

Abstract
Exofacial phosphatidylserine (PS) is an important ligand mediating apoptotic cell clearance by phagocytes. Oxidation of PS fatty acyl groups (oxPS) during apoptosis reportedly mediates recognition through scavenger receptors. Given the oxidative capacity of the neutrophil NADPH oxidase, we sought to identify oxPS signaling species in stimulated neutrophils. Using mass spectrometry analysis, only trace amounts of previously characterized oxPS species were found. Conversely, 18:1 and 18:0 lysophosphatidylserine (lyso-PS), known bioactive signaling phospholipids, were identified as abundant modified PS species following activation of the neutrophil oxidase. NADPH oxidase inhibitors blocked the production of lyso-PS in vitro, and accordingly, its generation in vivo by activated, murine neutrophils during zymosan-induced peritonitis was absent in mice lacking a functional NADPH oxidase (gp91phox-/-). Treatment of macrophages with lyso-PS enhanced the uptake of apoptotic cells in vitro, an effect that was dependent on signaling via the macrophage G2A receptor. Similarly, endogenously produced lyso-PS also enhanced the G2A-mediated uptake of activated PS-exposing (but non-apoptotic) neutrophils, raising the possibility of non-apoptotic mechanisms for removal of inflammatory cells during resolution. Finally, antibody blockade of G2A signaling in vivo prolonged zymosan-induced neutrophilia in wild-type mice, whereas having no effect in gp91phox-/- mice where lyso-PS are not generated. Taken together, we show that lyso-PS are modified PS species generated following activation of the NADPH oxidase and lyso-PS signaling through the macrophage G2A functions to enhance existing receptor/ligand systems for optimal resolution of neutrophilic inflammation.
Neutrophils are often robustly recruited early in inflammation. Within hours of their activation in tissues, they are removed by phagocytes, an event required for resolution of inflammation and the return to normalcy of tissue function. It is known that neutrophils undergoing apoptosis drive the production of anti-inflammatory mediators such as transforming growth factor-β that actively suppress production of inflammatory cytokines, chemokines, eicosanoids, and nitric oxide (1, 2). Indeed, enhanced induction of neutrophil apoptosis in vivo is potently anti-inflammatory (3, 4). However, if recognition and clearance fail, activated and dying neutrophils ultimately disintegrate releasing injurious intracellular constituents (e.g. serine proteases) (5). Failure of timely cell clearance is associated with both autoimmunity and enhanced inflammation (6, 7).
Phosphatidylserine (PS)2 exposed in the plasma membrane outer leaflet of apoptotic cells has long been known as a key ligand important for their recognition and removal. Interaction with various PS receptors, including the recently identified TIM4 (8, 9), BAI1 (10), and stabilin 2 (11) or PS-recognizing bridge molecule-receptor combinations (e.g. MFG-E8 and αv integrins or Gas6 and Mer (12)), have been demonstrated. In many, but not all cases, these interactions have been shown to have stereospecificity for the l-phosphoserine moiety, and not the d-isomer (13-15). Recently it has been demonstrated that oxidation of the sn-2 fatty acyl chain of PS (oxPS) makes it a more potent stimulus for the clearance of apoptotic cells involving scavenger receptors, particularly CD36 (16-18). In contrast to PS-derived signals that promote recognition and removal of apoptotic cells, other lipid-derived mediators released from cells, such as lipoxins, resolvins, and protectins, signaling through their cognate receptors, have been shown to play a role in orchestrating resolution of inflammation by enhancing these existing mechanisms for engulfment of apoptotic cells and tissue homeostasis (19).
The exact PS species signaling for the recognition and removal of infiltrating neutrophils have not been fully elucidated. Given that recruited neutrophils robustly activate their NADPH oxidase, we reasoned that activated neutrophils would be an excellent source for the identification of additional oxPS species signaling for apoptotic cell engulfment and specifically for the resolution of neutrophilic inflammation. Here, we show that neutrophils generate trace amounts of previously identified oxPS species, but more striking was the production of nanogram quantities of lyso-PS species both in vitro and in vivo following activation of the NADPH oxidase. Lyso-PS species have been shown to be biologically active, signaling via G protein-coupled receptors (e.g. GPR34 on mast cells and via G2A on neutrophils (20, 21)).
Our studies demonstrate that lyso-PS are modified PS species that signal via the macrophage G2A receptor to enhance existing receptor/ligand systems for the engulfment of PS exposing activated and apoptotic cells in vitro and in vivo. These data support inclusion of lyso-PS/G2A in the growing repertoire of lipid-derived signaling molecules and signaling receptors demonstrated to play a role in anti-inflammatory signaling and resolution of inflammation.
EXPERIMENTAL PROCEDURES
Materials—All lipids were purchased from Avanti Polar Lipids (Alabaster, AL) unless otherwise noted. Solvents, glucose, glucose oxidase, NaNO2, fMLP, phorbol myristate acetate (PMA), methoxylamine hydrochloride (MOX), cytochrome c, and zymosan were from Sigma. Myeloperoxidase (MPO), diphenyleneiodonium (DPI), cPLA2α inhibitor, and bromoenol lactone were from EMD Biosciences (Gibbstown, NJ). Aminopropyl Sep-Pak (NH2-SPE) columns were from Supelco (subsidiary of Sigma). Flash red carboxylate-modified beads (5 μm) were from Bangs Labs (Fishers, IN). ONO-RS-082 was from Biomol International (Plymouth Meeting, PA). HLB reverse solid phase columns were from Phenomenex (Torrance, CA). Anti-G2A M-20 was from Santa Cruz Biotechnology (Santa Cruz, CA). Annexin V, Alexa 488, and anti-goat IgG Alexa 488 were from Molecular Probes (Eugene, OR). Lyso-PS internal standard (17:1/OH-PS) was a generous gift from Dr. Walter Shaw at Avanti Polar Lipids (Alabaster, AL).
Animals—Male and female C57BL/6 and gp91phox-/- mice were purchased from Jackson Laboratories (Bar Harbor, ME) and also used from a breeding colony at National Jewish Health (Denver, Colorado). All animals received care in accordance with the guidelines of the Institutional Animal Care and Use Committee and were maintained on food and water ad libitum. Mice between the ages of 8 and 16 weeks provided a source of resident peritoneal and thioglycollate-elicited MΦ and were age and gender matched for all experiments.
Isolation and Culture of Murine Peritoneal MΦ—Thioglycollate (TG)-elicited peritoneal MΦ were obtained according to previously established methods (13). Briefly, mice were injected intraperitoneally with 1.5 ml of a 4% sterile and aged (3 month) solution of Brewer thioglycollate medium (Difco Laboratories, Detroit, MI). At 3 days post injection, mice were euthanized with CO2, and the peritoneal cavity lavaged with 5 ml of sterile Hanks' balanced salt solution (Cellgro, Kansas City, MO). Peritoneal cells were collected, centrifuged at 1,000 × g for 10 min at 4 °C, and plated at 2.5 × 105 cells/well in a 24-well tissue culture plate in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA), 2 mm l-glutamine, 100 μg/ml streptomycin, and 100 units/ml penicillin. Macrophages were allowed to adhere for 2 h at 37 °C in a 10% CO2 humidified incubator at which time non-adherent cells were removed and macrophages were cultured for an additional 48 h before use in phagocytosis assays. Resident peritoneal (RP) MΦ were isolated from mice using 5 ml of sterile Hanks' balanced salt solution to lavage the peritoneum following euthanization with CO2. Resident peritoneal cells were collected, centrifuged at 1,000 rpm for 10 min at 4 °C, and plated at 4 × 105 cells/well and cultured as described for thioglycollate-elicited macrophages. Murine macrophage RAW264.7 cell line (from ATCC, Manassas, VA) was cultured in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mm l-glutamine, 100 μg/ml streptomycin, and 100 units/ml penicillin at 37 °C in a 5% CO2 humidified incubator. Cells were plated at 4 × 104 cells per well in a 24-well tissue culture plate for 48 h prior to phagocytosis assays.
Induction of Sterile Peritonitis—Mice were injected intraperitoneal with 1 mg of zymosan (in 1 ml of PBS) and peritoneal cells were harvested by lavage with sterile Hanks' balanced salt solution supplemented with 1 mm EDTA and 10 mm HEPES (pH 7.2) at the times indicated. Cell counts and cytospins were done to determine cell differentials and absolute numbers. In some cases, peritoneal cells were separated into neutrophils and mononuclear cell fractions by Percoll density gradient centrifugation as described by Suratt et al. (22). Peritoneal cells were suspended in 6 ml of PBS and peritoneal lavage fluid (6 ml) was subjected to lipid extraction as described below. To test the role of lyso-PS signaling via G2A during resolution of inflammation, mice were injected intraperitoneally (100 μg/mouse) with either goat IgG or anti-G2A antibody (dialyzed against PBS) 24 h post-zymosan injection. Peritoneal cells were collected by lavage at the times indicated. Total cells counts and differentials were determined as described above.
