

Abstract
Two novel mutations (G159D and L29Q) in cardiac troponin C (CTnC) associate their phenotypic outcomes with dilated (DCM) and hypertrophic cardiomyopathy (HCM), respectively. Current paradigms propose that sarcomeric mutations associated with DCM decrease the myofilament Ca2+ sensitivity, whereas those associated with HCM increase it. Therefore, we incorporated the mutant CTnCs into skinned cardiac muscle in order to determine if their effects on the Ca2+ sensitivities of tension and ATPase activity coincide with the current paradigms and phenotypic outcomes. The G159D-CTnC decreases the Ca2+ sensitivity of tension and ATPase activation and reduces the maximal ATPase activity when incorporated into regulated actomyosin filaments. Under the same conditions, the L29Q-CTnC has no effect. Surprisingly, changes in the apparent G159D-CTnC Ca2+ affinity measured by tension in fibers do not occur in the isolated CTnC, and large changes measured in the isolated L29Q-CTnC do not manifest in the fiber. These counterintuitive findings are justified through a transition in Ca2+ affinity occurring at the level of cardiac troponin and higher, implying that the true effects of these mutations become apparent as the hierarchical level of the myofilament increases. Therefore, the contractile apparatus, representing a large cooperative machine, can provide the potential for a change (G159D) or no change (L29Q) in the Ca2+ regulation of contraction. In accordance with the clinical outcomes and current paradigms, the desensitization of myofilaments from G159D-CTnC is expected to weaken the contractile force of the myocardium, whereas the lack of myofilament changes from L29Q-CTnC may preserve diastolic and systolic function.
Cardiomyopathies are diseases of the myocardium that often lead to cardiac remodeling to compensate for deficiencies in cardiac output (1). In the case of dilated cardiomyopathy (DCM),2 heart failure is characterized by a systolic dysfunction (i.e. reduced ejection fraction), whereas hypertrophic (HCM) and restrictive cardiomyopathies are characterized as having diastolic dysfunctions (i.e. impaired relaxation) (2). In many cases, the cardiac contractile dysfunction is attributed to inherited sarcomeric gene mutations. The functional effects of more than 40 thin filament mutations associated with cardiomyopathies assessed in vitro suggest that these mutations affect the Ca2+ responsiveness of the myofilament in the absence of CTnI phosphorylation. As the number of in vitro-characterized mutations continues to grow, a developing paradigm emerges that associates decreases in the myofilament Ca2+ sensitivity with DCM (3–7) and associates increases with HCM (4, 6–12) and restrictive cardiomyopathy (13–17). This suggests that distinct effects on the Ca2+-dependent processes of the myofilament are critical determinants of the severity and molecular pathologies of these diseases.
The first cardiac troponin C (CTnC) mutation (E59D/D75Y) was found in an explanted heart from an adult male who died from idiopathic DCM (18); the second mutation (L29Q) was found in a living 60-year-old male diagnosed with HCM, despite having preserved diastolic and systolic function (19); and the third (G159D) was found by linkage analysis and cosegregation studies in more than three affected families with DCM (20, 21). The lack of affected relatives and cosegregation studies associated with the L29Q and E59D/D75Y CTnC mutations may or may not confirm these mutations as etiological causes for the diseases; therefore, the functional and biochemical effects of these mutations must be studied in order to predict the physiological consequences of these mutations. As such, our investigations will relate the changes in apparent CTnC Ca2+ affinity to the myofilament response and determine if these mutations coincide with the developing paradigm. The effects of the E59D/D75Y mutation were previously reported to coincide with the aforementioned paradigms (18, 22, 23); therefore, this paper will focus on the L29Q and G159D CTnC mutations.
Cardiac muscle contraction and relaxation is regulated by the intracellular
Ca2+ concentration via the thin filament regulatory protein, CTnC.
During systole, the rise in free calcium promotes Ca2+ binding to
CTnC. This leads to the translocation of CTnI and tropomyosin (Tm) away from
the outer domain of the actin filaments, allowing for subsequent cross-bridge
interaction and the generation of tension. With the decline of Ca2+
during diastole, Ca2+ dissociates from CTnC, and the inhibitory
actions of CTnI and Tm are restored (see Refs.
24 and
25 for a review). CTnC
contains three metal ion binding sites: two C-terminal sites that bind
Ca2+ and Mg2+ (“Ca2+-Mg2+
sites”) competitively (KCa = 1.4 ×
107 m–1;
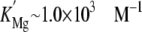 )
and one N-terminal, Ca2+-specific regulatory site
(KCa = 2.5 × 105
m–1) that is responsible for transmitting the
Ca2+ binding signal to the rest of the thin filament and switching
on contraction (26,
27).
)
and one N-terminal, Ca2+-specific regulatory site
(KCa = 2.5 × 105
m–1) that is responsible for transmitting the
Ca2+ binding signal to the rest of the thin filament and switching
on contraction (26,
27).
The effects of the L29Q and G159D mutations are of particular interest, because previous studies show that the apparent CTnC Ca2+ affinity as measured by tension and ATPase activity is unaffected unless CTnI (Ser23/Ser24) is phosphorylated (28–30). These studies identified a blunted protein kinase A-phosphorylated response as an underlying molecular pathology for these mutations. However, these findings represent challenges to the developing paradigm, because in the presence of phosphorylated CTnI, the myofilament Ca2+ sensitivities are similarly affected for both mutations yet lead to different effects on the structural opening of the CTnC N-lobe (31) and phenotypic outcomes (19, 20). In addition, the clinical data from the only L29Q proband results in a seemingly benign phenotype (19); however, subsequent in vitro studies have shown that in the absence of phosphorylated CTnI, the L29Q mutation can decrease (30), increase (32), or not affect (33) the myofilament Ca2+ sensitivity.
Our results show that in the absence of phosphorylated CTnI, skinned cardiac muscle preparations reconstituted with L29Q CTnC are all unaffected, possibly explaining the benign phenotypic outcome observed from this mutation. In contrast, the G159D mutation significantly decreases the Ca2+ sensitivities of tension and cardiac myofibrillar ATPase activation, coinciding with the effects of other DCM-associated thin filament mutations (6, 34–36). Surprisingly, the isolated G159D CTnC structure and apparent regulatory site Ca2+ affinity (as measured by IAANS fluorescence) are seemingly unaffected, whereas the structure and Ca2+ affinity of L29Q CTnC are significantly altered. The disparity of measurements between the isolated CTnC and skinned muscle have led us to pursue the key intermolecular interactions that dictate the final Ca2+ sensitivity of tension in the muscle fiber. Our results show that at the level of CTn, the G159D CTnC structure and Ca2+ affinity deviates from the WT, whereas the L29Q CTnC structure and Ca2+ affinity begin to converge with the WT. Therefore, the contractile apparatus, representing a large cooperative machine, can provide the potential for a change (G159D) or no change (L29Q) in the myofilament Ca2+ sensitivity if any of the proteins are mutated. Finally, our results show that the changes in myofilament Ca2+ sensitivity arising from these mutations do coincide with the developing paradigms and are indicative of the phenotypic outcomes.
EXPERIMENTAL PROCEDURES
Mutation, Expression, and Purification of CTnC—The CTnC sequence was derived from a human cardiac cDNA library and subcloned into the pET3-d vector with its sequence verified. Human CTnC mutants were generated by following Stratagene's guidelines for using the QuikChange site-directed mutagenesis kit. All mutant CTnCs were sequenced to verify the correct sequences prior to expression and purification. Standard methods previously used in this laboratory were utilized for expression and purification of the different CTnC mutants (37).
Fluorescence Labeling of CTnC—Monocysteine CTnC derivatives were engineered using cDNAs previously cloned for G159D, L29Q, and WT CTnC by substituting Cys35 for Ser in order to direct specific IAANS incorporation to Cys84 (denoted -CTnC(C84)IA). The CTnCs were also doubly labeled with IAANS at Cys35 and Cys84 (denoted -CTnCIA). Fluorescent incorporation and subsequent purification of labeled CTnC followed previous methods (38).
