

Abstract
Of all ligands of the transforming growth factor β superfamily, inhibins and activins are a physiologically relevant pair that are functional antagonists of each other. Activin stimulates whereas inhibin blocks follicle-stimulating hormone biosynthesis and secretion from pituitary gonadotrope cells, and together, inhibin and activin control the pituitary gonadal axis essential for normal reproductive function. Sharing a similar β-subunit, the secretion of inhibin heterodimers (α/β) or activin homodimers (β/β) as mature bioactive ligands depends, in part, on the proteolytic processing of precursor proteins. A short loop regulatory pathway controlling precursor processing and dimer secretion was discovered. Activin stimulates endogenous inhibin α- and βB-subunit mRNA, protein, and proteolytic processing. Simultaneously, activin stimulated the proconvertase furin through a Smad2/3-dependent process. The data provide a mechanism where the regulation of furin and inhibin subunits cooperates in an important positive short feedback loop. This regulatory loop augments the secretion of bioactive mature activin B, as well as inhibin B dimers, necessary for local follicle-stimulating hormone β regulation.
Members of the TGFβ2 superfamily of ligands are vital regulators of diverse cellular processes that include proliferation, differentiation, and migration. These processes are dependent on the proper assembly of dimeric secreted proteins that signal in an autocrine, paracrine, or endocrine manner (1–6). The regulation of pro-ligands in the TGFβ superfamily is an emerging area of interest, particularly because it is related to both developmental and reproductive phenotypes in animals deficient in protein cleavage enzymes (7). Inhibin and activin are unique members of the TGFβ superfamily. First, inhibin is an antagonist to activin signaling and has a more established role as an endocrine feedback modulator of the pituitary (8). The regulation of follicle-stimulating hormone (FSH) biosynthesis and release from the anterior pituitary gonadotrope cells rely heavily on the moment-by-moment regulation by both inhibin and activin from the ovary and pituitary. Second, inhibin and activin share a common β-subunit. Inhibin is the only TGFβ ligand with two dissimilar subunits, a unique α-subunit and one activin β-subunit to produce two isoforms of inhibin: inhibin A (α/βA) and inhibin B (α/βB). The activins are homodimers of two β subunits to give rise to activin A (βA/βA), activin B (βB/βB), or activin AB (βA/βB).
Very little is known about the intracellular assembly, processing, and secretion of inhibin and activin dimers. Mature inhibin and activin ligands are derived from large precursor proteins that undergo a series of steps to release mature bioactive hormones into the extracellular environment in a tightly regulated manner. An early step in the production of inhibin proteins involves the attachment of N-linked oligosaccharides to the inhibin α- and β-subunits in the lumen of the endoplasmic reticulum. We have previously demonstrated that specific oligosaccharide attachments to the subunits direct the differential assembly of inhibin and activin dimers (9). Additionally, misfolded inhibin subunit proteins exhibit secretion defects (9). It is evident that the biosynthesis and secretion of inhibin subunit precursors and mature dimers is a highly controlled process. Our findings led us to question what role proteolytic processing plays in the release of bioactive inhibin and activin ligands. Fig. 1A schematically represents the structures of the inhibin subunits. The consensus recognition motif for proconvertase enzyme cleavage is conserved in each precursor protein. Cleavage of activin A precursor (pro-βA) is necessary for the release of bioactive mature ligand (βA), whereas cleavage of precursor inhibin α-subunit (pro-αN-αC) to mature (αC) protein is not necessary because the precursor proteins have biological activity once secreted from the cell (10). The bioprocessing of inhibin and activin subunits has been given limited attention in favor of studies investigating the expression pattern of the genes and the secretion dynamics of ligands in the reproductive cycles from a variety of animals.
FIGURE 1.

A, schematic diagram of the human inhibin subunits, including the α-, βA-, and βB-subunits. The hatching denotes the mature portion of the subunit. The scissors denote the cleavage site with the amino acid recognition motif. ψ, N-linked oligosaccharides. B, basal expression levels of inhibin α- and βB-subunits in the mouse pituitary and in LβT2 cells determined by real time PCR. C, activin A increases the mRNA levels and protein secretion of the inhibin subunits in LβT2 cells. The cells were treated with activin A (20 ng/ml) for 48 h. The mRNA levels of inhibin α- and βB-subunits were measured using real time PCR following reverse transcription and are shown as a ratio over control. D, detection of activin B and inhibin B dimers following activin A treatment. The medium was trichloroacetic acid-precipitated and subjected to SDS-PAGE under nonreducing conditions. The dimers were detected using a rabbit polyclonal anti-inhibin βB-subunit antibody. E and F, increased secretion and processing of inhibin βB-subunit (E) or the inhibin α-subunit (F) was blocked by follistatin (FST). LβT2 cells were pretreated with follistatin (0, 50, or 250 ng/ml) followed by activin A treatment. The medium from treated cells was trichloroacetic acid-precipitated and subjected to immunoblotting procedures under reducing conditions. G, secretion of inhibin α- and βB-subunits proteins following activin A treatment. The cells were treated with activin A ligand (20 ng/ml) for 48 h. The immunoblot shows trichloroacetic acid-precipitated culture medium under reducing conditions. The proteins were detected with an anti-inhibin βB-subunit antibody and an anti-inhibin α-subunit antibody (I). Densitometric analysis shows an increase in processed mature βB-subunit (H) relative to unprocessed precursor protein. The asterisk depicts statistical significance in precursor processing with increased activin A concentration using two-way analysis of variance, followed by Bonferonni post-tests (p < 0.05). J, an increase in mature α-subunit (αC) was observed with increasing doses of activin A. The bars labeled with different letters are statistically different as determined by Tukey's multiple comparison test (p < 0.05).