Lipid Extraction and Derivatization—Lipids were extracted by the method of Folch et al. (23). Briefly, peritoneal cells or human neutrophils (2-5 × 106/ml) in 6 ml of PBS, 6 ml of culture supernatant or peritoneal lavage fluid were extracted in a separatory funnel with CHCl3/MeOH/dH2O, 20:10:6 (v/v/v), supplemented with 70 mm NaCl. Fifty ng of 17:1/OH-PS, 1 μg of 19:0/OH-PC, and 1 μg of 14:0/OH-PE were added to each sample as internal standards and in some samples 10 μg of 1,2-dimyristoyl-sn-glycero-3-phosphoserine was also added. The extractions were allowed to sit overnight at room temperature. The organic layer was collected and brought to dryness under a stream of nitrogen. Reactive aldehyde or ketone groups were derivatized in the gas phase with MOX as described (24). In brief, sodium hydroxide (1 ml) and MOX (50 mg) were added together and attached to an enclosed glass apparatus. During a 60-min incubation at 60 °C, the liberated CH3ONH2 gas derivatized the ketone or aldehyde groups present on the oxidized phospholipids. Following derivatization, the dried lipids were suspended in 200 μl of CHCl3 and applied to a hexane-conditioned aminopropyl Sep-Pak (NH2-SPE) column to separate lipids by class (25, 26). The neutral lipids were eluted with 4 ml of CHCl3, isopropyl alcohol, 2:1 (v/v). Polar lipids (phosphatidylcholine and phosphatidylethanolamine) were collected with 4 ml of MeOH and the acidic lipids (phosphatidylserine, phosphatidic acid, and phosphatidylglycerol) were collected with 4 ml of MeOH, CHCl3, 3.6 m ammonium acetate, 60:30:8 (v/v/v). The acidic lipid fraction from the NH2-SPE, which contained PS was dried under nitrogen until 200 μl of liquid remained. An equal volume of MeOH was added to suspend the lipids and water was added to reduce the MeOH content to less than 10% of final volume. The ammonium acetate present in this NH2-SPE fraction was removed by introducing the acidic lipid fraction onto a MeOH-conditioned and rinsed HLB reverse phase column. Bound phospholipids were washed with 4-5 volumes of dH2O and eluted with 2 ml of MeOH/CHCl3, 2:1 (v/v), dried under a stream of nitrogen and suspended in 65 μl of reverse phase buffer A. During the course of phospholipid extraction using known quantities of a standard lyso-PS, it was determined that the recovery efficiency for lyso-PS species reached 50%.
Reverse Phase Chromatography and Electrospray Ionization Tandem Mass Spectrometry—RP-HPLC/MS/MS analysis of PS present in the methanol, chloroform, 3.6 m ammonium acetate, 60:30:8 (v/v/v), elution of the NH2-SPE was performed using an Eclipse Plus 3.5 μmC18 (2.1 × 50 mm) column (Agilent, Santa Clara, CA) with mass spectrometric detection using a Sciex API 3000 triple quadrupole mass spectrometer (PE Sciex, Toronto, Canada). The HPLC was operated at a flow rate of 0.2 ml/min with a mobile phase of methanol/acetonitrile/water, 60:20:20 (v/v/v), with 1 mm ammonium acetate (solvent A) and 1 mm methanolic ammonium acetate (solvent B). The gradient for the PS analysis was 0% solvent B to 100% solvent B in 20 min and then an isocratic hold at 100% B for 10 min. In some cases, the PS species were detected in the negative ion mode by monitoring for the neutral loss of serine (neutral loss of 87 atomic mass unit) with a collision energy of -30 V (27). The mass range scanned for neutral loss of the 87 atomic mass unit scan was m/z 400-900 at a rate of 3 s/scan. In other cases, multiple reaction monitoring (MRM) in the negative ion mode of m/z 508.5 → 421.5 for 17:1/OH-PS (internal standard), m/z 522.5 → 435.5 for 18:1/OH-PS, m/z 524.5 → 437.5 for 18:0/OH-PS, and m/z 707.6 → 620.5 for MOX derivatized 18:0/9al-PS was used to detect the major oxPS species eluting from the RP-HPLC column. In addition, minor oxPS species observed previously (16-18) were monitored in the negative ion mode with their specific MRM transitions (see supplemental Table S1). For PS analysis in the negative ion mode, the electrospray voltage was -4000 V, the focusing potential was -200 V, and the declustering potential was -45 V. In addition PC and PE were also analyzed using the same chromatography conditions above and using a precursor of m/z 184 and neutral loss of 141 atomic mass units in the positive ion mode to specifically detect PC and PE lipids, respectively (27).
Quantitation—The quantitation of 18:1/OH-PS and 18:0/OH-PS in the samples was performed using a standard isotope dilution curve as previously described (28). The internal standard used for this quantitative analysis was 17:1/OH-PS. Because small amounts of lyso-PS were generated during the MOX derivatization process, quantitation of all lyso-PS was carried out on duplicate samples in the absence of MOX derivatization.
Vesicle Preparation and
Modification—1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
small unilamellar vesicles (SUVs), some containing either
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine or
1-oleoyl-2-hydroxy-sn-glycero-3-phosphoserine (lyso-PS) (at the
indicated molar ratios) at a final concentration of 100 mm were
prepared by evaporating the lipid to dryness under nitrogen in a glass tube.
Dried lipids were suspended in PBS or media without serum or protein
supplementation by vigorous vortexing, and small unilamellar vesicles were
created by sonication in a water bath sonicator
(29). Small unilamellar
vesicles were stored on ice and used within 1 h of preparation. For
phagocytosis assays, 100 μl (100 nmol of total lipid) were used per well.
For in vitro oxidation, 100 μg/ml synthetic
1-stearoyl-2-linoleoyl-sn-glycero-3-phosphoserine or
1-stearoyl-2-oleoyl-sn-glycero-3-phosphoserine suspended in PBS was oxidized
using the
MPO/H2O2/ generating system according to the methods of Greenburg et al.
(18). Oxidation of lipids was
also performed using 2,2′-azobis(2-methylpropionamidine)-dihydrochloride
(Sigma) at a final concentration of 10 mm at 37 °C for 2.5 h.
To detect oxidized species containing reactive aldehydes or ketones, lipids
were derivatized in the gas phase with MOX as stated above. Following
derivatization, lipids were processed as stated above and suspended at 250
μg/ml in CHCl3/MeOH, 2:1. For LC/MS/MS, 5 μg of the lipid was
dried under nitrogen and suspended in 65 μl of reverse phase buffer A.
generating system according to the methods of Greenburg et al.
(18). Oxidation of lipids was
also performed using 2,2′-azobis(2-methylpropionamidine)-dihydrochloride
(Sigma) at a final concentration of 10 mm at 37 °C for 2.5 h.
To detect oxidized species containing reactive aldehydes or ketones, lipids
were derivatized in the gas phase with MOX as stated above. Following
derivatization, lipids were processed as stated above and suspended at 250
μg/ml in CHCl3/MeOH, 2:1. For LC/MS/MS, 5 μg of the lipid was
dried under nitrogen and suspended in 65 μl of reverse phase buffer A.
Surface G2A Staining—Murine resident or thioglycollate-elicited peritoneal macrophages or the RAW264.7 murine macrophage cell line were incubated with 5 μg/ml anti-G2A or isotype control for 1 h at 4 °C. Cells were washed 2 times with ice-cold PBS and incubated with donkey anti-goat IgG Alexa 488 for 30 min at 4 °C. Cells were washed 2 times with ice-cold PBS and analyzed by flow cytometry.
Stimulation and Preparation of Neutrophils for Lipid Extraction and Engulfment Assays—Human neutrophils were obtained from normal, healthy donors in accordance with a protocol reviewed and approved by the Institutional Review Board. Using endotoxin-free reagents and plasticware, human neutrophils were isolated by the plasma Percoll method as described previously (30). For in vitro studies, human neutrophils were suspended at 5 × 106/ml in a HEPES buffer (137 mm NaCl, 2.7 mm KCl, 2 mm MgCl2, 5 mm glucose, 1 mm CaCl2, 10 mm HEPES (pH 7.4)) supplemented with 0.05% fatty acid-free bovine serum albumin. For stimulation, zymosan was opsonized with pooled human serum as described previously (31) and added to neutrophils at a concentration of 200 μg/ml. Phorbol myristate acetate (PMA) was used at a final concentration of 20 ng/ml and fMLP at 100 nm. Cells were stimulated for the times indicated at 37 °C and subjected to lipid extraction and LC/MS/MS analysis as described above.
For phagocytosis assays, human neutrophils were stimulated with 20 ng/ml PMA for 30 min at 37 °C or UV irradiated for 5 min on a trans-illuminator followed by incubation at 37 °C for 2 h. Surface PS exposure was detected by Annexin V binding and propidium iodide staining (as a test for permeability) and flow cytometry according to the manufacturers instructions. Under these conditions UV-irradiated neutrophils were greater than 50% apoptotic as determined by nuclear morphology and were 63.4 ± 5.8% Annexin V positive, and >97% propidium iodide negative as determined by flow cytometry. PMA-stimulated neutrophils were less than 5% apoptotic as determined by nuclear morphology and were 43.5 ± 6.5% Annexin V positive, and >96% propidium iodide negative as determined by flow cytometry.
In some experiments human neutrophils were preincubated with liposomes according to the methods described by Fadok et al. (14) with minor modifications. Briefly, unstimulated viable neutrophils or neutrophils UV irradiated to induce apoptosis (as described above) were suspended at 2 × 107 cells/ml in PBS supplemented with 0.01% fatty acid-free bovine serum albumin. An equal volume of liposomes containing the indicated lipids in PBS (as described above) was added to the cells at a final concentration of 1 μmol of total lipid per 1 × 107 cells (final concentration of 0.005% bovine serum albumin). Cells were incubated at 37 °C for 30 min and washed twice with PBS and used in phagocytosis assays as described below.
Following stimulation or induction of apoptosis, neutrophils were washed once and suspended at 2 × 107 cells/ml in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum for use in the phagocytosis assays (see below). Where indicated, unstimulated viable neutrophils were opsonized with mouse anti-human CD45. Briefly, cells at 5 × 106/ml were incubated with 10 μg/ml anti-CD45 for 30 min at 4 °C. Binding of anti-CD45 was confirmed by incubating the cells with anti-mouse IgG Alexa 488 (1:100) for 30 min on ice and analysis by flow cytometry (data not shown). Anti-CD45-opsonized neutrophils were washed as stated above for use in phagocytosis assays.
