamoto, N., Kawamura, T., Ioka-Nakamichi, T., Kitagawa, M., Tomita, M., Kanaya, S., Wada, C., and Mori, H. (2006) Genome Res. 16 686-691 [DOI] [PMC free article] [PubMed] [Google Scholar]