Members of a family of higher eukaryotic endoproteases, named proprotein convertases (PCs) (11, 12) are good candidates for endogenous inhibin subunit convertases. As with TGFβ ligands, the inhibin precursor molecules are apparent substrates for furin cleavage. Furin is the mammalian prototype for this family of enzymes. This membrane-associated, calcium-dependent serine protease is primarily concentrated in the trans-Golgi apparatus where precursor substrates containing the RXXR recognition motif are post-translationally converted into active forms (13). Furin is responsible for the maturation of several members of the TGFβ family, including TGFβ1 (14), BMP4 (15), nodal, lefty1 and lefty2 (16, 17), and Müllerian inhibiting substance (18). The inhibin α- and βA-subunit contain this furin recognition motif, and both proteins are believed to be processed by furin. The inhibin βB-subunit cleavage site is instead IRKR and does not contain this exact furin cleavage motif but satisfies the sequence rules that govern cleavage by furin (19).
Although the transcriptional response of LβT2 cells to activin has been demonstrated (20), there have been no reports that evaluate the intracellular assembly and secretion of endogenous inhibin subunits from pituitary gonadotrope cells. Here we sought to identify novel determinants that direct the assembly and secretion of activin homodimers and inhibin heterodimers. We demonstrate enhanced secretion and proteolytic processing of mature activin B and inhibin B following activin A treatment. We show that furin, stimulated by activin A, is a key enzyme responsible for subunit maturation. The data demonstrate a positive feedback loop between inhibin subunits and furin in pituitary gonadotrope cells.
EXPERIMENTAL PROCEDURES
Recombinant Ligands, Plasmids, and Chemical Reagents—Recombinant human activin A was purified as reported previously (21). Recombinant activin B (R & D Systems, Minneapolis, MN) was reconstituted in 0.1% bovine serum albumin in phosphate-buffered saline. The human inhibin α-subunit and βB-subunit cDNAs were provided by Genentech (South San Francisco, CA). The human furin promoter-luciferase construct (pGL2-furin P1) was generously provided by Dr. C. Dubois (Université de Sherbrooke, Quebec, Canada). The furin inhibitor, dec-RVKR-chloromethylketone (CMK) was purchased from Alexis Biochemicals (San Diego, CA).
Cell Lines—An immortalized mouse pituitary gonadotrope cell line, LβT2 cells (kindly provided by Dr. P. L. Mellon, University of California, San Diego, CA) and CHO cells were passaged as described (21, 22). A human colon adenocarcinoma cell line (LoVo cells) was obtained from the ATCC (Manassas, VA). LoVo and human embryonic kidney (HEK 293) cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and 1% antibiotic.
Cell Culture Treatments and Transient Transfections—For experimental treatments, LβT2 cells were plated in a 24-well or 6-well plates 1 day prior to transfection at a confluency of ∼80%. The cells were transfected the next day with the furin P1 promoter reporter construct. After 18 h, the cells were treated with the appropriate ligand at various doses for 24 h in phenol-free, serum-free F12:Dulbecco's modified Eagle's medium (Invitrogen) and then harvested for luciferase assays or RNA isolation. For siRNA transfections, the cells were plated at 60–70% confluency 1 day prior to transfection. The cells were transfected with nonspecific control siRNA or specific siRNAs targeted to each of the Smads (Smad2 and Smad3) for 24 h using Lipofectamine 2000 (Invitrogen). siRNAs targeted to furin (l-044065-01), Smad2 (M-040707-00), and Smad3 (M-040706-00), and a nonspecific control (D-001206-09-00) were purchased from (Dharmacon, Inc., Lafayette, CO). Transfections were performed in Opti-mem medium (Invitrogen). To demonstrate the effect of down-regulation of siRNA oligoduplexes on the furin P1 promoter activity, the cells were treated with control medium, activin A, or activin B at a concentration of 20 and 100 ng/ml, respectively, for 24 h. The furin inhibitor, dec-RVKR-CMK was added to cultures 24 h before treatment with activin ligands. Following the appropriate time points, the cells were harvested for luciferase assays, and the emitted luminescence was measured for 30 s using an AutoLumat Luminometer (Berthold Technologies Co., Oak Ridge, TN). The data are reported as fold induction representative of the means ± S.E. of at least three separate transfection or activin treatment experiments.