Where the phagocytic index (PI) is represented as percent of control, the PI for control resident peritoneal macrophages with UV-irradiated neutrophils was 20.1 ± 2.72, and with COOH beads was 44.4 ± 6.4. The PI for thioglycollate-elicited macrophages with UV neutrophils was 8.5 ± 1.7, and for RAW264.7 with COOH beads was 49.0 ± 4.5.
Superoxide Measurement—Release of
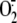 was determined by
cytochrome c reduction as described previously
(32).
was determined by
cytochrome c reduction as described previously
(32).
Phagocytosis Assays—Phagocytosis assays were performed as previously described (14). For antibody blocking experiments, 10 μg/ml anti-G2A or isotype control antibody was added for 30 min before liposomes or target cells were added. Liposomes (100 nmol in 100 μl) were added as indicted, either 1 h prior to or simultaneously with target cells. Target cells (either 2 × 106 UV-irradiated or PMA-stimulated neutrophils) or 2.5 × 105 COOH 5-μm beads in 100 μl were added per well. The macrophages/target cells were cocultured for 60 min at 37 °C in 10% CO2, washed three times with PBS, and stained with a modified Wright Giemsa stain (Fisher Scientific, Pittsburgh, PA). The phagocytic index (PI) was calculated by multiplying the percentage of MΦ that have phagocytosed by the average number of engulfed cells per MΦ (13). A minimum of 200 mΦ were counted blindly. Each condition was tested in duplicate using at least four mice per experiment and repeated 3-10 times as indicated.
Statistical Analysis—Statistical analyses and p value calculations were conducted using analysis of variance (JMP statistical program (SAS Institute, Cary, NC)). The Dunnett and Tukey-Kramer tests were used for single and multiple comparisons, respectively.
RESULTS
Lyso-PS Are Abundant Modified PS Species Produced in Neutrophils following Activation of the NADPH Oxidase—OxPS species have been identified following oxidation of synthetic PS liposomes and oxPS serves as a ligand for apoptotic cell recognition and removal involving CD36 on macrophages (18). We sought to identify oxPS species in human neutrophils stimulated to activate their NADPH oxidase. Analysis of the PS precursor pool from unstimulated neutrophils (Fig. 1A) was performed by LC/MS/MS monitoring for the neutral loss of 87 atomic mass units in negative ion mode, indicating loss of serine, a characteristic of PS species. The ionization efficiency of anionic phospholipids is nearly identical for dilute lipid solutions (33). Once the appropriate correction for 13C isotope effects is applied to the relative intensities of each of the diacyl-PS present in the electrospray mass spectrum (Fig. 1A), the mole percentage of various PS species present in neutrophils was determined (Fig. 1B). The presence of polyunsaturated fatty acids were demonstrated in 32% of the diacyl pool, whereas the remaining 68% contains monounsaturated or saturated fatty acid species (Fig. 1, A and B) and alkylacyl or alkenylacyl PS species were not identified.
FIGURE 1.

Human neutrophils are enriched with saturated and monounsaturated diacyl-PS precursor species. A, total ion mass spectrum from 15 to 25 min from the LC/MS/MS chromatogram of diacyl-PS species from control human neutrophils monitoring neutral loss of 87 atomic mass units in negative ion mode demonstrates a low abundance of diacyl-PS species containing polyunsaturated fatty acids (2 or more double bonds). The spectrum is representative if nine independent experiments, which are summarized and depicted in panel B. B, mole percentage of diacyl-PS from human neutrophils after 13C isotope correction. Percentage of diacyl-PS species containing 0-5 double bonds was determined in human neutrophils and murine resident peritoneal macrophages (inset; n = 6). C, collision-induced dissociation of the [M - H]- at m/z 786.7 shows this ion to be a mixture of 18:1/18:1-PS and 18:0/18:2-PS (inset: expanded view of m/z 260-300 atomic mass units showing the presence of 18:0 (m/z 283.1), 18:1 (m/z 281.1), and 18:2 (m/z 279.1) fatty acids). Bar graph shows the relative abundance of the fatty acid product ions (peak area of fatty acid/peak area of 1,2-dimyristoyl-sn-glycero-3-phosphoserine (internal standard)) from four independent experiments. All values of m/z are indicated as the nominal mass in this and subsequent figures.
The most abundant diacyl-PS species contains 1 double bond (36:1 at m/z 788.7) representing 57% of the total diacyl-PS pool. Among the PS species containing 2 or more double bonds, 36:2 (m/z 786.7) was the most abundant representing ∼20%. Collision-induced dissociation of the negative ion determined that 36:2-PS consisted of a mixture of 18:1/18:1-PS and 18:0/18:2-PS (Fig. 1C) reducing further the polyunsaturated fatty acid-containing diacyl-PS content from 32 to ∼20%. Also notable was the very low level of PS species containing four or more double bonds (e.g. arachidonate-containing species), representing less than 10% of the total PS pool. In contrast, neutrophil PC and PE pools demonstrate much greater enrichment with arachidonate (18 and 62%, respectively) (34). In contrast to the degree of saturation of the PS pool of human neutrophils, murine resident peritoneal macrophages (fig. 1B inset) or mouse liver cells (35) demonstrate that ∼80% of the PS pool has 2 or more double bonds of which 40-50% contain 4 or more.
Hypothesizing that stimulation of neutrophils was likely to yield oxPS
species in relatively low abundance, an aminopropyl Sep-Pak
(NH2-SPE) column was utilized to crudely separate phospholipid
classes thereby enhancing the sensitivity of oxPS detection. Additionally,
because reactive aldehydes and ketones were expected to covalently bind to
primary amines present on the NH2-SPE column, derivatization with
MOX was necessary for recovery
(24-26).
To confirm optimal recovery and detection of oxPS, synthetic PS containing
various fatty acyl groups was oxidized using
2,2′-azobis(2-methylpropiona-midine)-dihydrochloride as an oxidant
source and confirmed utilizing an
MPO/H2O2/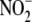 generating system to more closely approximate the oxidants found in
neutrophils stimulated to activate their NADPH oxidase
(18). Analysis by LC/MS/MS
monitoring, both at neutral loss of 87 atomic mass units in negative ion mode,
to detect the major oxPS species formed, as well as MRM of specific
transitions as a more sensitive method for detecting known oxidation products,
identified a number of expected oxPS species previously characterized
(18) (see supplemental Fig. S1
and Table S1). This confirmed that MRM could be used as a sensitive method to
detect the predicted oxPS species present upon stimulation of neutrophils.
generating system to more closely approximate the oxidants found in
neutrophils stimulated to activate their NADPH oxidase
(18). Analysis by LC/MS/MS
monitoring, both at neutral loss of 87 atomic mass units in negative ion mode,
to detect the major oxPS species formed, as well as MRM of specific
transitions as a more sensitive method for detecting known oxidation products,
identified a number of expected oxPS species previously characterized
(18) (see supplemental Fig. S1
and Table S1). This confirmed that MRM could be used as a sensitive method to
detect the predicted oxPS species present upon stimulation of neutrophils.
To identify products of PS oxidation in activated human neutrophils,
isolated neutrophils were stimulated with PMA resulting in robust activation
of the NADPH oxidase. Total lipids from control or stimulated neutrophils were
extracted and processed as described above and under “Experimental
Procedures” and analyzed by LC/MS/MS monitoring for the neutral loss of
87 atomic mass units as well as by MRM of specific modified PS transitions
likely to originate from the most abundant precursor diacyl-PS species
(e.g. 36:1-PS at m/z 788.5, 36:2-PS at
m/z 786.5, and 38:4-PS at m/z 810.5; also
see supplemental Table S1). The chromatogram and corresponding total ion mass
spectrum were obtained by monitoring the neutral loss of 87 atomic mass units
to detect PS species from control and PMA-stimulated neutrophils (30 min of
stimulation, the time of peak
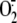 production with 79.9
± 0.2 compared with 1.6 ± 0.5 nmol of
production with 79.9
± 0.2 compared with 1.6 ± 0.5 nmol of
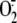 released/106
neutrophils from stimulated and unstimulated, respectively) is shown in
Fig. 2, A and
B. There was little detection of increased oxPS species
in PMA-stimulated neutrophils over that seen in control cells. More striking,
however, was the detection of increased lyso-PS species (m/z
522.5 representing 18:1/OH-PS) in stimulated neutrophils at ∼2.5-fold over
that seen in control cells (see supplemental Fig. S1 for a description of the
differences between diacyl-PS, oxPS, and lyso-PS).
released/106
neutrophils from stimulated and unstimulated, respectively) is shown in
Fig. 2, A and
B. There was little detection of increased oxPS species
in PMA-stimulated neutrophils over that seen in control cells. More striking,
however, was the detection of increased lyso-PS species (m/z
522.5 representing 18:1/OH-PS) in stimulated neutrophils at ∼2.5-fold over
that seen in control cells (see supplemental Fig. S1 for a description of the
differences between diacyl-PS, oxPS, and lyso-PS).
FIGURE 2.

Lyso-PS is an abundant modified PS species generated in human neutrophils stimulated to activate the NADPH oxidase. A, representative chromatography of control (top panel) or PMA-stimulated (bottom panel) human neutrophils monitoring neutral loss of 87 atomic mass units demonstrating the presence of chromatographic peaks corresponding to lyso-PS products with increases in 18:1/OH-PS at m/z 522.5 in stimulated over control cells. Very few peaks corresponding to oxidized PS products were evident. B, corresponding total ion mass spectrum from 0 to 25 min of the chromatography shown in panel A demonstrating increased lyso-PS production in stimulated (bottom panel) compared with control (top panel). Very few oxPS species were detectible. C, MRM of specific oxPS transitions, as described under “Experimental Procedures” and supplemental Table S1, in control (left panel) and PMA-stimulated (right panel) neutrophils. Very prominent is the increased production of lyso-PS in stimulated over control cells. Increases in the production of 18:0/9-al-PS is visible in PMA-stimulated neutrophils and is further demonstrated in the inset: the area between 8 and 20 min was expanded by ×50 to view oxPS species.