Purification of Tropomyosin—Porcine cardiac Tm was purified from the ammonium sulfate precipitate obtained in the course of native cardiac troponin subunit preparations (39). Briefly, the Tm was purified by isoelectric precipitation followed by ion exchange chromatography (Q-Sepharose). Purified Tm was dialyzed against 5 mm NH4(CO3)2, lyophilized, and stored at –20 °C.
Formation of Cardiac Troponin Complexes—cDNAs cloned in our laboratory from human cardiac tissue were used for the expression and purification of CTnI (40) and CTnT (39). Formation of fluorescent binary and ternary complexes and non-labeled ternary complexes was carried out using recently established protocols (37, 41) in the presence of 1.25 mm Mg2+.
Preparation of IAANS-labeled Regulated Thin Filaments—Regulatory complexes (actin-Tm-CTn) were prepared by mixing F-actin isolated from rabbit skeletal acetone powder (42) and Tm in a 7:1 molar ratio in a solution containing 60 mm KCl, 0.1 mm CaCl2, 2 mm MgCl2, 1 mm ATP, pH 7.0, on ice. The final concentration of actin was ∼0.8 mg/ml. After homogenization in a glass homogenizer, the F-actin-tropomyosin complex was combined with preformed IAANS-labeled CTn complex (tropomyosin/CTn = 1 mol/mol) in a solution containing 100 mm MOPS, 75 mm KCl, 1.25 mm MgCl2, 2 mm EGTA, 4 mm nitrilotriacetic acid (standard buffer conditions for our fluorometric titrations). After homogenization, the mixture was allowed to sit at room temperature for 10 min. The homogenate was centrifuged for 1 h at 150,000 × g, and the pellet resuspended in standard fluorescence buffer. The solution was centrifuged and resuspended again in order to avoid free Tm-CTn and free CTn trapped in the pellet. Regulated thin filaments (RTFs) were filtered through a 0.45-μm filter and stored on ice ready to use for experiments.
Determination of Apparent Ca2+ Affinities—Labeled proteins (isolated CTnC and binary and ternary complex) were dialyzed exhaustively in standard fluorescence buffer (see above). The isolated CTnCs were dialyzed in the absence of Mg2+ to prevent dimerization (43); therefore, Mg2+ was added just before the titration with Ca2+. All solutions were prepared at 21 °C. Steady state fluorescence measurements were made with a Jasco 6500 spectrofluorimeter. IAANS fluorescence was excited at 330 nm, and emission was monitored at 450 nm as incremental amounts of CaCl2 were added to a 2.0-ml mixture containing labeled proteins. The final concentrations of isolated CTnC, binary complex, ternary complex, and RTFs in each experiment were 0.25 μm, 0.5 μm, 0.5 μm, and 0.053 mg/ml, respectively. The concentration of free Ca2+ was calculated for actual titration conditions using the computer program pCa Calculator (44). This program corrected for dilution effects attributed to the incremental addition of Ca2+. The data were fitted to the Hill equation with the software suite of SigmaPlot 10.0. The Ca2+ affinities are reported as pCa50 (–log[Ca2+] at which 50% of maximal response is observed) values ± S.D. The following modified Hill equation was used to fit biphasic binding curves,
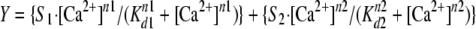 |
(Eq. 1) |
where Y represents the fluorescence intensity, S1 and S2 are the percentage contributions of each class of site to the fluorescence, Kd1 and Kd2 are the macroscopic dissociation constants for both classes of sites, and n1 and n2 are the respective Hill coefficients.
Reconstituted Myofilament ATPase Assays—Whole porcine cardiac myosin was isolated from left ventricles (45); F-actin, tropomyosin, and recombinant human CTn were prepared as described above. The ATPase assays were performed in a 96-well flat bottom plate using 0.1-ml reaction volumes. Preformed human cardiac Tn complexes were added (≤20 μl) to the 96 wells to achieve concentrations ranging from 0.0 to 2.0 μm in the final reaction mixture. F-actin, myosin, and tropomyosin were homogenized on ice in a glass tube and allowed to come to room temperature before the addition to each well (≤20 μl). The final concentrations of F-actin, myosin, and Tm in each well were 3.5, 0.6, and 1.0 μm, respectively. Two different ATPase buffers were prepared to adjust the final concentrations of all salts for high (pCa 4) and low (pCa > 9) Ca2+ conditions. The remainder of the 0.1-ml reaction volume comprised 50 μl of high or low Ca2+ ATPase buffer, double-distilled H2O, and 4 μl of ATP (3.14 mm, pH 7) to initiate the ATPase reaction. Methods for solving the free and bound metal ion equilibria were provided for by the computer program pCa Calculator (44). The final reaction conditions contained 50 mm KCl, 1.0 mm free Mg2+, 2.5 mm MgATP2–, 1.0 mm dithiothreitol, 20 mm MOPS, 1 mm EGTA (in low Ca2+-ATPase buffer) or 0.23 mm CaCl2 (in high Ca2+-ATPase buffer), pH 7, at 25 °C. The ATPase reaction was initiated by the addition of ATP in a row-wise fashion and immediately mixed with a 12-channel pipettor 4–5 times and allowed to incubate at 25 °C for 20 min; thereafter, stopping the reaction by adding cold trichloroacetic acid (4.75% final concentration). Each row was activated and deactivated in 30-s intervals. After sedimenting the precipitate by centrifugation, the supernatants were transferred to a new plate, and the liberated inorganic phosphate concentration in the supernatant was determined according to the method of Fiske and Subbarow (46). The ATPase rates were measured by single time points that were predetermined to be linear with time.
Preparation of Skinned Cardiac Myofibrils—Porcine cardiac myofibrils (CMF) were isolated form porcine left ventricles, as described by Solaro et al. (47), substituting MOPS for imidazole. All CMF preparations were stored in 50% glycerol (v/v) at –20 °C. CDTA treatment of cardiac myofibrils were performed by the methods of Morimoto and Ohtsuki (48) with the following modifications; CTnC extraction occurred in 5.0 mm CDTA, 5.0 mm dithiothreitol, pH 8.4, adjusted with Tris-base (solid) for 15 min, followed by centrifugation. This procedure was repeated four more times using 30 min CDTA incubations. CDTA was removed by suspending the pellet in wash buffer containing 10.0 mm MOPS, 10.0 mm KCl, 0.5% glycerol at pH 7 followed by centrifugation. These washes were repeated two more times in the absence of glycerol. CTnC reconstitution was performed by mixing CDTA-treated myofibrils (1.5–3.0 mg/ml) in wash buffer with recombinant CTnC (∼13 μm final concentration) on ice for 30 min. Unbound CTnC was removed by centrifugation, and the supernatant was discarded. Pellets were resuspended in wash buffer (without glycerol). This wash cycle was repeated and the final precipitate resuspended in wash buffer (without glycerol), stored on ice, and used for experiments.
Ca2+-activated Myofibrillar ATPase Assays—0.1-ml reaction mixtures were carried out in a 96-well plate. First, 50 μg (≤46-μl volume) of myofibrils in wash buffer (without glycerol) were aliquoted per well. The remainder of the 0.1-ml reaction volume comprised double-distilled H2O, ATPase buffers (50 μl) that have different amounts of calcium added to achieve the desired free calcium concentration, and 4 μl of ATP (3.14 mm, pH 7), which is added to initiate the ATPase reaction at 25 °C. The reactions were terminated 5 min later by the addition of trichloroacetic acid, and the inorganic phosphate was measured as above. Methods for solving the free and bound metal ion equilibria in our solutions were provided for by the computer program pCa Calculator (44). The final reaction condition contained 10–8.0 to 10–4.5 m free Ca2+, 1 mm free Mg2+, 2.5 mm Mg-ATP2–, 2 mm EGTA, 4 mm nitrilotriacetic acid, 20 mm MOPS, 1.0 mm dithiothreitol, 80 mm ionic strength (adjusted with KCl), pH 7, at 25 °C. The data were fitted to the Hill equation using the software suite of SigmaPlot 10.0.