RNA Isolation and Real Time PCR Analysis—Total RNA from
LβT2 cells was isolated using the RNeasy Mini kit (Qiagen). RNA samples
(1 μg) were primed with random hexamer primers using the Advantage Reverse
PCR kit (Clontech, Mountain View, CA). On-column DNase digestion was performed
using an RNase-free DNase set (Qiagen). Reverse transcription-PCR was
programmed according to the manufacturer's instructions. From the original
reverse transcription reaction, 1.5 μl was subjected to PCR amplification
in a 25-μl volume with TaqMan Universal PCR Master Mix (Applied Biosystems,
Foster City, CA) under the following conditions: 50 °C hold for 2 min, 95
°C hold for 10 min, the 40 cycles of 95 °C for 15 s, and 60 °C for
1 min. Primer and probe sets were 6-carboxy-fluorescein-labeled and acquired
from Applied Biosystems assays on demand and designed to span intron/exon
borders for inhibin βA (Mm00434338), βB
(Mm01286587), α (Mm00439683), and furin (Mm00440646). The data were
normalized with VIC-labeled glyceraldehyde-3-phosphate dehydrogenase as the
internal control (ΔCt). The threshold levels of
glyceraldehyde-3-phosphate dehydrogenase were not altered by any treatments.
Standard errors of the means from replicates are represented as
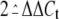 as described (23), which
represents the relative change in gene expression as analyzed by real-time
PCR.
as described (23), which
represents the relative change in gene expression as analyzed by real-time
PCR.
Mutagenesis of Inhibin βB-Subunit Cleavage Site—Site-directed mutagenesis was performed by PCR as described previously (9). The primers used for mutagenesis are listed in Table 1. The mutants were confirmed by DNA sequence analysis at the Northwestern University Biotech Core Facility.
TABLE 1.
Primers used for mutagenesis of inhibin βB-subunit cleavage site
| Mutation | Sequence (5′ → 3′) |
|---|---|
| IRKR → IRKA | atcgcactccaggccagccttgcgaatgcggtgcctgctgtc |
| gacagcaggcaccgcattcgcaaggctggcctggagtgcgat | |
| IRKR → IRAA | atcgcactccaggccagcagcgcgaatgcggtgcctgctgtc |
| gacagcaggcaccgcattcgcgctgctggcctggagtgcgat | |
| IRKR → IAAA | atcgcactccaggccagcagcagcaatgcggtgcctgctgtc |
| gacagcaggcaccgcattgctgctgctggcctggagtgcgat |
Immunoblot Analysis—The cells were lysed, and the media were collected for immunoblotting procedures as described (9). Inhibin subunit antibodies were a kind gift from Dr. W. Vale (The Salk Institute, La Jolla, CA). Phospho-Smad2 (pSmad2, 3101), phospho-Smad3/phospho-Smad1(pSmad3/pSmad1, 9514), and total Smad3 antibodies were purchased from Cell Signaling Technologies (Beverly, MA). A total Smad2 antibody was purchased from Zymed Laboratories Inc. An anti-actin rabbit polyclonal antibody (Sigma) was used as a loading control. Goat anti-rabbit horseradish peroxidase-conjugated secondary antibody was purchased from Zymed Laboratories Inc. Immunoblot results were visualized using an ECL detection reagent (Amersham Biosciences) and exposed at varying time points onto an x-ray film (Eastman Kodak Co.). Densitometric analysis of signals were quantified using the Kodak Imaging Software, version 4.0.1, and are presented as relative density of unprocessed precursor proteins to processed mature inhibin subunits.
Statistics—The values are reported as the means ± S.E. and analyzed using Prism (version 4.0a) (GraphPad Software, Inc., San Diego, CA). Analysis of variance, followed by the appropriate post-test (Tukey's, Bonferroni, or Kruskal-Wallis), was used to evaluate differences between treatments. The statistical significance was reported if p < 0.05.
RESULTS
Expression of Inhibin α-, βA-, and βB-Subunits in Adult Mouse Pituitary and in LβT2 Gonadotrope Cells—In the pituitary, as in many other tissues, activins are produced and act locally to regulate the function of their targets, including gonadotropes and other cell types (24). The expression of inhibin α-, βA-, and βB-subunits in adult mouse pituitary and LβT2 gonadotropes were quantitatively assessed by real time PCR of total RNA from these cells. In our characterization of adult mouse pituitary, real time PCR data showed expression of the inhibin α- and βB-subunits but undetectable levels of the βA-subunit (Fig. 1B). The level of the βB-subunit was 2-fold in excess of the α-subunit. A similar distribution of inhibin subunits was observed in the LβT2 gonadotropes, which also had undetectable levels of βA-subunit mRNA. There is approximately a 3-fold excess of inhibin βB-subunit mRNA to the inhibin α-subunit mRNA in this cell line. Based on this mRNA data, we infer that an excess of βB-subunits results in the assembly of activin B dimers rather than inhibin B dimers in the pituitary.
Activin A Stimulates the Inhibin α- and βB-Subunits in Mouse Pituitary Gonadotrope Cells—We hypothesized that activin augments the expression of inhibin subunits in pituitary cells. To investigate the effect of activin A on the transcription of the inhibin subunits, LβT2 gonadotropes were treated with activin A (20 ng/ml) for 48 h, and RNA was prepared for quantitative real time PCR analysis. Activin A treatment significantly increased the mRNA levels of both the inhibin α- and βB-subunits 6.7- and 14.2-fold, respectively, in LβT2 cells (Fig. 1C and supplemental Fig. S1). The level of glyceraldehyde-3-phosphate dehydrogenase expression was unchanged with activin A treatment. The up-regulation of inhibin βB-subunit mRNA transcripts following activin A treatment was ∼2-fold greater than that of inhibin α-subunit, suggesting that the homodimerization of inhibin βB-subunits to produce activin B (βB/βB) was more likely than the heterodimerization of inhibin B (αC/βB). To confirm this, media from these cells were collected and trichloroacetic acid-precipitated for analysis by SDS-PAGE under nonreducing conditions to determine whether dimeric activin B was in excess of inhibin B with activin A treatment. As shown in Fig. 1D, dimeric forms of activin B were detected with an inhibin βB-subunit antibody after activin A (20 ng/ml) treatment. Inhibin B dimers were observed following treatment with higher concentrations of activin A. These data show that activin B dimers are assembled in excess of inhibin B dimers when cells are treated with 20 ng/ml of activin A.