Neutral loss of 87 atomic mass units is not the most sensitive mass spectrometric method to detect oxPS, but is useful as a preliminary survey scan to identify all the major oxPS species formed upon stimulation of neutrophils. Therefore, the same control and PMA-stimulated neutrophils were also analyzed by MRM for specific transitions of predicted oxPS species, as MRM is more sensitive than neutral loss scanning. Although trace amounts of the predicted oxPS species were detected, there was no change in any of the species with stimulation except for a modest increase in 18:0/9-al-PS (m/z 707.7) and 18:0/5-al-PS (m/z 651.6) observed at this 30-min time point (Fig. 2C, see inset). These data suggest that in human neutrophils, lyso-PS species, known signaling phospholipids, are additional and abundant modified PS species generated following NADPH oxidase activation.
Generation of Lyso-PS Is Dependent on Activation of the NADPH Oxidase—Extending this observation to a more relevant NADPH oxidase activator, a time course of lyso-PS production between 5 and 120 min of opsonized zymosan (OPZ) stimulation is shown in Fig. 3, A and B. Using an internal standard (17:1/OH-PS) and a standard curve (see “Experimental Procedures”), both 18:1/OH-PS and 18:0/OH-PS were quantitated. It should be mentioned that whereas 17:1/OH-PS is not a stable isotope-labeled internal standard for 18:0/OH-PS or 18:1/OH-PS, the deuterated analogs of these lyso-PS species were not readily available. Therefore, an odd chain lyso-PS (17:1/OH-PS), which is not present endogenously, was used as the internal standard. An increase of lyso-PS was observed as early as 15 min and peaking at 60 min following addition of OPZ (Fig. 3A). Production over time expressed as total lyso-PS (both species added together) demonstrated a 2-3-fold increase in lyso-PS production after OPZ stimulation (Fig. 3B) with peak quantities reaching 250 ng (477 pmol) per 107 cells.
FIGURE 3.

Lyso-PS production is dependent on NADPH oxidase activation. A, human neutrophils were stimulated with OPZ for the times indicated and the major lyso-PS species (18:1/OH-PS and 18:0/OH-PS) were quantitated (see “Experimental Procedures”). Data represent mean ± S.E.; n = 4 experiments. B, lyso-PS species and superoxide production (C) were quantitated in neutrophils stimulated with OPZ for the times indicated in the absence (black circles) or presence (gray circles) of 6.6 μm DPI to inhibit the NADPH oxidase (mean ± S.E.; n = 5, *, p < 0.0001 versus control; #, p < 0.002 versus control, Ctr). D, human neutrophils were stimulated with 200 μg/ml OPZ for 15 min, 20 ng/ml PMA for 30 min, 100 nm fMLP for 15 min, or UV-irradiated to induce apoptosis, in the absence or presence of 6.6 μm DPI and lyso-PS was quantitated (mean ± S.E.; n = 3, *, p < 0.0001 versus control).
Lyso-PS generation in OPZ-stimulated neutrophils was dependent on
activation of the NADPH oxidase as pretreatment with DPI, an inhibitor of the
NADPH oxidase, suppressed both lyso-PS
(Fig. 4B) and
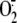 production
(Fig. 3C).
Quantitation of lyso-PS following PMA stimulation reached quantities of 350 ng
(677 pmol) per 107 cells (Fig.
3D). Dependence on activation of the NADPH oxidase for
lyso-PS generation in response to PMA was similarly demonstrated in
neutrophils pretreated with DPI. Conversely, stimulation with fMLP, which
resulted in minimal activation of the NADPH oxidase (4.7 ± 0.3 compared
with 1.6 ± 0.5 nmol of
production
(Fig. 3C).
Quantitation of lyso-PS following PMA stimulation reached quantities of 350 ng
(677 pmol) per 107 cells (Fig.
3D). Dependence on activation of the NADPH oxidase for
lyso-PS generation in response to PMA was similarly demonstrated in
neutrophils pretreated with DPI. Conversely, stimulation with fMLP, which
resulted in minimal activation of the NADPH oxidase (4.7 ± 0.3 compared
with 1.6 ± 0.5 nmol of
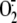 released/106
neutrophils, stimulated and unstimulated, respectively) failed to generate a
detectible increase in lyso-PS. As an additional control, UV-irradiated
apoptotic neutrophils, which do not activate the NADPH oxidase, were analyzed,
and no increased production of lyso-PS over control cells was found
(Fig. 3D).
released/106
neutrophils, stimulated and unstimulated, respectively) failed to generate a
detectible increase in lyso-PS. As an additional control, UV-irradiated
apoptotic neutrophils, which do not activate the NADPH oxidase, were analyzed,
and no increased production of lyso-PS over control cells was found
(Fig. 3D).
FIGURE 4.

Lyso-PS is produced by neutrophils in vivo during sterile peritonitis and requires activation of the NADPH oxidase. A, C57BL/6 WT mice were injected intraperitoneally with 1 mg of zymosan and peritoneal lavages were collected at the times indicated. Total neutrophils, monocyte/macrophage, and apoptotic neutrophil numbers were determined. B, a representative LC/MS/MS chromatogram of oxPS obtained from monitoring the neutral loss of 87 atomic mass units in peritoneal cells from 6-h zymosan-treated mice (top panel) and 24-h zymosan-treated mice (middle panel). Prominent chromatographic peaks corresponding to the lyso-PS product 18:0/OH-PS at m/z 524.5 were observed at both time points. The bottom panel shows the corresponding total ion mass spectrum from 0 to 25 min for both the 6- and 24-h time points. Note the presence of lyso-PS compared with other oxPS products. C, lyso-PS was quantitated from total peritoneal cells or cells that were gradient separated into polymorphonuclear leukocytes (PMN) and mononuclear cell fractions collected at 6 h following zymosan injection. D, lyso-PS was quantitated at the times indicated following intraperitoneal injection of zymosan in wild-type mice or in gp91phox-/- mice lacking a functional NADPH oxidase (n = 3-10 mice per time point investigated; *, p < 0.0001 versus time 0; α, p < 0.02 versus time 0). E, gp91phox-/- mice were injected intraperitoneally with 1 mg of zymosan. Peritoneal lavages were collected at the times indicated and total neutrophils and monocyte/macrophage numbers were determined.
In all instances only cell-associated lyso-PS species were detected and lyso-PS were undetectable in culture supernatants in the presence of 0.05% bovine serum albumin. Only trace amounts of oxPS species were detected by MRM in cells over a time course of stimulation with PMA, OPZ, or UV (Fig. 2C and data not shown). Additionally, evidence for production of other lysophospholipids (lyso-PC and lyso-PE) was sought, but no increase was detected over baseline controls under any of the conditions assayed (data not shown).
Activation of a PLA2 in tandem with the NADPH oxidase was considered a potential source of lyso-PS, either directly acting on diacyl-PS, or utilizing oxPS as a substrate and thereby limiting the accumulation and detection of oxPS species. To test the role of various PLA2s in lyso-PS production, inhibitors were employed at standard concentrations shown to inactivate their respective enzyme. OPZ stimulation of neutrophils activates cPLA2α in concert with the NADPH oxidase (36, 37), and was therefore considered first, although very little of the potential PS precursor pool appeared to contain arachidonic acid (Fig. 1B) for which this enzyme is specific.
Lyso-PS production in OPZ-stimulated neutrophils was unchanged in the presence of 5 μm pyrrolidine, a cPLA2α inhibitor, at a concentration that completely suppressed cPLA2α activity (data not shown) (38). Similarly, neither bromoenol lactone, an inhibitor of Group VI PLA2s including iPLA2, nor ONO-RS-082, a general phospholipase inhibitor (for sPLA2 inhibition), had any effect on lyso-PS production (data not shown). Taken together, these data suggest that lyso-PS is generated either via activity of another PLA2 (e.g. PAF acetylhydrolase) or a PS-PLA1 (39-42) for which inhibitors are unavailable, or alternatively by non-enzymatic ester hydrolysis of diacyl-PS or oxPS by the reactive oxygen species from the NADPH oxidase (see “Discussion”).
Lyso-PS Accumulates following Activation of the Neutrophil NADPH Oxidase in Vivo—To determine whether production of lyso-PS occurred in vivo, intraperitoneal injection of zymosan in C57BL/6 mice was used to induce self-limited, sterile peritonitis characterized by initial neutrophilia followed by monocytic influx with resolution of the neutrophil phase over 48 h (43). At designated times, peritoneal cells were collected by lavage and total cell counts and differentials were determined. The lipids from both the cells and the lavage fluid were extracted, derivatized, class separated, and analyzed by LC/MS/MS monitoring for the neutral loss of 87 atomic mass units and MRM for the presence of lyso-PS and oxPS, respectively. As shown in Fig. 4A, neutrophilic infiltration was evident by 3 h, peaked at 6 h where it was maintained through 24 h, and resolved thereafter. By 18 h, apoptotic neutrophils (identified by nuclear morphology (44)) were evident, but infrequent, an observation in keeping with efficient removal of apoptotic cells (45, 46). Similar to the spectra from stimulated human neutrophils, very few oxPS species were detected (e.g. 18:0/9-al-PS at m/z 707.5) at any of the time points as analyzed by neutral loss of 87 atomic mass units (Fig. 4B) or MRM (data not shown). Conversely, lyso-PS species (18:0/OH-PS at m/z 524.5 being the predominant product in murine peritoneal cells, with 18:1/OH-PS at m/z 522.5 the minor species) were easily detected (6 and 24 h neutral loss of the 87 atomic mass unit chromatogram and corresponding total ion mass spectrum are shown in Fig. 4B).