Skinned Fiber Preparations—Skinned fibers were prepared using established protocols (37) with the following modifications. Muscle fibers were isolated from porcine left ventricular papillary muscle; pCa solutions were generated using the pCa Calculator program (44); and endogenous CTnC was extracted with 5 mm CDTA, pH 8.4 (adjusted with solid Tris-base) until the fibers developed <20% of the initial force (residual force) in pCa 4.0 solution. CDTA-treated fibers were reconstituted with CTnC (WT and mutant), and the Ca2+ dependence of force generation and maximal recovered force (in the pCa 4.0) was measured. Data were analyzed using the following equation,
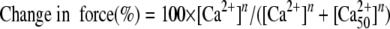 |
(Eq. 2) |
where [Ca2+50] represents the free Ca2+ concentration that produces 50% force, and n is the Hill coefficient (37, 49).
Statistical Analysis—Data are expressed as the average of ≥5 experiments ± S.D. Significant differences were determined using an unpaired Student's t test (Sigma Plot version 8.0), with significance defined as follows: *, p < 0.05; #, p < 0.001.
RESULTS
Effect of CTnC Mutations on the Ca2+ Sensitivity of Tension—Skinned papillary muscle fibers were treated with CDTA to remove the endogenous porcine CTnC, followed by reconstituting them back with the recombinant CTnCs (WT, G159D, and L29Q). Fig. 1A shows that reconstituting fibers with G159D CTnC significantly decreases the Ca2+ sensitivity of tension by –0.08 pCa units. This is made evident as a right-ward shift in the curve with respect to the WT. Moreover, this mutant can generate less tension in the pCa 6.0–5.6 range (Fig. 1, A and C) but does not affect the ability to restore the maximal activated tension in pCa 4.0 when compared with the WT1 control (Fig. 1C). Alternatively, the L29Q CTnC has no effect on the Ca2+ sensitivity (Fig. 1B) or maximal tension (Fig. 1C) when reconstituted into skinned fibers. Fig. 1C shows that all CTnC-reconstituted fibers are able to restore ∼70% of the maximal tension in accordance with similarly published values (37, 50). After CDTA treatment, the residual forces (in pCa 4) of muscle fibers from two heart preparations are 19.4% (WT1 and L29Q) and 12.3% (WT2 and G159D), respectively. SDS-PAGE followed by Western blot analysis showed that a 15–20% residual force corresponds to 14.0% ± 5.3% of the endogenous CTnC remaining after CDTA treatment. Moreover, incubation of fibers with WT or mutant CTnC is able to restore the full complement of CTnC back into the CDTA-treated fibers (data not shown). Therefore, the changes in the Ca2+ sensitivity or lack of changes are due to the specific CTnC mutation, rather than the inability to incorporate the CTnC into the muscle fibers. The force and pCa relationships are summarized in Table 1.
FIGURE 1.

The effects of CTnC mutations associated with cardiomyopathies on the Ca2+ sensitivity and maximum tension in skinned muscle fibers. Shown are the Ca2+ dependences of tension from porcine muscle fibers reconstituted with G159D CTnC (A) and L29Q CTnC (B). C, a comparison of the relative tensions from muscle fibers after CDTA treatment in pCa 4.0 (residual tension; white), and after CTnC reconstitution in pCa 4.0 (black) and 5.8 (gray). 100% relative tension indicates the maximal tension (in pCa 4.0) of fibers before CDTA treatment. All CTnC concentrations were 0.5 mg/ml. Data are summarized in Table 1.
TABLE 1.
Summary of pCa-tension relationships of skinned cardiac fibers reconstituted with mutant CTnCs
| CTnC-reconstituted fiber | pCa50a | ΔpCa50b | nH | Residual tension | Restored tension |
|---|---|---|---|---|---|
| % | % | ||||
| WT1 | 5.69 ± 0.05 | 4.07 ± 0.68 | 21.4 ± 3.5 | 74.9 ± 8.2 | |
| L29Q | 5.70 ± 0.01 | +0.01 | 3.89 ± 0.18 | 19.4 ± 6.3 | 66.0 ± 5.6 |
| WT2 | 5.70 ± 0.04 | 2.26 ± 0.29 | 12.3 ± 2.5 | 66.1 ± 4.6 | |
| G159D | 5.62 ± 0.02c | -0.08c | 2.23 ± 0.10 | 12.3 ± 1.4 | 73.4 ± 7.7 |
The pCa50 values were obtained by fitting the data in the range from pCa 7 to 4.5.
ΔpCa50 = pCa50 of mutant CTnC fiber — pCa50 of WT CTnC fiber.
p < 0.05, unpaired t test versus WT.
Effect of CTnC Mutations on Cardiac Myofibrillar and Regulated Actomyosin ATPase Activities—CMF isolated from porcine left ventricles were treated with CDTA followed by reconstitution with recombinant CTnCs. Fig. 2A shows the Ca2+ dependence of ATPase activity from the WT, G159D, and L29Q CTnC-reconstituted CMF. The maximal output for each mutant and WT CTnC were normalized to 100% in Fig. 2A to better illustrate the ability of the G159D reconstituted CMF to reduce the Ca2+ sensitivity of ATPase activity by –0.08 pCa units and the inability of the L29Q mutant to generate statistical differences from the WT controls (ΔpCa = +0.02, p > 0.05). Fig. 2B shows that after CDTA treatment, the myofibrillar ATPase activity in pCa 4.5 is nearly inactivated (1.59-fold activation), whereas, after the mutant CTnCs are reconstituted, the maximal myofibrillar ATPase is activated ∼4-fold and statistically insignificant in comparison with the WT (Fig. 2B and Table 2).
FIGURE 2.

The effects of skinned cardiac myofibrils and regulated actomyosin filaments reconstituted with CTnC mutants. A, Ca2+ sensitivity of ATPase activity from skinned cardiac myofibrils reconstituted with the G159D and L29Q CTnC. B, a comparison of the CDTA-treated and CTnC-reconstituted myofibrillar ATPase activities in pCa 8.0 (black) and pCa 4.5 (gray) solutions. C, effect of CTnC mutations on the activation (+Ca2+) and inhibition (–Ca2+) of regulated actin-Tm-activated myosin ATPase activity. CTn containing G159D, L29Q, or WT CTnC was mixed with actin, Tm, and myosin to reconstitute the regulated actomyosin filaments. The protein concentrations used are as follows: 3.5 μm F-actin, 1.0 μm Tm, 0.6μm myosin, and 0–2.0μm CTn. At all concentrations of CTn (0–2.0μm), the specific ATPase activities are measured in the presence of 0.1 mm free Ca2+ (+Ca2+) or 1.0 mm EGTA (–Ca2+). All experiments were done in the presence of 1.0 mm free Mg2+. Data are summarized in Table 2.
TABLE 2.
Summary of cardiac myofibrillar and regulated actomyosin ATPase activity data from Fig. 2
| WT | L29Q | G159D | |
|---|---|---|---|
| pCa50a | 5.95 ± 0.03 | 5.97 ± 0.02 | 5.87 ± 0.03b |
| nHa | 1.44 ± 0.09 | 1.38 ± 0.13 | 1.46 ± 0.16 |
| Minimum activity (-Ca2+)c | 39.5 ± 4.6 | 38.5 ± 3.3 | 41.7 ± 4.6 |
| Maximum activity (+Ca2+)c | 157.3 ± 11.3 | 168.4 ± 12.3 | 157.5 ± 12.2 |
| Fold activationd | 4.02 ± 0.43 | 4.37 ± 0.22 | 3.79 ± 0.20 |
| Minimum activity (-Ca2+)e | 0.088 ± 0.011 | 0.093 ± 0.014 | 0.096 ± 0.015 |
| Maximum activity (+Ca2+)e | 0.535 ± 0.021 | 0.528 ± 0.031 | 0.485 ± 0.034b |
| Percentage activation (-Ca2+)f | 26.8 ± 3.3 | 28.4 ± 4.3 | 29.3 ± 4.4 |
| Percentage activation (+Ca2+)f | 162.5 ± 6.3 | 160.4 ± 9.5 | 143.5 ± 5.9b |
Values obtained from fitting cardiac myofibrillar data in the range of pCa 8 to 4.5.
p < 0.05; unpaired t test versus WT.