RNA and conditioned media from LβT2 cells pretreated with follistatin to block both exogenous and endogenous activins were also analyzed. Co-treatment with follistatin abolished the observed increase in inhibin subunit mRNA transcripts (data not shown). Follistatin treatment blocked the increase in protein secretion of the inhibin βB-subunit (Fig. 1E) as well as inhibin α-subunit (Fig. 1F). The data suggest that endogenous mouse inhibin α- and βB-subunits are induced by activin and can be efficiently blocked by follistatin.
The secretion of inhibin subunits following activin A treatment was also assessed under reducing conditions. We analyzed the conditioned media from LβT2 gonadotropes treated with activin A for 48 h. Consistent with the mRNA data, both the inhibin α- and βB-subunits were secreted with activin A treatment (Fig. 1, G and I). This increase correlated with an increase in inhibin pro-βB-subunit and αC accumulation and subunit maturation. Densitometric quantification of immunoreactive mature βB protein over unprocessed protein levels revealed a 1.5-fold increase in pro-βB processing (Fig. 1H, asterisk). The amount of unprocessed pro-βB is not significantly different when stimulated with different concentrations of activin A. An immunoreactive mature αC band was ∼4-fold greater with activin A treatment (20 ng/ml) when compared with no ligand treatment (Fig. 1J). An increase in inhibin subunit maturation prompted us to question whether there was an increase in proconvertase processing enzymes such as furin, because furin was identified as a key relevant TGFβ1-converting enzyme (14).
Effect of Activin A Treatment on Furin mRNA Accumulation—The increase in mature inhibin α- and βB-subunits suggests that activin treatment might regulate the cellular levels of endogenous furin convertase. To test this, LβT2 cells were treated with increasing doses of activin A ligand for 24 h, and RNA was collected for quantitative real time PCR analysis. A concentration-dependent effect of activin A on inhibin α- and βB-subunits was confirmed (Fig. 2, A and B). Additionally, furin mRNA levels increased with activin A treatment, up to a 3.5-fold increase observed with 400 ng/ml of activin A (Fig. 2C).
FIGURE 2.

Dose-dependent effect of activin A on mRNA accumulation for inhibin α-subunit (A), βB-subunit (B), and furin (C). LβT2 cells were treated with varying doses of activin A (Act A) for 24 h as indicated. D–F, time course study of activin A stimulation of the mRNA levels of inhibin α-subunit (D) and βB-subunit (E) and furin (F). LβT2 cells were treated with activin A (100 ng/ml) for different time periods. mRNA levels were measured with real time PCR following reverse transcription. G, effect of TGFβ superfamily ligands on furin mRNA levels. LβT2 cells were treated with activin A, inhibin A (Inh A), BMP3, BMP4, TGFβ-1, -2, and -3 (100 ng/ml of each) for 24 h. Furin mRNA levels were measured with real time PCR following reverse transcription. The results are averages of three independent experiments each performed in triplicate. The bars labeled with different letters are statistically different as determined by Tukey's multiple comparison test (p < 0.05).
We next compared the effect of activin A treatment on these mRNAs at different time points. Stimulation of inhibin α-subunit mRNA levels was observed following 9 h of treatment with activin A (100 ng/ml) (Fig. 2D), whereas a significant increase in inhibin βB subunits was observed by 6 h (Fig. 2E). No significant change in furin mRNA levels were observed until 9 h (Fig. 2F), with a maximum 1.5-fold stimulation occurring at 24 h. The level of glyceraldehyde-3-phosphate dehydrogenase expression was unchanged with activin A treatment. Our results demonstrate that activin A can augment the mRNA levels of the inhibin α- and βB-subunits as well as furin. Treatment of LβT2 cells with actinomycin D (RNA synthesis inhibitor) or cycloheximide (protein synthesis inhibitor) abolished the increase in furin mRNA following activin A treatment (supplemental Fig. S2). These data confirm that the regulation of endogenous furin mRNA levels by activin is at the transcriptional level.
Because many TGFβ superfamily ligands are processed by furin, we sought to test whether other ligands of this family could regulate cellular levels of furin convertase mRNA. LβT2 cells were treated with activin A, inhibin A, bone morphogenic proteins (BMP3 and BMP4), and TGFβ-1, -2, and -3 ligands for 24 h, and furin mRNA levels were measured by PCR (Fig. 2G). Inhibin A, BMP, and TGFβ ligands had no significant stimulatory effect on furin mRNA levels. These data demonstrate that in the mouse pituitary gonadotrope cell line, only activin A can regulate endogenous furin mRNA levels.