To determine whether neutrophils were the source of lyso-PS, peritoneal cells lavaged at 6 h (a time when neutrophils comprise 80% of the cells (Fig. 4A)), were subsequently gradient separated into neutrophil and mononuclear cell fractions. As shown in Fig. 4C, neutrophils were determined to be the source of essentially all the detectable lyso-PS. A complete time course to assess accumulation showed that lyso-PS production was detected by 3 h following zymosan injection, production continued to increase over time with peak production detected at 24 h (Fig. 4D). In all instances lyso-PS was only detected in cells and was not detected in the lavage fluid, whereas lyso-PC (16: 0/OH-PC, 18:1/OH-PC, and 18:0/OH-PC) was transiently detected in the lavage fluid early (only at 1 h, a time before detectable neutrophil recruitment) during the time course of inflammation (data not shown).
Requirement for NADPH oxidase activity in the production of lyso-PS in vivo was also assessed utilizing the gp91phox-/- mouse lacking a functional NADPH oxidase. In contrast to wild-type mice, lyso-PS production in the gp91phox-/- mouse was suppressed throughout the time course (Fig. 4D) despite robust and sustained neutrophilia characteristic of this murine model of chronic granulomatous disease (Fig. 4E) (47). These data demonstrate that lyso-PS production also occurs in vivo in neutrophils and requires activation of a functional NADPH oxidase.
Lyso-PS Enhances PS-dependent Engulfment of Apoptotic Cells—Given the timing of lyso-PS accumulation in the relationship to the resolution of neutrophilic inflammation in zymosan-induced peritonitis and the rarity of morphologically apoptotic neutrophils (Fig. 4, A and D), a role for lyso-PS in apoptotic neutrophil clearance was hypothesized. To test this hypothesis, phagocytosis assays of UV-irradiated neutrophils (which do not produce an increase in detectible lyso-PS or oxPS (Fig. 3D)) by resident peritoneal macrophages (RPMΦ) were performed in vitro. Following either pretreatment (1 h) or simultaneous addition of liposomes containing lyso-PS, diacyl-PS, or carrier lipid alone, UV-irradiated apoptotic neutrophils (demonstrating greater than 50% apoptosis by nuclear morphology) were cocultured with macrophages and their engulfment assessed. Both pretreatment and simultaneous addition of lyso-PS liposomes (but not PC carrier lipids) enhanced engulfment in a dose-dependent manner with 30 mol % resulting in the greatest enhancement (Fig. 5A).
FIGURE 5.

Lyso-PS enhances engulfment of apoptotic cells or surrogate carboxylate-modified beads. A, resident peritoneal macrophages (RPMΦ) were treated with liposomes containing carrier lipid (PC) or the indicated mole percent of either PS or lyso-PS for 1 h prior to, or simultaneously with, the addition of UV-irradiated apoptotic neutrophils and the phagocytic index was determined after 1 h. Data represent mean ± S.E.; n = 10 experiments; *, p < 0.0001 versus control; π, p < 0.05 versus control. B, thioglycollate-elicited peritoneal macrophages (TGMΦ) were pretreated with liposomes (carrier PC alone or 30 mol % lyso-PS or PS) and the phagocytic index determined as in A. Data represent mean ± S.E.; n = 3 experiments; *, p < 0.0001 versus control. C, engulfment of apoptotic cell mimics (COOH modified 5 μm beads) in both RPMΦ and RAW264.7 murine macrophage cell lines was determined following pretreatment with liposomes as in B. Data represent mean ± S.E.; n = 6 experiments; α, p < 0.02 versus control; π, p < 0.05 versus control. D, the effect of liposomes on phagocytosis of apoptotic, viable or anti-CD45 opsonized cells in RPMΦ pretreated as in B was also determined. Data represent mean ± S.E.; n = 4 experiments; *, p < 0.0001 versus control (Ctr).
The observed increase in engulfment accompanying lyso-PS treatment was in contrast to treatment of macrophages with liposomes containing 30 mol % diacyl-PS, which inhibited the uptake of apoptotic cells as has been shown in previous investigations and presumed to be a result of blockade or internalization of receptors directly recognizing PS (13, 18). To test whether the effect of lyso-PS was specific for RPMΦ, thioglycollate-elicited peritoneal macrophages (TGMΦ) were also assessed (Fig. 5B) with identical results obtained for these inflammatory cells.
Although analysis by LC/MS/MS demonstrated that UV-irradiated neutrophils produced no detectible increase in the oxPS or lyso-PS species (Fig. 3D), to eliminate any possible contribution of signaling lipids from the apoptotic cells themselves, the effect of lyso-PS on the engulfment of carboxylate-modified (COOH) beads, which act as mimics of PS-exposing cells (48, 49), was examined. Fig. 5C shows that pretreatment with lyso-PS, but not diacyl-PS or PC liposomes (carrier lipid), enhanced engulfment of COOH beads by both RPMΦ and RAW264.7 cells similarly to that seen with apoptotic cells.
We also investigated whether the enhanced uptake by lyso-PS was specific for apoptotic cell engulfment or was nonspecific for other types of phagocytosis. For these studies, unstimulated viable and IgG opsonized viable neutrophils were utilized as targets for engulfment. Neither lyso-PS, nor diacyl-PS (as previously published) had any effect on these other forms of phagocytosis (fig. 5D).
Lyso-PS Enhances PS-dependent Engulfment of Apoptotic Cells through Macrophage G2A Signaling—We have recently reported that lyso-PS can signal through the G protein-coupled receptor, G2A, for calcium flux (20), and G2A has been recently described as a receptor on macrophages responsible for mediating macrophage chemotactic responses (50, 51). Accordingly, G2A appeared to be a reasonable candidate for lyso-PS-dependent enhancement of apoptotic cell engulfment. RPMΦ, TGMΦ, and RAW264.7 cells were stained with anti-G2A antibody (or isotype control antibody) and analyzed by flow cytometry for surface G2A. As shown in Fig. 6, G2A was clearly present on the surface of each of these macrophage populations. To test the requirement for G2A in lyso-PS-dependent enhancement of apoptotic cell engulfment, the macrophage populations were pretreated with blocking antibody to G2A (or isotype control) for 30 min and then given liposomes containing lyso-PS, PS, or carrier lipid alone followed by UV-irradiated apoptotic neutrophils or COOH. Anti-G2A blocking antibody inhibited the lyso-PS-enhanced engulfment of both apoptotic cells as well as COOH by all macrophages (Fig. 6). No appreciable effect of G2A blocking antibody was seen on PC- or PS-treated macrophages. These data demonstrate that lyso-PS signals via the G protein-coupled receptor G2A for the enhanced engulfment of apoptotic cells.
FIGURE 6.

Lyso-PS enhances engulfment of apoptotic cells by various macrophages via signaling through the G protein-coupled receptor G2A. Surface G2A was determined on RPMΦ (A), RAW264.7 murine macrophages (B), or TGMΦ (C) by staining with anti-G2A or isotype control antibody and analysis by flow cytometry (shown is a representative histogram from three independent experiments). The role of G2A in phagocytosis was determined for each macrophage population as described in the legend to Fig. 5, following incubation with 5 μg of isotype control antibody or anti-G2A blocking antibody for 30 min prior to liposome pretreatment. Macrophages were fed either apoptotic cells (A and C) or COOH beads (apoptotic cell mimics) (B) and their phagocytic index was determined after 1 h. All data represent mean ± S.E.; n = 3-6; *, p < 0.0001 versus control; #, p < 0.002 versus control; α, p < 0.02 versus control (Ctr).
Endogenous Production of Lyso-PS Enhances PS-dependent Clearance of Activated Neutrophils—Lagasse and Weissman (52) showed that neutrophils infiltrating into the inflamed peritoneum were cleared in vivo with identical kinetics whether they were apoptotic as defined by DNA degradation (wild-type) or not (in those overexpressing Bcl-2). Additionally, we noted that peritoneal neutrophils, although rarely exhibiting apoptotic nuclear features (fig. 4A), frequently exposed PS (Annexin V+ and PI-; data not shown) confirming our own and others work demonstrating PS exposure on activated cells (53, 54). Given these observations, we hypothesized that PS-exposed, activated neutrophils might be recognized similarly to apoptotic cells, and that lyso-PS production might enhance their optimal engulfment via G2A-mediated signaling. A series of experiments employing neutrophils stimulated with PMA (30 min) in the absence and presence of DPI to inhibit the NADPH oxidase and consequent lyso-PS production were performed. As shown in representative dot plots, PMA-stimulated neutrophils expose PS, although less so than UV-irradiated apoptotic neutrophils (Fig. 7A) while demonstrating no other features of apoptosis (e.g. nuclear condensation and hypodiploid DNA; data not shown). Engulfment by resident peritoneal macrophages of PMA-stimulated neutrophils and UV-irradiated apoptotic neutrophils was similar, with activated neutrophils taken up slightly better (Fig. 7B). Pretreatment with DPI to inhibit the NADPH oxidase had little effect on PS exposure (Fig. 7A), but suppressed lyso-PS production in PMA-treated neutrophils (Fig. 3D). Importantly, DPI pretreatment also reduced the engulfment of PMA-stimulated neutrophils to equal that observed with UV-irradiated apoptotic neutrophils as targets demonstrating that signals generated in an NADPH oxidase-dependent manner (e.g. lyso-PS) enhanced PS-dependent phagocytosis. DPI had no effect on the engulfment of UV-irradiated apoptotic neutrophils (Fig. 7B) in which the NADPH oxidase is not activated and lyso-PS is not produced (Fig. 3D). Taken together, these data support the hypothesis that activated PS-exposed neutrophils in the absence of apoptosis are recognized and engulfed by macrophages, and that lyso-PS production enhances this process.
FIGURE 7.

