

Abstract
Transcriptional dysregulation is a central pathogenic mechanism in Huntington's disease, a fatal neurodegenerative disorder associated with polyglutamine (polyQ) expansion in the huntingtin (Htt) protein. In this study, we show that mutant Htt alters the normal expression of specific mRNA species at least partly by disrupting the binding activities of many transcription factors which govern the expression of the dysregulated mRNA species. Chromatin immunoprecipitation (ChIP) demonstrates Htt occupation of gene promoters in vivo in a polyQ-dependent manner, and furthermore, ChIP-on-chip and ChIP subcloning reveal that wild-type and mutant Htt exhibit differential genomic distributions. Exon 1 Htt binds DNA directly in the absence of other proteins and alters DNA conformation. PolyQ expansion increases Htt–DNA interactions, with binding to recognition elements of transcription factors whose function is altered in HD. Together, these findings suggest mutant Htt modulates gene expression through abnormal interactions with genomic DNA, altering DNA conformation and transcription factor binding.
Keywords: transcription factor, chromatin immunoprecipitation, DNA microarrays, polyglutamine, gene expression, DNA conformation
Introduction
Huntington's disease (HD) is an autosomal dominant, late-onset neurodegenerative disorder caused by a CAG repeat expansion in exon 1 of the HD gene, which translates into a polyglutamine (polyQ) tract in the huntingtin (Htt) protein (The Huntington's Disease Collaborative Research Group, 1993). Thus, HD belongs to a group of neurodegenerative disorders caused by polyQ expansion, which include spinal and bulbar muscular atrophy (SBMA), dentatorubral pallidoluysian atrophy, and the spinocerebellar ataxias (SCA) types 1, 2, 3, 6, 7, and 17 (Bates et al., 2002). HD neuropathology is characterized by generalized brain atrophy, selective neuronal cell death, and the widespread occurrence of polyQ aggregates (DiFiglia et al., 1997).
Htt cleavage promotes nuclear localization and nucleocytoplasmic shuttling motifs have been identified within the Htt protein, suggesting that Htt can be transported in and out of the nucleus (Takano and Gusella, 2002; Xia et al., 2003). The first 17 aa of Htt can function as a cytosolic retention signal (Steffan et al., 2004; Cornett et al., 2005). Whereas some wild-type Htt is normally localized in the nucleus, polyglutamine-expanded Htt exhibits more nuclear localization than nonexpanded protein (Dorsman et al., 1999; Kegel et al., 2002). Nuclear-localized expanded polyglutamine protein is highly detrimental in vitro and in vivo (Klement et al., 1998; Saudou et al., 1998; Jackson et al., 2003; Schilling et al., 2004; Benn et al., 2005). In transgenic mice, exclusive nuclear localization of mutant exon 1 Htt in the nucleus is sufficient for the onset and progression of behavioral phenotypes, neurodegeneration, and transcriptional dysregulation (Benn et al., 2005).
Transcriptional dysregulation is a central pathogenic mechanism in HD (Luthi-Carter and Cha, 2003). Human HD and mouse models of HD demonstrate downregulation of the mRNA of specific genes (Luthi-Carter et al., 2000; Hodges et al., 2006). Indeed, comparison of gene expression studies has revealed remarkable concordance among mouse models and within human HD brain (Kuhn et al., 2007). Downregulation is not attributable to post-transcriptional mRNA stability, but rather to decreased transcription from gene promoters (Hu et al., 2004; McCaw et al., 2004; Cui et al., 2006). The polyQ motif occurs in many transcription factors and can function as a transcriptional activation domain. Interestingly, polyQ repeat expansions in TATA-binding protein (TBP) and androgen receptor (AR) cause the disorders SCA17 and SBMA, respectively.
Although mutant Htt disrupts transcription in neurons, the underlying molecular mechanism is unknown. In this study, we used mRNA expression profiling to identify genes expressed exclusively in the presence of wild-type or mutant Htt. Additionally, many transcription factor activities were perturbed in response to mutant Htt. DNA immunoprecipitation using Htt-specific antibodies demonstrated an increase in occupancy of mutant Htt at gene promoters in vivo. Furthermore, we have used chromatin immunoprecipitation (ChIP) combined with DNA microarray analyses to identify the genomic binding sites of wild-type and mutant Htt. Finally, wild-type and mutant Htt bind directly to DNA in vitro in the absence of other proteins, and differentially alter DNA conformation. Thus, increased binding of mutant Htt to DNA modulates DNA conformation and alters transcription factor function, and ultimately results in transcriptional dysregulation.
Materials and Methods
Transgenic R6/2 mouse striatum.
R6/2 transgenic mice and wild-type littermate controls (Mangiarini et al., 1996) were killed, and brains rapidly removed, striata were dissected and flash-frozen in chilled isopentane and stored at −80°C until use. The guidelines for animal care and use were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care.
Immortalized striatal HD cell lines.
Striatal cell lines established from wild-type (Q7/7) and homozygote mutant (Q111/111) Hdh knock-in embryonic mice (Trettel et al., 2000) were used in passages 5–16. STHdh cell lines express full-length murine Htt with either 7 (STHdh7/7) or 111 (STHdh111/111) glutamines. Cells were kept at 33°C for propagation and were placed at 39°C for 48 h to stop proliferation.