Relationship between Furin Gene Expression and Mature Inhibin α- and βB-Subunits—We next investigated the relationship between furin and inhibin subunit processing. LβT2 cells were pretreated with a potent furin and PC inhibitor, decanoyl-RVKR-CMK for 1 h. The cells were then treated with different concentrations of activin A for 48 h. Medium was collected, trichloroacetic acid-precipitated, and subjected to Western blot analysis under reducing conditions. As shown in Fig. 3, increasing concentrations of activin A resulted in the enhanced secretion of both inhibin α-subunit (Fig. 3A) and inhibin βB-subunits (Fig. 3B). Treatment with CMK inhibited the ability of all proconvertase enzymes to cleave the inhibin subunits. Accumulation of unprocessed proteins suggests that this inhibitor affects cleavage and not secretion. These results imply a direct relationship between activin A-dependent expression of furin and the maturation of the inhibin subunits expressed and secreted from LβT2 gonadotropes.
FIGURE 3.

Involvement of processing enzymes in the maturation and secretion of inhibin α- and βB-subunits in LβT2 cells. The cells were cultured for 48 h in the presence or absence of increasing doses of activin A and the furin inhibitor dec-RVRK-CMK (CMK). Trichloroacetic acid-precipitated media were separated by SDS-PAGE and immunoblotted using an anti-inhibin α-subunit antibody (A) and an anti-inhibin βB-subunit antibody (B). The results are representative of three independent experiments. DMSO, dimethyl sulfoxide.
To assess whether the inhibin α- and βB-subunits are targets for processing by furin, lysates from CHO cells transfected with the human inhibin α-subunit or the βB-subunit were subjected to an in vitro digestion assay using recombinant furin. These lysates contain an abundance of inhibin precursor proteins that are not completely cleaved by the proteoloytic enzymes endogenously expressed in CHO cells. Inhibin subunits were incubated with purified furin in the solution and proteolytic cleavage of the precursor was assayed after 24 h of incubation at 37 °C. As shown in the left panel of Fig. 4A, inhibin α-subunit and inhibin βB-subunit precursors incubated in the presence of recombinant furin were cleaved to their respective mature forms. The furin-deficient LoVo cell line (25) was utilized to determine whether overexpression of a furin expression vector would cleave the inhibin subunits. LoVo cells were transiently transfected with the inhibin α- and βB-subunits and a furin expression vector. Overexpression of furin results in the cleavage of the pro-αN-αC and pro-βB proteins (Fig. 4A, right panel). The size of these mature products is consistent with cleavage at the minimal furin consensus sequence in each protein sequence. Together, these data confirm that furin can cleave the inhibin subunits.
FIGURE 4.

The inhibin α- and βB-subunits are targets of furin processing. A, processing of subunits in CHO and furin-deficient LoVo cells. Lysates of CHO cells transfected with inhibin α-subunit (top panels) or inhibin βB-subunit (lower panels) were harvested and treated with and without recombinant furin for 24 h and then examined by immunoblot analysis. LoVo cells were transiently transfected with inhibin α-subunit or inhibin βB-subunit in the presence or absence of an expression vector for furin. Media from these cells were then trichloroacetic acid-precipitated and examined by immunoblot analysis. B, expression of the wild type inhibin βB-subunit (IRKR) or the βB-subunit with modified cleavage recognition sites (IRKA, IRAA, and IAAA) in HEK 293 cells. The media were examined by immunoblot analysis using an anti-inhibinα-subunit antibody. C, LβT2 were transfected with furin siRNA to furin. The cells were treated with activin A (Act A, 0, 20, 100 ng/ml) for 24 h. RNA was harvested from LβT2 cells transfected with furin siRNA and treated with activin A to quantify the level of furin gene silencing from siRNA. The media were examined by immunoblot analysis using an anti-inhibin α-subunit antibody (D) and an anti-inhibin βB-subunit antibody (E).
To verify that the predicted IRKR site in the inhibin βB-subunit is the proteolytic processing site, the cleavage motif was mutated. Three βB-subunit mutants (IRKA, IRAA, and IAAA) were generated and expressed in HEK 293 cells. Mutagenesis of Arg292 in the IRKA mutant displayed ∼40% total cleavage activity, and unprocessed precursor protein accumulated (Fig. 4B). The IRAA and IAAA mutants had very low to undetectable levels of mature βB-subunit. These results confirm that cleavage of the βB-subunit occurs at the consensus furin motif and the requirement of basic residues at specific positions.
The effects of furin inhibition were also assessed using mouse furin-specific siRNA oligoduplexes. LβT2 cells were transiently transfected with furin siRNA followed by treatment with activin A for 24 h. RNA was harvested from these cells, and the specificity of the furin siRNA duplex was assessed by real time PCR. We determined that endogenous furin mRNA levels were decreased by 72% with the furin siRNA following activin A treatment, and no significant decrease in endogenous furin mRNA was observed with nonspecific control siRNA oligoduplexes (Fig. 4C). The medium collected from these cells was also analyzed by immunoblot analysis, and the knockdown of endogenous furin mRNA resulted in decreased mature inhibin αC (Fig. 4D) as well as mature βB-subunit (Fig. 4E). Similar to the CMK experiments presented in Fig. 6, these data demonstrate that furin is a key processing enzyme for the inhibin α- and βB-subunits.
FIGURE 6.