Endogenously produced lyso-PS enhances engulfment of activated neutrophils via signaling through G2A. A, human neutrophils were pretreated in the absence or presence of DPI to inhibit the NADPH oxidase and stimulated with 20 ng/ml PMA for 30 min or UV-irradiated to induce apoptosis. Surface PS exposure was assessed by Annexin V staining and cell permeability with propidium iodide as analyzed by flow cytometry (shown is a representative dot plot from six independent experiments). B, RPMΦ were fed human neutrophils described in A (PMA stimulated, left panel; Apoptotic, right panel) in the absence or presence of isotype control or anti-G2A blocking antibody pretreated for 30 min prior to feeding human neutrophils. The phagocytic index was determined after 1 h. Data represent mean ± S.E.; n = 6; *, p < 0.0001 versus the no antibody control. C, RPMΦ were pretreated with isotype control antibody or anti-G2A blocking antibody as described above prior to pretreatment with lyso-PS liposomes as described in the legend to Fig. 5. Macrophages were then fed neutrophils stimulated with PMA in the absence or presence of DPI and the phagocytic index determined after 1 h. Data represent mean ± S.E.; n = 4; *, p < 0.0001 versus control; #, p < 0.002 versus control. D, UV-irradiated apoptotic or unstimulated viable neutrophils were pre-treated with the indicated liposomes prior to being co-cultured with RPMΦ. The phagocytic index was determined in the presence of either isotype control antibody or blocking antibody to G2A. Data represent mean ± S.E.; n = 4; *, p < 0.0001 versus PBS control (Ctr).
Endogenous Lyso-PS Enhances Clearance of Activated Neutrophils through G2A-mediated Signaling—As in experiments above in which lyso-PS signaling via G2A enhanced the clearance of apoptotic cells, we asked whether macrophage G2A played a role in the clearance of activated neutrophils. As predicted, pretreatment of macrophages with blocking antibody to G2A (but not isotype control) reduced the engulfment of PMA-stimulated neutrophils by a similar magnitude as DPI pretreatment (Fig. 7B). Anti-G2A had no effect on the engulfment of UV-irradiated cells or neutrophils stimulated with PMA in the presence of DPI in which there was no detectable increase in lyso-PS production. Finally, addition of lyso-PS liposomes restored the enhancement of engulfment of DPI/PMA-treated neutrophils, an effect that was prevented by anti-G2A blocking antibody (Fig. 7C). These data demonstrate that endogenous production of lyso-PS in PMA-stimulated neutrophils signals via the macrophage G2A receptor thereby enhancing their engulfment (see “Discussion”).
Although either simultaneous or pretreatment of macrophages with liposomes containing lyso-PS were shown to enhance the engulfment of UV-irradiated apoptotic neutrophils but not unstimulated viable cells (Fig. 5D), we asked whether pre-loading of the target cell plasma membrane with lyso-PS would also enhance their engulfment via G2A signaling. UV-irradiated apoptotic neutrophils or unstimulated viable neutrophils were incubated with PBS or liposomes containing lyso-PS or carrier lipid alone for 30 min followed by vigorous washing prior to coculturing with macrophages. All cells were assessed for permeabilization by propidium iodide staining, and in all cases, less than 3% of the cells were propidium iodide positive indicating that the lipid treatments were well tolerated (data not shown). PS exposure (as measured by Annexin V staining) was not increased for either the UV-irradiated apoptotic or unstimulated viable neutrophils following incubation with either PBS or lyso-PS liposomes. Fig. 7D shows the phagocytic index of RPMΦ cocultured with either UV-irradiated apoptotic or unstimulated viable neutrophils preincubated with the indicated liposomes in the presence of either isotype control antibody or blocking anti-G2A antibody to assess the role of G2A signaling. As with the addition of lyso-PS in liposomes to macrophages (Fig. 5), pre-loading of the target cell with liposomes containing lyso-PS enhanced only the engulfment of UV-irradiated PS-exposing apoptotic cells, and not unstimulated viable cells, and did so in a G2A-dependent manner. Taken together, these data demonstrate that endogenous production of lyso-PS significantly enhances existing PS-dependent uptake mechanisms via signaling through macrophage G2A.
The Timely Clearance of Activated and Apoptotic Neutrophils in Vivo Is Assisted by G2A Signaling—Finally, we asked whether blockade of G2A signaling would delay clearance of recruited neutrophils in vivo. Blocking antibody to G2A (or isotype control antibody) was injected intraperitoneally at 24 h following zymosan at a time when neutrophil numbers are at a maximum and just before they are rapidly cleared from the peritoneum. As hypothesized, anti-G2A slowed resolution of neutrophilic inflammation, whereas isotype control antibody had no effect (Fig. 8A). Furthermore, neutrophils demonstrating obvious apoptotic nuclear morphology were also increased in the presence of anti-G2A antibody (Fig. 8B). Neither antibody had any effect on lyso-PS levels present in the peritoneal cells (data not shown) or on numbers of mononuclear cells recovered from the peritoneum (Fig. 8C). The specificity of this effect for lyso-PS was determined by injecting either isotype control antibody or anti-G2A blocking antibody into gp91phox-/- mice that do not generate lyso-PS during zymosan-induced peritonitis. No effect on neutrophil or monocyte/macrophage numbers was seen with either antibody. Together, these data support the hypothesis that activated neutrophils assist in driving their own clearance by the production of lyso-PS, which facilitates signaling via macrophage G2A (see “Discussion”).
FIGURE 8.

G2A blockade significantly delays clearance of recruited neutrophils during zymosan-induced peritonitis. C57BL/6 wild-type mice were injected intraperitoneally with zymosan at time 0 followed by intraperitoneal injection of 100 μg/mouse anti-G2A or isotype control antibody at 24 h (indicated by the arrow). Cells were collected by peritoneal lavage at the times indicated. Total numbers of neutrophils (A), apoptotic neutrophils (B), and mononuclear cells (C) were determined. The dashed line is the time course without antibody treatment (from Fig. 4). All data represent mean ± S.E.; n = 4; *, p < 0.0004 versus isotype control; #, p < 0.006 versus isotype control; α, p < 0.02 versus isotype control.
DISCUSSION
Lyso-PS Are Abundant Modified PS Species Generated in Neutrophils Stimulated to Activate the NADPH Oxidase—The signaling potential of lyso-PS was first recognized in its description as a potent mast cell secretagogue, and it has subsequently been given signaling roles in other settings (20, 21, 39, 55, 56). Here, we have shown that lyso-PS species were generated in vitro and in vivo as major modified PS species in neutrophils stimulated to activate the NADPH oxidase. Quantitation of lyso-PS levels in human neutrophils stimulated with either OPZ or PMA were estimated to reach up to 350 ng/107 neutrophils (equal to 670 ± 49 pmol versus 190 ± 25 pmol in untreated cells). Similarly, up to 80 ng/107 cells of lyso-PS was recovered from the peritoneal cells in vivo (150 ng of total lyso-PS/peritoneal lavage, equal to 285 ± 25 pmol versus 3 ± 2 pmol in controls). The entire PS pool represents ∼4 ± 1.9% of the total membrane phospholipid in human neutrophils (3320 pmol per 107 cells) (34), and based on these values, we estimate that up to 20% of total PS was converted to lyso-PS species under our conditions of stimulation.
Lyso-PS Production Requires a Functional NADPH Oxidase—Ex vivo or in vivo stimulation of neutrophils in which the NADPH oxidase was inhibited or genetically deleted did not result in a detectible increase in lyso-PS over baseline (Figs. 3 and 4). Baseline differences in lyso-PS levels in unstimulated human neutrophils were noted and were somewhat variable averaging ∼100 ng/107 cells (Fig. 3A). Whether baseline levels of lyso-PS were generated during the 3-h isolation procedure (and inadvertent activation) is unclear. Nonetheless, under conditions of deliberate activation, only stimuli that activated the NADPH oxidase resulted in increased production of lyso-PS; conditions failing to activate it (e.g. fMLP which activates cPLA2α, or UV irradiation-induced apoptosis) did not generate lyso-PS (Fig. 3D).
Although NADPH oxidase activation appears to be required for lyso-PS production, we have not demonstrated whether it alone is sufficient. Involvement of cPLA2α or iPLA2 is unlikely given the lack of effect of inhibitors (data not shown). Furthermore, the relative lack of arachidonate-containing precursor PS species (Fig. 1B) would limit substrate availability for cPLA2α. Appropriate inhibitors (selective and cell permeable) for neutrophil secretory PLA2s(e.g. sPLA2 V and X), PAF-AH or the PS-PLA1 are not available, and genetic manipulation of these in terminally differentiated cells is difficult. However, whereas involvement of various phospholipases cannot be ruled out entirely, it has been previously reported that hypochlorous acid, generated from MPO oxidation of hydrogen peroxide, results in the production of lyso-PCs from some diacyl-PC species in vitro (57, 58) and may therefore be a mechanism for lyso-PS generation as well.
Lyso-PS Enhances PS-dependent Engulfment of Apoptotic and Activated Viable Cells—In our investigation of a peritonitis model, apoptotic neutrophils with obvious morphological features were identified, although rarely (Fig. 4A). These observations are in keeping with data demonstrating the highly efficient manner in which apoptotic cells are cleared. Indeed, given the rapidity by which clearance occurs, accumulation of significant numbers of apoptotic cells in tissues is unusual unless engulfment is defective (45, 46).
We hypothesize that lyso-PS are significant bioactive PS species generated by activated neutrophils that ultimately signal via macrophage G2A for their enhanced clearance from inflamed tissues. Our in vitro experiments allowed a clear demonstration that lyso-PS specifically enhanced the engulfment of both stimulated viable as well as apoptotic neutrophils that expose PS, but not unstimulated or IgG opsonized viable cells. Almost half of the signal for optimal engulfment of activated PS-exposing neutrophils was attributable to lyso-PS produced endogenously during activation of the NADPH oxidase as compared with neutrophils stimulated in the presence of DPI where increased lyso-PS production was suppressed (Fig. 7B).