Minimum and maximum specific ATPase activities from CMF in pCa 8 (-Ca2+) and pCa 4.5 (+Ca2+), respectively (nmol of Pi/mg) × min-1).
Fold CMF ATPase activation = maximum activity/minimum activity (-fold activation of CDTA-treated CMF = 1.59 ± 0.27).
Minimum and maximum specific ATPase activities of reconstituted actomyosin when 2.0 μm CTn is present in pCa 9 (-Ca2+) and pCa 4 (+Ca2+), respectively (mol of Pi/mol of myosin) × s-1.
Percentage of ATPase activation of reconstituted actomyosin in pCa 9 (-Ca2+) and pCa 4 (+Ca2+). 100% activation is defined as the percentage of ATPase activation in the presence of F-actin, Tm, and myosin only.
Preformed cardiac troponin complexes reconstituted with WT, G159D, or L29Q CTnC were mixed with F-actin, tropomyosin, and myosin in order to determine if the different CTnC mutations can affect the ability to inhibit or activate the regulated actomyosin ATPase activity. The ATPase activity of the unregulated filaments (i.e. in the absence of CTn) is defined as 100%. Fig. 2C shows that in the absence of Ca2+ (–Ca2+), the ATPase activities of regulated actomyosin filaments containing the G159D and L29Q mutants are statistically insignificant in comparison with the WT (∼27%) at all CTn concentrations. However, in pCa 4.0 solution (+Ca2+), the incorporation of G159D CTnC into regulated actomyosin filaments significantly lowers the ATPase activation (144%) in comparison with the WT counterpart (163%) when CTn is sufficiently present to saturate the thin filament. Table 2 summarizes the values obtained in Fig. 2.
Effect of Metal Ion Binding on Fluorescence from IAANS-labeled CTnC—WT, G159D, and L29Q CTnC clones were used to generate monocysteine mutants to direct specific incorporation of the IAANS fluorophore onto residue Cys84 (CTnC(C84)IA), which solely reports metal ion binding to the regulatory site of CTnC(C84)IA in isolation and in binary complex (38). In addition, both cysteine residues (positions 35 and 84) were labeled with IAANS (CTnCIA), which reports metal ion binding to the Ca2+-specific and Ca2+-Mg2+ sites in isolated CTnCIA and to the Ca2+-specific site within CTn (38, 51).
Fig. 3A represents the Ca2+-dependent changes in fluorescence arising from the WT, G159D, and L29Q CTnCIA in the presence (inset) and absence of Mg2+. As the free Ca2+ increases from pCa 8.0 to 3.6, the graphs are biphasic, representing metal ion binding to two classes of binding sites. Therefore, the pCa50 and Hill values obtained with the CTnCIA configuration were fitted to a two-site Hill equation and are summarized in Table 3. As the free Ca2+ increases from pCa 8.0 to 6.6 in the absence of Mg2+, the isolated WT CTnCIA fluorescence decreases –11.3% with a pCa50 of 7.04, indicating Ca2+ binding to the Ca2+-Mg2+ sites. In the presence of Mg2+ (inset), the WT CTnCIA Ca2+-Mg2+ sites decrease their fluorescence intensity (–4.8%) with a concomitant increase in affinity for Ca2+ by +0.08 pCa units. Increasing the concentration of free Ca2+ from pCa 6.6 to 3.8 leads to an increase in the fluorescence intensity for the WT CTnCIA protein, indicating Ca2+ binding to the Ca2+-specific (regulatory) site with a pCa50 of 5.23 (–Mg2+) and 4.94 (+Mg2+).
FIGURE 3.

The Ca2+-dependent changes in fluorescence arising from IAANS-labeled CTnC mutants associated with cardiomyopathy. A, a comparison of the Ca2+-dependent changes in fluorescence from the double-labeled L29Q and G159D CTnCIA. B, the Ca2+-dependent changes in fluorescence from the monolabeled L29Q and G159D CTnC(C84)IA. Incremental amounts of Ca2+ were added to 2.0 ml of labeled CTnCs (0.25 μm) in the presence and absence of 1.25 mm Mg2+ (insets). Fluorescence changes are expressed as a percentage of the total change. Data are summarized in Table 3.
TABLE 3.
Fitted pCa50 values and Hill coefficients for isolated IAANS-labeled CTnC mutants in Fig. 3
|
Preparation
|
[Mg2+]a
|
High affinity binding
|
Low affinity binding
|
||
|---|---|---|---|---|---|
| pCa50 | nH | pCa50 | nH | ||
| mm | |||||
| WT-CTnC(C84)IAb | 5.18 ± 0.05 | 0.859 | |||
| WT-CTnC(C84)IAb | 1.25 | 4.84 ± 0.01 | 0.843 | ||
| G159D-CTnC(C84)IAb | 5.20 ± 0.05 | 0.909 | |||
| G159D-CTnC(C84)IAb | 1.25 | 4.88 ± 0.02 | 0.896 | ||
| L29Q-CTnC(C84)IAc | 6.68 ± 0.02 | 1.847 | 5.30 ± 0.07d | 0.724 | |
| L29Q-CTnC(C84)IAc | 1.25 | 6.59 ± 0.03 | 0.996 | 4.85 ± 0.06 | 1.212 |
| WT-CTnCIAc | 7.04 ± 0.02 | -1.826 | 5.23 ± 0.06 | 0.791 | |
| WT-CTnCIAc | 1.25 | 7.12 ± 0.03 | -2.052 | 4.94 ± 0.01 | 0.849 |
| G159D-CTnCIAc | 6.87 ± 0.09d | -1.949 | 5.21 ± 0.04 | 0.820 | |
| G159D-CTnCIAc | 1.25 | 6.93 ± 0.02d | -2.097 | 4.97 ± 0.05 | 0.868 |
| L29Q-CTnCIAc | 6.99 ± 0.01 | -1.522 | 5.08 ± 0.06d | 0.697 | |
| L29Q-CTnCIAc | 1.25 | 7.06 ± 0.06 | -2.097 | 4.88 ± 0.01e | 0.822 |
The contaminating Mg2+ levels were less than 1.0 μm in experiments lacking Mg2+. In the presence of 1.25 mm Mg2+, the free concentration was estimated to be ∼0.6 mm.
The pCa50 values were obtained by fitting only the data in the range from pCa 6.4 to 3.8 from each individual curve to the Hill equation and averaging them as a group to obtain the S.D.
The pCa50 values were obtained by fitting the data in the range from pCa 8.0 to 3.6 from each individual curve to a two-site Hill equation and averaging them as a group to obtain the S.D.
p < 0.05, unpaired t test versus WT.
p < 0.001, unpaired t test versus WT.
Upon Ca2+ binding to the Ca2+-Mg2+ sites of isolated L29Q CTnCIA (Fig. 3A), there are larger decreases in fluorescence in the absence (–72.5%) and presence (–28.1%) of Mg2+; however, the fitted pCa50 values for the Ca2+-Mg2+ sites are unaffected in comparison with the WT counterpart. Moreover, the pCa50 value for the regulatory site is markedly reduced in the absence of Mg2+ (ΔpCa50 = –0.15) and, to a lesser extent, in the presence of Mg2+ (ΔpCa50 = –0.06) when compared with the WT counterpart. Upon Ca2+ binding to the Ca2+-Mg2+ sites of G159D CTnCIA, the pCa50 values are reduced in the presence (ΔpCa50 = –0.19) and absence (ΔpCa50 = –0.17) of Mg2+. However, from pCa 6.6 to 3.8, the G159D CTnCIA regulatory site affinity is unaffected (ΔpCa50 ≤ –0.03). Table 3 summarizes the fitted values obtained in Fig. 3A.