Furin action in pituitary gonadotrope cells. Activin and furin participate in a positive feedback loop. Activin stimulation of pituitary gonadotrope cells stimulates the production of both activin B and inhibin B dimers. A differential induction of activin B (bold arrows) over inhibin B (thin arrow) is observed with activin A treatment. Only activin can stimulate the endogenous expression of furin mRNA through a Smad-dependent pathway. The increased levels of furin lead to an increase in both inhibin α- and βB-subunit protein maturation and secretion.
Activins Increase Furin Gene Activity and Stimulates the Furin Promoter—To further understand the regulation of the furin gene by activin, a vector with the human furin promoter region upstream of a luciferase reporter gene (pGL2-furin P1) was utilized. LβT2 cells were transiently transected with the pGL2-furin P1 luciferase reporter construct. The cells were treated with either activin A or activin B ligand for 24 h. We observed a dose-dependent effect with activin A treatment, where at 5 ng/ml maximal luciferase activity was observed with a 5-fold increase over unstimulated controls (Fig. 5A). Activin B could not as efficiently activate the furin-luciferase reporter construct, and a dose of 20 ng/ml resulted in only a 2.7-fold increase over unstimulated controls (Fig. 5B). This may be due to the fact that activin B and follistatin are endogenously expressed within these cells. These results suggest that the activin signal transduction pathway transactivates the furin gene.
FIGURE 5.

Activin transactivates the human furin promoter in a Smad2- and Smad3-dependent pathway in LβT2 cells. A, cells were transiently transfected with pGL2 or pGL2-furin P1 construct. Transfected cells were then treated with varying doses of activin A (A) or activin B (B) for 24 h. C, LβT2 cells were transiently transfected with pGL2 or pGL2-furin P1 construct and siRNA specific for Smad2 or Smad3 or a nonspecific control siRNA overnight. Transfected cells were then treated with activin A (5 ng/ml) or activin B (20 ng/ml) (D) for 24 h. Luciferase activity was determined and expressed as fold induction relative to the control vector alone. The data are expressed as the means ± S.E. of three independent experiments performed in triplicate. Bars with different letters indicate statistical differences, p < 0.05. E, Western blot analysis was performed the show the specificity and degree of knockdown of phosphorylated and total Smad2 or Smad3 protein by siRNA. Actin was used as a loading control of cell lysates for these immunoblots.
Involvement of Smads in Activin-induced Furin Transcription—To gain insight into the molecular mechanism underlying the furin mRNA up-regulation following activin stimulation, we analyzed the involvement of Smad regulatory proteins. Previous work demonstrated the presence of three Smad-binding elements in the furin promoter known to be responsive to Smad2/4 and Smad3/4 complexes (26). Here we investigated whether endogenous Smad2 and Smad3 are necessary for the activin-induced activation of the furin promoter in LβT2 cells. Smad2 and Smad3 siRNA were utilized to individually suppress these two Smad molecules in LβT2 cells. The cells were transiently co-transfected with siRNA oligoduplexes to either Smad2 or Smad3 and the furin P1-luc reporter construct. The cells were then treated with activin A or activin B ligands for 24 h, and luciferase activity was assessed. The results of the luciferase assays show that the down-regulation of Smad2 or Smad3 by siRNA reduced activin-mediated induction of the furin promoter back to unstimulated control levels (Fig. 5, C and D). These data suggest that the receptor-restricted Smads are both necessary to mediate the activin-induced transactivation of the furin promoter. Specificity of Smad2 or Smad3 knockdown was assessed by immunoblot analysis of phosphorylated Smad2 and phosphorylated Smad3, which are the activated forms of Smad proteins that mediate activin signaling (Fig. 5, E and F, respectively). An antibody to actin protein was utilized as a loading control for cell lysates. Quantitation of protein band intensity after normalization against the actin controls revealed that endogenous phosphorylated Smad2 protein was decreased by 54%, whereas phosphorylated Smad3 protein was decreased by 57% with Smad2 and Smad3 siRNA, respectively. Total Smad proteins were also examined following siRNA transfection, and Smad2 and Smad3 proteins were reduced to 42 and 45%, respectively. We observed a significant decrease in furin-reporter activation with Smad2 and Smad3 siRNA, implying that different promoter sites may bind different Smad proteins. These results suggest that Smad2 and Smad3 are both indispensable for the activin-induced transcriptional activation of the furin promoter in LβT2 cells.
DISCUSSION
The gonadotrope cells of the anterior pituitary synthesize and secrete FSH, a major physiological regulator of the female reproductive axis. Activin is the main positive regulator of FSHβ subunit expression, where inhibin dimers target these cells and antagonize the actions of activin. In this report, we provide evidence that of all TGFβ ligands tested, only activin stimulates the biosynthesis, secretion, and proteolytic maturation of the inhibin α- and β-subunits. This is, in part, due to an activin-induced increase in furin expression. Overall, these data provide a mechanism where the regulation of furin and inhibin subunits cooperate in a novel positive feedback loop that augments the regulation and secretion of bioactive mature activin B and inhibin B dimers necessary for local FSHβ regulation.