One possibility suggested by our observations is that some of the neutrophils in an inflammatory reaction are actually cleared before they become apoptotic as a result of PS becoming exposed on the cell surface following stimulation. Lyso-PS that is generated following activation of the NADPH oxidase may therefore serve to maintain the engulfment capacity of macrophages. Whether neutrophils exposing PS and generating lyso-PS in vivo are truly “eaten alive” or are beginning to undergo early apoptosis is at this point unclear and there is likely a continuum between PS exposure during activation and the appearance of other markers signifying cell death (59). We and others have previously published that PS exposure occurs on activated cells including neutrophils (53, 54), and that PS on living cells can act as an “eat me” signal in mammalian systems (14) and possibly in Caenorhabditis elegans (60). In addition, this possibility may also explain the findings of Lagasse and Weissman (52) who demonstrated that activated neutrophils overexpressing Bcl-2, which were devoid of classic features of apoptosis (e.g. nuclear fragmentation), were cleared equivalently to normal wild-type neutrophils that were presumably undergoing apoptosis.
Enhanced Clearance by Lyso-PS Is Mediated by Signaling through the Macrophage G2A Receptor—Although other signaling lipids known to enhance clearance of apoptotic cells and/or resolution of neutrophilic inflammation (e.g. lipoxins, resolvins, and protectins) are generated and released (19, 61, 62), lyso-PS appears to remain cell-associated. This would imply a localized enhancing effect only on the phagocytes that come into contact with the lyso-PS exposing cell. Blockade of G2A on macrophages inhibited lyso-PS enhancement of uptake of both PS-exposing activated and apoptotic neutrophils (Figs. 6 and 7). Furthermore, blockade of G2A signaling in vivo significantly delayed clearance of peritoneal neutrophils (Fig. 8). We speculate that the propensity for G2A knock-out mice to develop late-onset autoimmunity reminiscent of systemic lupus erythematosus (a disease associated with defective clearance of apoptotic cells) (63) may be secondary to an overall engulfment deficit of neutrophils from inflamed tissues. Analogous to lipoxins and other lipid mediators shown to enhance resolution of inflammation, the results presented here demonstrate that lyso-PS signaling through the macrophage G2A receptor also enhances existing pathways of recognition and removal likely involving multiple receptors and bridge molecule combinations (64).
Whether lyso-PS production is specific to neutrophils or may be generated in other cells undergoing activation or apoptosis, in particular those expressing NADPH oxidases, remains to be determined. We hypothesize that depending on cell type and conditions, signals for cell clearance will differ, although redundancy in responses is expected given the importance of this process in tissue homeostasis. Furthermore, activated and apoptosing cells may present a mix of signaling PS species: diacyl-PS, oxPS, and lyso-PS in addition to other signaling phospholipids (62, 65, 66), and non-lipid ligands (67, 68). Such complex signaling and its integration in cellular and tissue responses will require a systematic approach toward the study of a growing number of signals from bioactive lipids important in resolution of inflammation.
Supplementary Material
Acknowledgments
We thank Dr. Aideen Byrne for helpful discussion and Dr. Moumita Ghosh, Jenai Kailey, and Dianne Ashby for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants AI058228 (to D. L. B.), HL34303 (to D. L. B., C. C. L., and R. C. M.), and GM61031 and HL81151 (to P. M. H.). This work was also supported by a grant from the Immunodeficiency Foundation (to D. L. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and Figs. S1 and S2.
Footnotes
The abbreviations used are: PS, phosphatidylserine; lysophosphatidylserine(s); oxPS, oxidized phosphatidylserine; PC, phosphatidylcholine; PE, phosphatidylethanolamine; COOH, carboxylate-modified beads; MRM, multiple reaction monitoring; MOX, methoxylamine hydrochloride; MPO, myeloperoxidase; OPZ, opsonized zymosan; DPI, diphenyleneiodonium; PI, phagocytic index; RPMΦ, resident peritoneal macrophage; TGMΦ, thioglycollate-elicited peritoneal macrophages; 18:0/9-al-PS, 1-stearoyl-2-(9′-oxo-nonanoyl)-glycerophosphoserine; 18:0/5-al-PS, 1-stearoyl-2-(5′-oxovaleroyl)-glycerophosphoserine; PLA2, phospholipase A2; PMA, phorbol 12-myristate 13-acetate; PBS, phosphate-buffered saline; fMLP, formylmethionylleucylphenylalanine; RP-HPLC, reverse phase-high performance liquid chromatography.
References
- 1.Freire-de-Lima, C. G., Xiao, Y. Q., Gardai, S. J., Bratton, D. L., Schiemann, W. P., and Henson, P. M. (2006) J. Biol. Chem. 281 38376-38384 [DOI] [PubMed] [Google Scholar]
- 2.Fadok, V. A., Bratton, D. L., Konowal, A., Freed, P. W., Westcott, J. Y., and Henson, P. M. (1998) J. Clin. Investig. 101 890-898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossi, A. G., Hallett, J. M., Sawatzky, D. A., Teixeira, M. M., and Haslett, C. (2007) Biochem. Soc. Trans. 35 288-291 [DOI] [PubMed] [Google Scholar]
- 4.Hallett, J. M., Leitch, A. E., Riley, N. A., Duffin, R., Haslett, C., and Rossi, A. G. (2008) Trends Pharmacol. Sci. 29 250-257 [DOI] [PubMed] [Google Scholar]
- 5.Fadok, V. A., Bratton, D. L., Guthrie, L., and Henson, P. M. (2001) J. Immunol. 166 6847-6854 [DOI] [PubMed] [Google Scholar]
- 6.Mevorach, D., Mascarenhas, J. O., Gershov, D., and Elkon, K. B. (1998) J. Exp. Med. 188 2313-2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor, P. R., Carugati, A., Fadok, V. A., Cook, H. T., Andrews, M., Carroll, M. C., Savill, J. S., Henson, P. M., Botto, M., and Walport, M. J. (2000) J. Exp. Med. 192 359-366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyanishi, M., Tada, K., Koike, M., Uchiyama, Y., Kitamura, T., and Nagata, S. (2007) Nature 450 435-439 [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi, N., Karisola, P., Pena-Cruz, V., Dorfman, D. M., Jinushi, M., Umetsu, S. E., Butte, M. J., Nagumo, H., Chernova, I., Zhu, B., Sharpe, A. H., Ito, S., Dranoff, G., Kaplan, G. G., Casasnovas, J. M., Umetsu, D. T., Dekruyff, R. H., and Freeman, G. J. (2007) Immunity 27 927-940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park, D., Tosello-Trampont, A. C., Elliott, M. R., Lu, M., Haney, L. B., Ma, Z., Klibanov, A. L., Mandell, J. W., and Ravichandran, K. S. (2007) Nature 450 430-434 [DOI] [PubMed] [Google Scholar]
- 11.Park, S. Y., Jung, M. Y., Kim, H. J., Lee, S. J., Kim, S. Y., Lee, B. H., Kwon, T. H., Park, R. W., and Kim, I. S. (2008) Cell Death Differ. 15 192-201 [DOI] [PubMed] [Google Scholar]
- 12.Hanayama, R., Tanaka, M., Miwa, K., Shinohara, A., Iwamatsu, A., and Nagata, S. (2002) Nature 417 182-187 [DOI] [PubMed] [Google Scholar]
- 13.Fadok, V. A., Voelker, D. R., Campbell, P. A., Cohen, J. J., Bratton, D. L., and Henson, P. M. (1992) J. Immunol. 148 2207-2216 [PubMed] [Google Scholar]
- 14.Fadok, V. A., de Cathelineau, A., Daleke, D. L., Henson, P. M., and Bratton, D. L. (2001) J. Biol. Chem. 276 1071-1077 [DOI] [PubMed] [Google Scholar]
- 15.Hoffmann, P. R., Kench, J. A., Vondracek, A., Kruk, E., Daleke, D. L., Jordan, M., Marrack, P., Henson, P. M., and Fadok, V. A. (2005) J. Immunol. 174 1393-1404 [DOI] [PubMed] [Google Scholar]
- 16.Arroyo, A., Modriansky, M., Serinkan, F. B., Bello, R. I., Matsura, T., Jiang, J., Tyurin, V. A., Tyurina, Y. Y., Fadeel, B., and Kagan, V. E. (2002) J. Biol. Chem. 277 49965-49975 [DOI] [PubMed] [Google Scholar]
- 17.Kagan, V. E., Gleiss, B., Tyurina, Y. Y., Tyurin, V. A., Elenstrom-Magnusson, C., Liu, S. X., Serinkan, F. B., Arroyo, A., Chandra, J., Orrenius, S., and Fadeel, B. (2002) J. Immunol. 169 487-499 [DOI] [PubMed] [Google Scholar]