Fig. 3B represents the calcium-dependent changes in fluorescence from isolated WT, L29Q, and G159D CTnC(C84)IA in the absence and presence (inset) of Mg2+. As the free Ca2+ increases from pCa 6.8 to 3.6, the WT CTnC(C84)IA fluorescence intensity increases monophasically, indicating Ca2+ binding to the regulatory site. The pCa50 values are 5.18 (–Mg2+) and 4.84 (+Mg2+). In this same pCa range, the G159D CTnC(C84)IA regulatory site affinity is unaffected. This is made evident by the WT and G159D CTnC(C84)IA curves overlapping each other in the presence (inset) and absence of Mg2+. Alternatively, upon Ca2+ binding to L29Q CTnC(C84)IA, the fluorescence intensity unexpectedly increases biphasically, revealing that this labeled protein is sensitive to Ca2+ binding to the two classes of sites (in +Mg2+ or –Mg2+). Therefore, the L29Q CTnC(C84)IA structural effects differ from the previous CTnCs tested herein and may preclude its comparison with the WT control. Despite the inconsistencies between the L29Q and WT CTnC(C84)IA proteins, these data were fitted with a two-site Hill equation in order to calculate the pCa50 values of both classes of sites (summarized in Table 3). These results show that when fitted, the pCa50 for the L29Q CTnC(C84)IA regulatory site increases +0.12 pCa units (–Mg2+) when compared with the WT counterpart.
Effect of Ca2+ Binding on Fluorescence from IAANS-labeled CTnC in Complex—WT, L29Q, and G159D CTnC(C84)IA were mixed with recombinant CTnI to form fluorescent binary complexes. Fig. 4A shows the Ca2+-dependent changes in fluorescence arising from the WT and mutant binary CTnC(C84)IA complexes. As the free Ca2+ rises from pCa 7.6 to 4.8, the WT binary complex increases its fluorescence intensity 1.24-fold with a pCa50 of 6.24. The results from the G159D binary complex are statistically insignificant in comparison with the WT, whereas the fluorescence intensity of the L29Q binary complex increases 1.33-fold with a significant reduction in Ca2+ affinity (ΔpCa50 = –0.19). Table 4 summarizes the pCa50 and Hill values calculated from the data obtained in Fig. 4A.
FIGURE 4.

The Ca2+-dependent changes in fluorescence arising from IAANS-labeled CTnC mutants in complex. Shown is the effect of thin filament proteins on the Ca2+ dependence of fluorescence from L29Q and G159D mutations in CTnC(C84)IA binary complexes (A), CTnCIA ternary complexes (B), and CTnC(C84)IA regulated thin filaments (C). Incremental amounts of Ca2+ were added to 2.0 ml of labeled regulatory complexes in the presence of 1.25 mm Mg2+. The protein concentrations used were as follows: 0.5 μm binary complex, 0.5 μm ternary complex, and 0.053 mg/ml regulated thin filaments. Fluorescence changes are expressed as a percentage of the total change. Data are summarized in Table 4.
TABLE 4.
Comparison of pCa50 and Hill coefficients used to fit the data obtained in Fig. 4
|
WT
|
G159D
|
L29Q
|
||||
|---|---|---|---|---|---|---|
| pCa50 | nH | pCa50 | nH | pCa50 | nH | |
| Binary (CTnC(C84)IA)a | 6.24 ± 0.02 | 1.11 | 6.29 ± 0.02 | 1.06 | 6.05 ± 0.02b | 1.09 |
| Ternary (CTnCIA)c | 6.68 ± 0.02 | -0.94 | 6.53 ± 0.05b | -1.00 | 6.61 ± 0.00b | -0.95 |
| Regulated thin filament (CTnC(C84)IA)d | 6.12 ± 0.03 | 1.22 | 5.97 ± 0.05b | 1.23 | 6.17 ± 0.03 | 1.25 |
Binary values obtained by fitting the data in the range of pCa 7.7 to 4.5.
p < 0.001 unpaired t test versus WT.
Ternary values obtained by fitting the data in the range of pCa 8.7 to 4.9.
Regulated thin filament values obtained by fitting the data in the range of pCa 7.6 to 4.2.
WT, L29Q, and G159D CTnCIA were mixed with recombinant CTnI and CTnT to generate fluorescent cardiac troponin complexes (CTnIA). Fig. 4B illustrates the Ca2+-dependent changes in fluorescence arising from CTnIA complexes. In the pCa range of 8.8 to 5.2, Ca2+ binding to the WT and mutant CTnIA is accompanied by a 1.4-fold decrease in fluorescence intensity. The G159D CTnIA Ca2+ affinity is significantly reduced (ΔpCa50 = –0.15) in comparison with WT CTnIA, whereas the changes in Ca2+ affinity arising from L29Q CTnIA are not as significant (ΔpCa50 = –0.07, p < 0.05) (see Table 4). In accordance with previous studies (37), the addition of Ca2+ to binary CTnCIA and ternary CTnC(C84)IA complexes yielded no change in fluorescence (data not shown).
WT, L29Q, and G159D CTnC(C84)IA were mixed with recombinant CTnI and CTnT to form CTn(C84)IA complexes. These preformed complexes were mixed with F-actin and Tm to generate fluorescent RTFs. Fig. 4C shows the Ca2+ dependence of fluorescence from IAANS-labeled RTFs. The WT RTF increases its fluorescence intensity 1.62-fold upon increasing the free Ca2+ from pCa 8.0 to 4.5 with a pCa50 of 6.12. In the same pCa range, the G159D RTF fluorescence intensity increases to a lesser extent (1.48-fold) with a concomitant reduction in Ca2+ affinity (ΔpCa50 = –0.15). Alternatively, the L29Q RTF Ca2+ affinity and fluorescence intensities are statistically insignificant in comparison with the WT control (ΔpCa = +0.05, p > 0.05). Table 4 summarizes the pCa50 and Hill values that are derived from the data in Fig. 4.
DISCUSSION
Since the heart is a dynamic pump, thoracic imaging techniques are essential in identifying the specific systolic and diastolic dysfunctions underlying the pathologies of familial cardiomyopathies. However, different individuals affected by the same mutation can show variable penetrance (e.g. graded wall thickening or dilation), suggesting that cardiomyopathies are complex multifactorial diseases. Therefore, steady state measurements have the benefit of observing the influence of these mutations on contractility in the absence of long term neurohumoral and autonomic stimulation. This has led to the in vitro investigation of numerous thin filament mutations linked to cardiomyopathies and the discovery of their characteristic effects on the Ca2+-dependent processes of contraction in the absence of CTnI phosphorylation. Within the context of RTF mutations, a unifying paradigm emerges that associates decreases in the myofilament Ca2+ sensitivity with DCM-linked mutations and increases with HCM- and restrictive cardiomyopathy-linked mutations (4, 7, 14). However, does this imply that insignificant changes in the myofilament Ca2+ sensitivity arising from a mutation are going to be associated with a benign phenotype? This poses a difficult challenge to the developing paradigm, because these mutations are rare polymorphisms, making it difficult to determine if the clinical outcome is primarily due to a disease causing mutation in the myocardium or physiological adaptations to conditions such as hypertension, diabetes, obesity, and alcoholism, to name a few (2).