We have utilized the immortalized mouse pituitary gonadotrope cell line, LβT2, to investigate the molecular mechanisms regulating the post-translational processing of inhibin and activin proproteins. Although a previous report has also demonstrated that the inhibin α- and βB-subunit genes are induced with activin treatment in LβT2 cells (20), no reports have shown that activin can stimulate the proprotein convertase, furin mRNA. Real time PCR experiments have shown that of the TGFβ superfamily ligands tested in our assay, only activin A positively regulates furin mRNA levels. BMP4 is proteolytically activated by furin (15); however, BMP ligands do not regulate furin expression in mouse pituitary gonadotrope cells that express both BMP type I and type II receptors (27). Although it has been demonstrated that TGFβ1 can regulate furin gene expression in rat synoviocytes (28), this ligand cannot regulate furin mRNA in mouse pituitary gonadotrope cells because LβT2 cells lack endogenous TGFβ type II receptor and therefore are not responsive to TGFβ ligand (29). This selective auto-regulation of furin by activin suggests that furin plays an important role in regulation of hormone biosynthesis and secretion from pituitary gonadotrope cells. Additionally, it provides evidence that furin may be involved in the increased bio-availability of FSHβ mediators such as mature inhibin and activin dimers.
Our results with the furin inhibitor CMK, furin-deficient LoVo cells, and furin siRNA oligoduplexes establish that the inhibin α- or βB-subunits are targets of furin processing and that furin is one of the key enzymes responsible for the cleavage and maturation of the inhibin α- and the βB-subunits observed by immunoblotting procedures. Our results confirm that cleavage occurs at the consensus furin motifs. Furthermore, we predict that the consensus motif is also a cleavage site for other proconvertase proteins in different cells.3 Inhibin βB production following pituitary stimulation with activin A suggests that its processing is tightly regulated in these cells in an autocrine manner. Endogenous furin expression may be the limiting factor in the amounts of mature bioactive activin B the gonadotrope cells synthesize.
We have shown that furin is a novel activin transcriptional target in LβT2 pituitary gonadotrope cells. The furin P1 promoter construct is transactivated by both activin isoforms, A and B. To understand the molecular mechanism involved in the activin regulation of the furin promoter, we assessed the involvement of Smad regulatory proteins. The furin P1 proximal promoter region contains SMAD-binding elements (26). The down-regulation of both Smad2 and Smad3 by specific siRNA oligoduplexes abolished the effect of activin on the furin promoter. Activin-mediated stimulation of the endogenous furin and the furin P1-luciferase reporter gene is dependent on both the intracellular Smads. The furin gene is only regulated by activin ligands in pituitary gonadotrope cells and not any other TGFβ ligands and provides an additional level of control in the regulation of pituitary function and hormone biosynthesis that has not been previously discovered with respect to reproductive functions.
Few reports have uncovered the physiological functions of proprotein convertases in the regulation of female reproductive function (12). The generation of null mice for the furin gene results in embryonic lethality approximately at embryonic day 11. Embryos failed to undergo axial rotation and ventral closure (32). Defects in embryo implantation were observed when the PC5/6 gene, PCSK5, was inactivated, resulting in death at embryonic days 4.5–6.5 (33). The PC1 plays a crucial role in the post-translational modification of prohormones and neuropeptides, and a compound heterozygous mutation in PC1 has been described in a woman with obesity, hypocortisolemia, and hypogonadotropic hypogonadism (34). The reproductive defect in this case may be the result of impaired gonadotropin-releasing hormone processing and/or abnormalities in neuropeptides related to GnRH secretion. Our data suggest a novel correlation between the proprotein convertase, furin, and regulation of the reproductive hormones, inhibin and activin, that are vital regulators of the pituitary-gonadal axis (Fig. 6).
In conclusion, our results identify furin as a tightly regulated activin target gene. Furin may be an important modulator of the inhibin subunit maturation and activation involved in autocrine signaling in the pituitary gonadotrope cell line, LβT2. The activin-specific regulation of furin could represent a novel intracellular mechanism by which activin regulates the reproductive processes involved in cell proliferation, differentiation, and hormone biosynthesis. We predict that the biosynthesis and secretion of the inhibin α- and β-subunits are tightly regulated and finely tuned and that proteolytic processing is an additional step important for the actions of these hormones.
Supplementary Material
Acknowledgments
We thank Dr. I. Boime (Washington University School of Medicine) and Dr. K. Mayo (Northwestern University) for critical discussion in the preparation of this manuscript, Dr. C. M. Dubois (Université de Sherbrooke) for the furin promoter plasmid, Dr. P. L. Mellon (University of California, San Diego) for LβT2 cells, and Dr. W. Vale (The Salk Institute) for rabbit polyclonal antibodies to the inhibin subunits.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 HD37096. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: TGF, transforming growth factor; FSH, follicle-stimulating hormone; BMP, bone morphogenetic protein; PC, proprotein convertase; CMK, dec-RVKR-chloromethylketone; CHO, Chinese hamster ovary; siRNA, small interfering RNA.