- 18.Greenberg, M. E., Sun, M., Zhang, R., Febbraio, M., Silverstein, R., and Hazen, S. L. (2006) J. Exp. Med. 203 2613-2625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwab, J. M., Chiang, N., Arita, M., and Serhan, C. N. (2007) Nature 447 869-874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frasch, S. C., Zemski-Berry, K., Murphy, R. C., Borregaard, N., Henson, P. M., and Bratton, D. L. (2007) J. Immunol. 178 6540-6548 [DOI] [PubMed] [Google Scholar]
- 21.Sugo, T., Tachimoto, H., Chikatsu, T., Murakami, Y., Kikukawa, Y., Sato, S., Kikuchi, K., Nagi, T., Harada, M., Ogi, K., Ebisawa, M., and Mori, M. (2006) Biochem. Biophys. Res. Commun. 341 1078-1087 [DOI] [PubMed] [Google Scholar]
- 22.Suratt, B. T., Young, S. K., Lieber, J., Nick, J. A., Henson, P. M., and Worthen, G. S. (2001) Am. J. Physiol. 281 L913-L921 [DOI] [PubMed] [Google Scholar]
- 23.Folch, J., Lees, M., and Sloane Stanley, G. H. (1957) J. Biol. Chem. 226 497-509 [PubMed] [Google Scholar]
- 24.Zemski Berry, K. A., and Murphy, R. C. (2005) Antioxid. Redox Signal. 7 157-169 [DOI] [PubMed] [Google Scholar]
- 25.Kim, H. Y., and Salem, N., Jr. (1990) J. Lipid Res. 31 2285-2289 [PubMed] [Google Scholar]
- 26.Bodennec, J., Pelled, D., and Futerman, A. H. (2003) J. Lipid Res. 44 218-226 [DOI] [PubMed] [Google Scholar]
- 27.Pulfer, M., and Murphy, R. C. (2003) Mass Spectrom. Rev. 22 332-364 [DOI] [PubMed] [Google Scholar]
- 28.Hall, L. M., and Murphy, R. C. (1998) J. Am. Soc. Mass Spectrom. 9 527-532 [DOI] [PubMed] [Google Scholar]
- 29.Fadok, V. A., Savill, J. S., Haslett, C., Bratton, D. L., Doherty, D. E., Campbell, P. A., and Henson, P. M. (1992) J. Immunol. 149 4029-4035 [PubMed] [Google Scholar]
- 30.Haslett, C., Guthrie, L. A., Kopaniak, M. M., Johnston, R. B., Jr., and Henson, P. M. (1985) Am. J. Pathol. 119 101-110 [PMC free article] [PubMed] [Google Scholar]
- 31.Yamamori, T., Inanami, O., Sumimoto, H., Akasaki, T., Nagahata, H., and Kuwabara, M. (2002) Biochem. Biophys. Res. Commun. 293 1571-1578 [DOI] [PubMed] [Google Scholar]
- 32.Guthrie, L. A., McPhail, L. C., Henson, P. M., and Johnston, R. B., Jr. (1984) J. Exp. Med. 160 1656-1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han, X., and Gross, R. W. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 10635-10639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mueller, H. W., O'Flaherty, J. T., Greene, D. G., Samuel, M. P., and Wykle, R. L. (1984) J. Lipid Res. 25 383-388 [PubMed] [Google Scholar]
- 35.Van Hoeven, R. P., Emmelot, P., Krol, J. H., and Oomen-Meulemans, E. P. (1975) Biochim. Biophys. Acta 380 1-11 [PubMed] [Google Scholar]
- 36.Hazan, I., Dana, R., Granot, Y., and Levy, R. (1997) Biochem. J. 326 867-876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao, X., Bey, E. A., Wientjes, F. B., and Cathcart, M. K. (2002) J. Biol. Chem. 277 25385-25392 [DOI] [PubMed] [Google Scholar]
- 38.Suram, S., Brown, G. D., Ghosh, M., Gordon, S., Loper, R., Taylor, P. R., Akira, S., Uematsu, S., Williams, D. L., and Leslie, C. C. (2006) J. Biol. Chem. 281 5506-5514 [DOI] [PubMed] [Google Scholar]
- 39.Hosono, H., Aoki, J., Nagai, Y., Bandoh, K., Ishida, M., Taguchi, R., Arai, H., and Inoue, K. (2001) J. Biol. Chem. 276 29664-29670 [DOI] [PubMed] [Google Scholar]
- 40.Kono, N., Inoue, T., Yoshida, Y., Sato, H., Matsusue, T., Itabe, H., Niki, E., Aoki, J., and Arai, H. (2008) J. Biol. Chem. 283 1628-1636 [DOI] [PubMed] [Google Scholar]
- 41.Kriska, T., Marathe, G. K., Schmidt, J. C., McIntyre, T. M., and Girotti, A. W. (2007) J. Biol. Chem. 282 100-108 [DOI] [PubMed] [Google Scholar]
- 42.Stremler, K. E., Stafforini, D. M., Prescott, S. M., and McIntyre, T. M. (1991) J. Biol. Chem. 266 11095-11103 [PubMed] [Google Scholar]
- 43.Chatterjee, B. E., Yona, S., Rosignoli, G., Young, R. E., Nourshargh, S., Flower, R. J., and Perretti, M. (2005) J. Leukocyte Biol. 78 639-646 [DOI] [PubMed] [Google Scholar]
- 44.Frasch, S. C., Nick, J. A., Fadok, V. A., Bratton, D. L., Worthen, G. S., and Henson, P. M. (1998) J. Biol. Chem. 273 8389-8397 [DOI] [PubMed] [Google Scholar]
- 45.Guzik, K., and Potempa, J. (2008) Biochimie (Paris) 90 405-415 [DOI] [PubMed] [Google Scholar]
- 46.Serhan, C. N., and Savill, J. (2005) Nat. Immun. 6 1191-1197 [DOI] [PubMed] [Google Scholar]
- 47.Pollock, J. D., Williams, D. A., Gifford, M. A., Li, L. L., Du, X., Fisherman, J., Orkin, S. H., Doerschuk, C. M., and Dinauer, M. C. (1995) Nat. Genet. 9 202-209 [DOI] [PubMed] [Google Scholar]
- 48.Erwig, L. P., McPhilips, K. A., Wynes, M. W., Ivetic, A., Ridley, A. J., and Henson, P. M. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 12825-12830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kiss, R. S., Elliott, M. R., Ma, Z., Marcel, Y. L., and Ravichandran, K. S. (2006) Curr. Biol. 16 2252-2258 [DOI] [PubMed] [Google Scholar]
- 50.Peter, C., Waibel, M., Radu, C. G., Yang, L. V., Witte, O. N., Schulze-Osthoff, K., Wesselborg, S., and Lauber, K. (2008) J. Biol. Chem. 283 5296-5305 [DOI] [PubMed] [Google Scholar]
- 51.Yang, L. V., Radu, C. G., Wang, L., Riedinger, M., and Witte, O. N. (2005) Blood 105 1127-1134 [DOI] [PubMed] [Google Scholar]
- 52.Lagasse, E., and Weissman, I. L. (1994) J. Exp. Med. 179 1047-1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frasch, S. C., Henson, P. M., Nagaosa, K., Fessler, M. B., Borregaard, N., and Bratton, D. L. (2004) J. Biol. Chem. 279 17625-17633 [DOI] [PubMed] [Google Scholar]
- 54.Elliott, J. I., Sardini, A., Cooper, J. C., Alexander, D. R., Davanture, S., Chimini, G., and Higgins, C. F. (2006) Blood 108 1611-1617 [DOI] [PubMed] [Google Scholar]
- 55.Smith, G. A., Hesketh, T. R., Plumb, R. W., and Metcalfe, J. C. (1979) FEBS Lett. 105 58-62 [DOI] [PubMed] [Google Scholar]
- 56.Kawamoto, K., Aoki, J., Tanaka, A., Itakura, A., Hosono, H., Arai, H., Kiso, Y., and Matsuda, H. (2002) J. Immunol. 168 6412-6419 [DOI] [PubMed] [Google Scholar]
- 57.Panasenko, O. M., Spalteholz, H., Schiller, J., and Arnhold, J. (2003) Free Radic. Biol. Med. 34 553-562 [DOI] [PubMed] [Google Scholar]
- 58.Panasenko, O. M., Spalteholz, H., Schiller, J., and Arnhold, J. (2006) Biochemistry (Mosc.) 71 571-580 [DOI] [PubMed] [Google Scholar]
- 59.Fuchs, T. A., Abed, U., Goosmann, C., Hurwitz, R., Schulze, I., Wahn, V., Weinrauch, Y., Brinkmann, V., and Zychlinsky, A. (2007) J. Cell Biol. 176 231-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Darland-Ransom, M., Wang, X., Sun, C. L., Mapes, J., Gengyo-Ando, K., Mitani, S., and Xue, D. (2008) Science 320 528-531 [DOI] [PubMed] [Google Scholar]
- 61.Campbell, E. L., Louis, N. A., Tomassetti, S. E., Canny, G. O., Arita, M., Serhan, C. N., and Colgan, S. P. (2007) FASEB J. 21 3162-3170 [DOI] [PubMed] [Google Scholar]
- 62.Lauber, K., Bohn, E., Krober, S. M., Xiao, Y. J., Blumenthal, S. G., Lindemann, R. K., Marini, P., Wiedig, C., Zobywalski, A., Baksh, S., Xu, Y., Autenrieth, I. B., Schulze-Osthoff, K., Belka, C., Stuhler, G., and Wesselborg, S. (2003) Cell 113 717-730 [DOI] [PubMed] [Google Scholar]
- 63.Le, L. Q., Kabarowski, J. H., Weng, Z., Satterthwaite, A. B., Harvill, E. T., Jensen, E. R., Miller, J. F., and Witte, O. N. (2001) Immunity 14 561-571 [DOI] [PubMed] [Google Scholar]
- 64.Bratton, D. L., and Henson, P. M. (2008) Curr. Biol. 18 R76-R79 [DOI] [PubMed] [Google Scholar]
- 65.Kim, S. J., Gershov, D., Ma, X., Brot, N., and Elkon, K. B. (2002) J. Exp. Med. 196 655-665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chang, M. K., Bergmark, C., Laurila, A., Horkko, S., Han, K. H., Friedman, P., Dennis, E. A., and Witztum, J. L. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 6353-6358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gardai, S. J., Bratton, D. L., Ogden, C. A., and Henson, P. M. (2006) J. Leukocyte Biol. 79 896-903 [DOI] [PubMed] [Google Scholar]
- 68.Gardai, S. J., McPhillips, K. A., Frasch, S. C., Janssen, W. J., Starefeldt, A., Murphy-Ullrich, J. E., Bratton, D. L., Oldenborg, P. A., Michalak, M., and Henson, P. M. (2005) Cell 123 321-334 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.