Echocardiographic examination of the only L29Q CTnC proband shows that the septal and free ventricular walls are concentrically hypertrophied (15 mm; cut-off = 13 mm) (2, 19). Notably, in the absence of any cardiomyopathy, many clinical studies strongly correlate concentric hypertrophy with hypertension (52, 53). Moreover, the amount of left ventricle hypertrophy may be overestimated in the L29Q proband, because the authors did not adjust the left ventricle mass to body surface area or mass indices in order to assess the possible impact of obesity and hypertension (54, 55). Despite the mild hypertrophy, cardiac catheterization showed a normal ejection fraction with preserved diastolic heart function (19). Based upon the available clinical data and current paradigms, we hypothesized that if the L29Q mutation is benign, then significant changes in the Ca2+ sensitivity should not arise in the skinned muscle. When we assessed the effects of L29Q CTnC on tension and myofibrillar ATPase activation, neither the Ca2+ sensitivities nor the maximal generating capabilities were significantly different from the WT controls. Moreover, regulated actomyosin reconstituted with preformed CTn containing the recombinant L29Q mutant did not affect the ability to inhibit or maximally activate the ATP hydrolysis at all levels of CTn. By taking into account the clinical pathology of the L29Q proband, the authors concluded that the L29Q mutation may “simply be a rare polymorphism without any phenotypical relevance” (19), in which case, our in vitro results would coincide with their findings. Therefore, the combined clinical and in vitro data should be weighed equally in order to determine more accurately the potential severity of specific mutations. Under this premise, the E244D CTnT mutation associated with HCM would be considered a benign polymorphism, because it does not affect the Ca2+ regulation in vitro (56, 57) or present with any cardiac dysfunction in the only proband (58).
Two reports have shown that the L29Q mutation can alter the myofilament Ca2+ sensitivity in a reconstituted S1-ATPase system (30) and in the skinned rat cardiomyocyte (32). Unexpectedly, the first investigation reports a paradigm contradiction, because the HCM-associated L29Q mutation decreases the myofilament Ca2+ sensitivity (30). However, their use of skeletal actin, Tm, and myosin may disrupt specific cardiac myofilament interactions that govern the Ca2+ regulation of contraction (or ATPase activity).
The second study shows that L29Q CTnC is able to increase the apparent CTnC Ca2+ affinity in the isolated state and in the skinned rat cardiomyocyte (32). Although in our hands, the changes in the Ca2+ sensitivities measured in fibers and cardiac myofibrils reconstituted with L29Q CTnC are statistically insignificant with respect to the WT, we do observe tendencies to increase the Ca2+ sensitivity of tension, myofibrillar ATPase activity, and RTF fluorescence (summarized in Table 5). It is expected that the different protein expression profiles among higher mammals and lower rodents will affect the biophysical properties that govern Ca2+ binding to CTnC. Furthermore, the cardiomyocyte system is a more sensitive technique to measure changes in Ca2+ affinity that could otherwise go undetected at the scale of the skinned fiber.
TABLE 5.
Summary of mutant CTnC regulatory site ΔpCa50 values from various preparations
| WT pCa50 | G159D ΔpCa50a | L29Q ΔpCa50a | |
|---|---|---|---|
| Isolated (CTnCIA)b | 5.18 | +0.02 | -0.15c |
| Isolated (CTnC(C84)IA)b | 5.23 | -0.02 | +0.12c |
| Binary (CTnC(C84)IA) | 6.24 | +0.05 | -0.19d |
| Ternary (CTnCIA) | 6.68 | -0.15d | -0.17d |
| Regulated thin filament (CTnC(C84)IA) | 6.12 | -0.15d | +0.05 |
| Cardiac myofibrils | 5.95 | -0.08c | +0.02 |
| Skinned fiber | 5.70e | -0.08c | +0.01 |
| 5.69f |
Values are given in the absence of Mg2+.
p < 0.05; unpaired t test versus WT.
p < 0.001; unpaired t test versus WT.
Mean pCa50 of control fibers associated with G159D.
Mean pCa50 of control fibers associated with L29Q.
Measuring the structural and Ca2+ dependent changes from the isolated L29Q (-CTnCIA and -CTnC(C84)IA) proteins confirms that this mutation has the ability to alter the regulatory site binding properties and N- and C-domain intramolecular interactions. Interestingly, the L29Q CTnCIA reports a significant decrease in affinity at the regulatory site, whereas the L29Q CTnC(C84)IA protein unexpectedly reports Ca2+ binding to two classes of sites with an increased affinity at the regulatory site (Fig. 3). Although these results suggest that the combined effects of the L29Q mutation and IAANS fluorophore on Cys84 may dramatically perturb the structure of isolated L29Q CTnC(C84)IA, it also provides the opportunity to determine if the addition of different myofilament proteins and their interactions have the ability to reverse these processes. Formation of L29Q ternary complexes shows that the changes in Ca2+ affinity are smaller and become insignificant in the RTF when compared with the WT counterparts (see Table 5). The above results show that the RTF, representing a highly cooperative system can restore the Ca2+ affinity of L29Q CTnC to that of the WT via myofilament intermolecular interactions and can explain why significant changes in the Ca2+ sensitivity of tension do not arise.
With regard to G159D CTnC in the skinned muscle, this mutation reduces the Ca2+ sensitivity of tension and myofibrillar ATPase activation (Table 5) without affecting the capability to generate maximal output (pCa ≤ 4.5). Despite the inability of this mutant to reduce the maximal output, the myofilament Ca2+ desensitization is expected to recruit fewer strongly attached cross-bridges at submaximal Ca2+ concentrations, leading to the reduction of both ATPase activation and the subsequent generation of tension (1, 3, 59). Since muscle relaxation is directly correlated with Ca2+ dissociation from the CTnC regulatory site (60), mutations that directly (or indirectly) decrease the calcium affinity of CTnC would increase the rate of cardiac muscle relaxation. Therefore, an improvement in muscle relaxation would facilitate the entry of blood from the atria into the ventricles by providing less resistance to stretch (i.e. fewer attached cross-bridges from previous systole), which can result in higher end diastolic dimensions. In support of this argument, independent studies have shown that the reconstitution of ΔK210-CTnT (associated with DCM) into skinned fibers also reduces the myofilament Ca2+ sensitivity without affecting the maximal tension (3, 36). Moreover, the ΔK210-CTnT mutation is still able to recapitulate the DCM phenotype (e.g. reduced ejection fraction and dilation) in a knock-in mouse model (3).
Previous investigations have reported that the G159D mutation does not alter the Ca2+ sensitivity of tension or ATPase activation in the absence of phosphorylated CTnI. These reports show that ∼50% (28) to ∼68% (29) of the endogenous CTnC is replaced by the mutant in comparison with the ∼86% that is exchanged in our fibers. Therefore, an increase in the ratio of incorporated mutant to endogenous CTnC along the thin filament may explain why changes in the Ca2+ sensitivity arise in our system. In addition, the pCa50 values for ATPase activity reported by Biesiadecki et al. (29) suggests that small significant changes in the myofilament Ca2+ sensitivity may not be uncovered by the Ca2+-buffering capabilities of their solutions. Therefore, we employed the combinatorial use of nitrilotriacetic acid and EGTA to improve the Ca2+-buffering capabilities of our solutions, which has previously been shown to increase the sensitivity and reproducibility of the experiment (44).
Considering the indirect two-way communication between cross-bridge attachment and Ca2+ binding to CTnC, it is expected that a perturbation due to a CTnC mutation can affect the Ca2+ sensitivity of contraction through the following mechanisms: 1) direct modification of the regulatory site; 2) modifying CTnC intramolecular interactions; and/or 3) modifying CTnC intermolecular interactions. Measuring the shifts in the Ca2+ sensitivity and maximum tension (or ATPase) is not sufficient to suggest the mechanism(s). Therefore, covalently attached fluorophores on CTnC were used to identify key intra- and intermolecular interactions that dictate the final Ca2+ sensitivity of tension in the muscle fiber.