M. Antenos and T. K. Woodruff, manuscript in preparation.
References
- 1.Woodruff, T. K., Lyon, R. J., Hansen, S. E., Rice, G. C., and Mather, J. P. (1990) Endocrinology 127 3196–3205 [DOI] [PubMed] [Google Scholar]
- 2.Mather, J. P., Attie, K. M., Woodruff, T. K., Rice, G. C., and Phillips, D. M. (1990) Endocrinology 127 3206–3214 [DOI] [PubMed] [Google Scholar]
- 3.Schwartz, N. B., and Channing, C. P. (1977) Proc. Natl. Acad. Sci. U. S. A. 74 5721–5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorenzen, J. R., Dworkin, G. H., and Schwartz, N. B. (1981) Am. J. Physiol. 240 E209–E215 [DOI] [PubMed] [Google Scholar]
- 5.de Kretser, D. M., and Robertson, D. M. (1989) Biol. Reprod. 40 33–47 [DOI] [PubMed] [Google Scholar]
- 6.Rivier, C., Meunier, H., Roberts, V., and Vale, W. (1990) Recent Prog. Horm. Res. 46 231–257 [DOI] [PubMed] [Google Scholar]
- 7.Thomas, G. (2002) Nat. Rev. Mol. Cell. Biol. 3 753–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling, N., Ying, S. Y., Ueno, N., Esch, F., Denoroy, L., and Guillemin, R. (1985) Proc. Natl. Acad. Sci. U. S. A. 82 7217–7221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antenos, M., Stemler, M., Boime, I., and Woodruff, T. K. (2007) Mol. Endocrinol. 21 1670–1684 [DOI] [PubMed] [Google Scholar]
- 10.Mason, A. J., Farnworth, P. G., and Sullivan, J. (1996) Mol. Endocrinol. 10 1055–1065 [DOI] [PubMed] [Google Scholar]
- 11.Steiner, D. F. (1998) Curr. Opin. Chem. Biol. 2 31–39 [DOI] [PubMed] [Google Scholar]
- 12.Scamuffa, N., Calvo, F., Chretien, M., Seidah, N. G., and Khatib, A. M. (2006) FASEB J. 20 1954–1963 [DOI] [PubMed] [Google Scholar]
- 13.Molloy, S. S., Anderson, E. D., Jean, F., and Thomas, G. (1999) Trends Cell Biol. 9 28–35 [DOI] [PubMed] [Google Scholar]
- 14.Dubois, C. M., Blanchette, F., Laprise, M. H., Leduc, R., Grondin, F., and Seidah, N. G. (2001) Am. J. Pathol. 158 305–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui, Y., Jean, F., Thomas, G., and Christian, J. L. (1998) EMBO J. 17 4735–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Constam, D. B., and Robertson, E. J. (2000) Genes Dev. 14 1146–1155 [PMC free article] [PubMed] [Google Scholar]
- 17.Constam, D. B., and Robertson, E. J. (2000) Development 127 245–254 [DOI] [PubMed] [Google Scholar]
- 18.Nachtigal, M. W., and Ingraham, H. A. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 7711–7716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakayama, K. (1997) Biochem. J. 327 625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang, H., Bailey, J. S., Coss, D., Lin, B., Tsutsumi, R., Lawson, M. A., Mellon, P. L., and Webster, N. J. (2006) Mol. Endocrinol. 20 2909–2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pangas, S. A., and Woodruff, T. K. (2002) J. Endocrinol. 172 199–210 [DOI] [PubMed] [Google Scholar]
- 22.Suszko, M. I., Lo, D. J., Suh, H., Camper, S. A., and Woodruff, T. K. (2003) Mol. Endocrinol. 17 318–332 [DOI] [PubMed] [Google Scholar]
- 23.Livak, K. J., and Schmittgen, T. D. (2001) Methods 25 402–408 [DOI] [PubMed] [Google Scholar]
- 24.Bilezikjian, L. M., Blount, A. L., Leal, A. M., Donaldson, C. J., Fischer, W. H., and Vale, W. W. (2004) Mol. Cell. Endocrinol. 225 29–36 [DOI] [PubMed] [Google Scholar]
- 25.Takahashi, S., Kasai, K., Hatsuzawa, K., Kitamura, N., Misumi, Y., Ikehara, Y., Murakami, K., and Nakayama, K. (1993) Biochem. Biophys. Res. Commun. 195 1019–1026 [DOI] [PubMed] [Google Scholar]
- 26.Blanchette, F., Rudd, P., Grondin, F., Attisano, L., and Dubois, C. M. (2001) J. Cell. Physiol. 188 264–273 [DOI] [PubMed] [Google Scholar]
- 27.Nicol, L., Faure, M. O., McNeilly, J. R., Fontaine, J., Taragnat, C., and McNeilly, A. S. (2008) J. Endocrinol. 196 497–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blanchette, F., Day, R., Dong, W., Laprise, M. H., and Dubois, C. M. (1997) J. Clin. Investig. 99 1974–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suszko, M. I., and Woodruff, T. K. (2006) J. Mol. Endocrinol. 36 591–600 [DOI] [PubMed] [Google Scholar]
- 30.Deleted in proof
- 31.Deleted in proof
- 32.Roebroek, A. J., Umans, L., Pauli, I. G., Robertson, E. J., van Leuven, F., Van de Ven, W. J., and Constam, D. B. (1998) Development 125 4863–4876 [DOI] [PubMed] [Google Scholar]
- 33.Essalmani, R., Hamelin, J., Marcinkiewicz, J., Chamberland, A., Mbikay, M., Chretien, M., Seidah, N. G., and Prat, A. (2006) Mol. Cell. Biol. 26 354–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson, R. S., Creemers, J. W., Ohagi, S., Raffin-Sanson, M. L., Sanders, L., Montague, C. T., Hutton, J. C., and O'Rahilly, S. (1997) Nat. Genet. 16 303–306 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.