The isolated G159D (-CTnCIA and -CTnC(C84)IA) regulatory site apparent affinities are statistically insignificant in comparison with their respective WT counterparts. Therefore, the existence of mechanism 1 may be excluded. Nevertheless, a reduction in the affinity for the Ca2+-Mg2+ sites (±Mg2+) indicates that intramolecular interactions within the C-domain are altered (mechanism 2). Measuring the Ca2+ dependent changes in G159D binary complexes did not uncover the changes seen in the Ca2+ sensitivity of tension or ATPase. Rather, within the ternary complex, significant reductions in the regulatory site Ca2+ affinity begin to emerge (ΔpCa = –0.15), which persist up to the level of the RTF and, to a lesser extent, in the skinned fiber (summarized in Table 5). Since the CTnC C-lobe is “rigidly” integrated into the IT arm of CTn (61), the effect of the G159D mutation on the Ca2+-Mg2+ sites may destabilize these interactions. This implies that a CTnC C-domain mutation can indirectly affect the N-domain through altered CTnC and CTnT interactions, which becomes apparent at the level of CTn (i.e. mechanisms 2 and 3).
The cardiac contractile apparatus, representing a large cooperative machine, can provide the potential for changes (G159D) or no change (L29Q) in the myofilament Ca2+ sensitivity if any of the proteins are mutated. The true effects of the mutations become apparent as the hierarchical level of the myofilament increases. This is illustrated by the tendency of the RTF (in the absence of myosin) to retain its regulatory role and best indicate changes (+ or –) in the apparent CTnC Ca2+ affinity that are measured in the skinned muscle. Considering that the in vitro characterizations of over 40 RTF mutations assessed (in the absence of CTnI phosphorylation) have led to the emergence of current paradigms related to familial cardiomyopathies, we measured the various Ca2+-dependent processes of contraction to determine if these mutations challenge or support the current paradigms. Our results indicate that the myofilament Ca2+ desensitization arising from G159D CTnC and the lack of effects arising from L29Q CTnC do coincide with the current paradigms and are indicative of their respective clinical outcomes.
This work was supported, in whole or in part, by National Institutes of Health Grants HL67415 and HL42325 (to J. D. P.) and T32-HL07188. This work was also supported by American Heart Association Grant AHA 0315164B (to D. D.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: DCM, dilated cardiomyopathy; CDTA, trans-1,2-cyclohexane-N,N,N′,N′-tetraacetic acid; CMF, cardiac myofibril(s); CTn, cardiac troponin; CTnIA, cardiac troponin reconstituted from CTnCIA, CTnI, and CTnT; CTnC, cardiac troponin C; CTnC(C84)IA, CTnC with one IAANS molecule covalently attached to Cys84; CTnCIA, CTnC with two IAANS molecules covalently attached to Cys35 and Cys84; CTnI, cardiac troponin I; CTnT, cardiac troponin T; HCM, hypertrophic cardiomyopathy; IAANS, 2-(4′-(iodoacetamido)anilino)-naphthalene-6-sulfonic acid; MOPS, 4-morpholinepropanesulfonic acid; pCa, –log[Ca2+]; RTF, regulated thin filament; Tm, tropomyosin; WT, wild type.
References
- 1.Fatkin, D., and Graham, R. M. (2002) Physiol. Rev. 82 945–980 [DOI] [PubMed] [Google Scholar]
- 2.Griffin, B. P., and Topol, E. J. (2004) Manual of Cardiovascular Medicine, 2nd Ed., pp. 101–142, Lippincott Williams and Wilkins, Philadelphia
- 3.Du, C. K., Morimoto, S., Nishii, K., Minakami, R., Ohta, M., Tadano, N., Lu, Q. W., Wang, Y. Y., Zhan, D. Y., Mochizuki, M., Kita, S., Miwa, Y., Takahashi-Yanaga, F., Iwamoto, T., Ohtsuki, I., and Sasaguri, T. (2007) Circ. Res. 101 185–194 [DOI] [PubMed] [Google Scholar]
- 4.Gomes, A. V., and Potter, J. D. (2004) Ann. N. Y. Acad. Sci. 1015 214–224 [DOI] [PubMed] [Google Scholar]
- 5.Rajan, S., Ahmed, R. P., Jagatheesan, G., Petrashevskaya, N., Boivin, G. P., Urboniene, D., Arteaga, G. M., Wolska, B. M., Solaro, R. J., Liggett, S. B., and Wieczorek, D. F. (2007) Circ. Res. 101 205–214 [DOI] [PubMed] [Google Scholar]
- 6.Chang, A. N., Harada, K., Ackerman, M. J., and Potter, J. D. (2005) J. Biol. Chem. 280 34343–34349 [DOI] [PubMed] [Google Scholar]
- 7.Robinson, P., Griffiths, P. J., Watkins, H., and Redwood, C. S. (2007) Circ. Res. 101 1266–1273 [DOI] [PubMed] [Google Scholar]
- 8.Gomes, A. V., Harada, K., and Potter, J. D. (2005) J. Mol. Cell. Cardiol. 39 754–765 [DOI] [PubMed] [Google Scholar]
- 9.Karibe, A., Tobacman, L. S., Strand, J., Butters, C., Back, N., Bachinski, L. L., Arai, A. E., Ortiz, A., Roberts, R., Homsher, E., and Fananapazir, L. (2001) Circulation 103 65–71 [DOI] [PubMed] [Google Scholar]
- 10.Heller, M. J., Nili, M., Homsher, E., and Tobacman, L. S. (2003) J. Biol. Chem. 278 41742–41748 [DOI] [PubMed] [Google Scholar]
- 11.Takahashi-Yanaga, F., Morimoto, S., Harada, K., Minakami, R., Shiraishi, F., Ohta, M., Lu, Q. W., Sasaguri, T., and Ohtsuki, I. (2001) J. Mol. Cell Cardiol. 33 2095–2107 [DOI] [PubMed] [Google Scholar]
- 12.Chandra, M., Tschirgi, M. L., and Tardiff, J. C. (2005) Am. J. Physiol. 289 H2112–H2119 [DOI] [PubMed] [Google Scholar]
- 13.Davis, J., Wen, H., Edwards, T., and Metzger, J. M. (2007) Circ. Res. 100 1494–1502 [DOI] [PubMed] [Google Scholar]
- 14.Gomes, A. V., Liang, J., and Potter, J. D. (2005) J. Biol. Chem. 280 30909–30915 [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi, T., and Solaro, R. J. (2006) J. Biol. Chem. 281 13471–13477 [DOI] [PubMed] [Google Scholar]
- 16.Yumoto, F., Lu, Q.-W., Morimoto, S., Tanaka, H., Kono, N., Nagata, K., Ojima, T., Takahashi-Yanaga, F., Miwa, Y., Sasaguri, T., Nishita, K., Tanokura, M., and Ohtsuki, I. (2005) Biochem. Biophys. Res. Commun. 338 1519–1526 [DOI] [PubMed] [Google Scholar]
- 17.Pinto, J. R., Parvatiyar, M. S., Jones, M. A., Liang, J., and Potter, J. D. (2008) J. Biol. Chem. 283 2156–2166 [DOI] [PubMed] [Google Scholar]
- 18.Liao, R., Gwathmey, J. K., and Wang, C. (1998) Circulation 98 I-625 [Google Scholar]
- 19.Hoffmann, B., Schmidt-Traub, H., Perrot, A., Osterziel, K. J., and Gessner, R. (2001) Hum. Mutat. 17 524. [DOI] [PubMed] [Google Scholar]
- 20.Mogensen, J., Murphy, R. T., Shaw, T., Bahl, A., Redwood, C., Watkins, H., Burke, M., Elliott, P. M., and McKenna, W. J. (2004) J. Am Coll. Cardiol. 44 2033–2040 [DOI] [PubMed] [Google Scholar]
- 21.Kaski, J. P., Burch, M., and Elliott, P. M. (2007) Cardiol. Young 17 675–677 [DOI] [PubMed] [Google Scholar]
- 22.Dweck, D., Gomes, A. V., and Potter, J. D. (2005) Biophys. J. 88 317a15501937 [Google Scholar]
- 23.Lim, C. C., Yang, H., Yang, M., Wang, C. K., Shi, J., Berg, E. A., Pimentel, D. R., Gwathmey, J. K., Hajjar, R. J., Helmes, M., Costello, C. E., Huo, S., and Liao, R. (2008) Biophys. J. 94 3577–3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon, A. M., Homsher, E., and Regnier, M. (2000) Physiol. Rev. 80 853–924 [DOI] [PubMed] [Google Scholar]
- 25.Li, M. X., Wang, X., and Sykes, B. D. (2004) J. Muscle Res. Cell Motil. 25 559–579 [DOI] [PubMed] [Google Scholar]
- 26.Potter, J. D., and Gergely, J. (1975) J. Biol. Chem. 250 4628–4633 [PubMed] [Google Scholar]
- 27.Holroyde, M. J., Robertson, S. P., Johnson, J. D., Solaro, R. J., and Potter, J. D. (1980) J. Biol. Chem. 255 11688–11693 [PubMed] [Google Scholar]
- 28.Preston, L. C., Ashley, C. C., and Redwood, C. S. (2007) Biochem. Biophys. Res. Commun. 360 27–32 [DOI] [PubMed] [Google Scholar]
- 29.Biesiadecki, B. J., Kobayashi, T., Walker, J. S., Solaro, R. J., and de Tombe, P. P. (2007) Circ. Res. 100 1486–1493 [DOI] [PubMed] [Google Scholar]
- 30.Schmidtmann, A., Lindow, C., Villard, S., Heuser, A., Mugge, A., Gessner, R., Granier, C., and Jaquet, K. (2005) FEBS J. 272 6087–6097 [DOI] [PubMed] [Google Scholar]
- 31.Dong, W. J., Xing, J., Ouyang, Y., An, J., and Cheung, H. C. (2008) J. Biol. Chem. 283 3424–3432 [DOI] [PubMed] [Google Scholar]
- 32.Liang, B., Chung, F., Qu, Y., Pavlov, D., Gillis, T. E., Tikunova, S. B., Davis, J. P., and Tibbits, G. F. (2008) Physiol. Genomics 33 257–266 [DOI] [PubMed] [Google Scholar]
- 33.Baryshnikova, O. K., Li, M. X., and Sykes, B. D. (2008) J. Mol. Biol. 375 735–751 [DOI] [PubMed] [Google Scholar]
- 34.Lu, Q. W., Morimoto, S., Harada, K., Du, C. K., Takahashi-Yanaga, F., Miwa, Y., Sasaguri, T., and Ohtsuki, I. (2003) J. Mol. Cell Cardiol. 35 1421–1427 [DOI] [PubMed] [Google Scholar]
- 35.Venkatraman, G., Gomes, A. V., Kerrick, W. G., and Potter, J. D. (2005) J. Biol. Chem. 280 17584–17592 [DOI] [PubMed] [Google Scholar]
- 36.Morimoto, S., Lu, Q. W., Harada, K., Takahashi-Yanaga, F., Minakami, R., Ohta, M., Sasaguri, T., and Ohtsuki, I. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 913–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szczesna, D., Guzman, G., Miller, T., Zhao, J., Farokhi, K., Ellemberger, H., and Potter, J. D. (1996) J. Biol. Chem. 271 8381–8386 [DOI] [PubMed] [Google Scholar]
- 38.Putkey, J. A., Liu, W., Lin, X., Ahmed, S., Zhang, M., Potter, J. D., and Kerrick, W. G. (1997) Biochemistry 36 970–978 [DOI] [PubMed] [Google Scholar]
- 39.Potter, J. D. (1982) Methods Enzymol. 85 241–263 [DOI] [PubMed] [Google Scholar]
- 40.Zhang, R., Zhao, J., and Potter, J. D. (1995) J. Biol. Chem. 270 30773–30780 [DOI] [PubMed] [Google Scholar]
- 41.Szczesna, D., Zhang, R., Zhao, J., Jones, M., Guzman, G., and Potter, J. D. (2000) J. Biol. Chem. 275 624–630 [DOI] [PubMed] [Google Scholar]
- 42.Pardee, J. D., and Spudich, J. A. (1982) Methods Enzymol. 85 164–181 [DOI] [PubMed] [Google Scholar]
- 43.Jaquet, K., and Heilmeyer, L. M., Jr. (1987) Biochem. Biophys. Res. Commun. 145 1390–1396 [DOI] [PubMed] [Google Scholar]
- 44.Dweck, D., Reyes-Alfonso, A., Jr., and Potter, J. D. (2005) Anal. Biochem. 347 303–315 [DOI] [PubMed] [Google Scholar]
- 45.Murakami, U., Uchida, K., and Hiratsuka, T. (1976) J. Biochem. (Tokyo) 80 611–619 [DOI] [PubMed] [Google Scholar]
- 46.Fiske, C. H., and Subbarow, Y. (1925) J. Biol. Chem. 66 375–400 [Google Scholar]
- 47.Solaro, R. J., Pang, D. C., and Briggs, F. N. (1971) Biochim. Biophys. Acta 245 259–262 [DOI] [PubMed] [Google Scholar]
- 48.Morimoto, S., and Ohtsuki, I. (1987) J. Biochem. (Tokyo) 101 291–301 [DOI] [PubMed] [Google Scholar]
- 49.Guth, K., and Potter, J. D. (1987) J. Biol. Chem. 262 13627–13635 [PubMed] [Google Scholar]
- 50.Gulati, J., Sonnenblick, E., and Babu, A. (1991) J. Physiol. 441 305–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson, J. D., Collins, J. H., Robertson, S. P., and Potter, J. D. (1980) J. Biol. Chem. 255 9635–9640 [PubMed] [Google Scholar]
- 52.Lilly, L. S. (2006) Pathophysiology of Heart Disease: A Collaborative Project of Medical Students and Faculty, 4th Ed., Lippincott Williams and Wilkins, Philadelphia
- 53.de Simone, G., Kitzman, D. W., Chinali, M., Oberman, A., Hopkins, P. N., Rao, D. C., Arnett, D. K., and Devereux, R. B. (2005) Eur. Heart J. 26 1039–1045 [DOI] [PubMed] [Google Scholar]
- 54.de Simone, G., Kizer, J. R., Chinali, M., Roman, M. J., Bella, J. N., Best, L. G., Lee, E. T., and Devereux, R. B. (2005) Am. J. Hypertens 18 191–196 [DOI] [PubMed] [Google Scholar]
- 55.Palmieri, V., de Simone, G., Arnett, D. K., Bella, J. N., Kitzman, D. W., Oberman, A., Hopkins, P. N., Province, M. A., and Devereux, R. B. (2001) Am J. Cardiol. 88 1163–1168 [DOI] [PubMed] [Google Scholar]
- 56.Harada, K., and Potter, J. D. (2004) J. Biol. Chem. 279 14488–14495 [DOI] [PubMed] [Google Scholar]
- 57.Yanaga, F., Morimoto, S., and Ohtsuki, I. (1999) J. Biol. Chem. 274 8806–8812 [DOI] [PubMed] [Google Scholar]
- 58.Watkins, H., McKenna, W. J., Thierfelder, L., Suk, H. J., Anan, R., O'Donoghue, A., Spirito, P., Matsumori, A., Moravec, C. S., Seidman, J. G., and Seidman, C. E. (1995) N. Engl. J. Med. 332 1058–1064 [DOI] [PubMed] [Google Scholar]
- 59.Fabiato, A. (1981) J. Gen. Physiol. 78 457–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller, T., Szczesna, D., Housmans, P. R., Zhao, J., de Freitas, F., Gomes, A. V., Culbreath, L., McCue, J., Wang, Y., Xu, Y., Kerrick, W. G., and Potter, J. D. (2001) J. Biol. Chem. 276 3743–3755 [DOI] [PubMed] [Google Scholar]
- 61.Takeda, S., Yamashita, A., Maeda, K., and Maeda, Y. (2003) Nature 424 35–41 [DOI] [PubMed] [Google Scholar]