Postmortem human brain tissue.
Postmortem human brain tissue from HD patients (six cases) and neurologically normal control patients (seven cases) were kindly provided by the Alzheimer's Disease Research Center at Massachusetts General Hospital under the auspices of Partners HealthCare Human Subjects Committee, as approved by the institutional review board. Postmortem intervals ranged from 13 to 24 h for the HD cases [corresponding to Vonsattel grade II (two cases), grade III (two cases), and grade IV (two cases)], and from 10 to 21 h for the control cases. Unfixed frontal cortex was obtained from each case. Brain samples were stored at −80°C.
Nuclear extracts.
Nuclear proteins were extracted from mouse brain tissue using a Sigma CellLytic NuCLEAR extraction kit according to the manufacturer's instructions (Sigma-Aldrich).
GST fusion proteins.
Glutathione S-transferase (GST)-Htt clones (HD20Q, HD32Q, and HD53Q) were a generous gift from Dr. Erich Wanker (Max Planck Institute, Berlin, Germany). GST-p53 was generously supplied by Dr. Joan Steffan (University of California, Irvine, CA), whereas the GST/cAMP response element-binding protein (CREB)-binding protein (CBP) fusion proteins 1–5 (1 is 1–450 aa, 2 is 419–1098 aa, 3 is 1069–1459 aa, 4 is 1459–1891 aa, and 5 is 1892–2441 aa of CBP) were kind gifts from Riki Kurokawa from the laboratory of Christopher Glass (University of California, San Diego, CA) (Kurokawa et al., 1998). GST fusion proteins were prepared as described previously (Yohrling et al., 2003). Verification of protein synthesis was performed with SDS-PAGE and Coomassie blue staining (data not shown).
Gene expression profiling analysis.
The gene expression data from the comparison study of STHdh7/7 and STHdh111/111 cell lines were analyzed as described previously (Sadri-Vakili et al., 2007) (GenBank accession number GSE11358).
Transcription factor array.
The TranSignal Protein/DNA array kit profiles binding activities of up to 345 transcription factors simultaneously and was used according to the manufacturer's instructions (Panomics). Twenty micrograms of nuclear extract or 1 μg of GST protein were incubated with biotin-labeled DNA probe mix in binding buffer. Unbound probes were washed away, leaving protein–DNA complexes. Bound DNA probes were recovered from the complex and denatured before hybridizing to the array membrane. The biotin-labeled probe was detected with streptavidin-HRP and was chemiluminescently enhanced before imaging and densitometry with the Alpha Innotech FluorChem Imager (Alpha Innotech).
Correlating microarray gene expression profiling with transcription factor binding activity profiles.
The gene expression profiling of the STHdh cell lines generated four lists of genes based on presence/absence and up/down criteria labeled as follows: (1) absent in STHdh7/7, present in STHdh111/111; (2) present in STHdh7/7, absent in STHdh111/111; (3) present in both lines, with relative upregulation in STHdh111/111; and (4) present in both lines, with relative downregulation in STHdh111/111. To identify the relevant networks and pathways and related transcription factors for these sets of genes, we performed literature mining. The Genomatix Bibliosphere was used to mine PubMed abstracts for gene–gene and gene–transcription factor cocitations between each of the genes from the filtered lists. The criteria for the cocitation of the genes were as follows: (1) input genes plus transcription factors, (2) genes cocited with at least two input genes, (3) genes that are at least cocited two times with one input gene, and (4) genes restricted to sentences with the order “gene-function word-gene.” We generated tables and interaction network maps with all of the mined information. Transcription factors found involved in the network as a direct interaction with the input genes were compared with transcription factor binding activity profiles. Common transcription factors in both studies were combined.
ChIP assay.
The ChIP technique was performed as described previously (Chen-Plotkin et al., 2006). Antibodies used were S830 (raised against Htt exon 1 fusion protein; a kind gift from Gillian P. Bates, Kings College, London, UK), N18 (Santa Cruz Biotechnology), monoclonal antibody 1 (MAb1; recognizes Htt amino acids 1–17; a kind gift from Marian DiFiglia, Massachusetts General Hospital, Boston, MA), MAb2166 (raised against amino acids 181–810 of Htt), and MAb2170 (raised against amino acids 1247–1646 of Htt; Millipore Bioscience Research Reagents). Antibodies against RAP30 (catalog #sc-236) and RAP74 (catalog #sc-235) were obtained from Santa Cruz Biotechnology. Positive control ChIP was performed with anti-acetylated histone H3 (Millipore), and negative controls included IgG (Jackson ImmunoResearch) and no antibody.
DNA quantitation.
Immunoprecipitated DNA [from ChIP or DNA immunoprecipitation (DIP)] was quantitated using the DNA Quantitation System, which detects 10–500 pg/μl DNA (Promega), according to the manufacturer's instructions. Briefly, a premix was made with a buffer solution containing sodium pyrophosphate, nucleoside diphosphate kinase enzyme solution, and T4 DNA polymerase, aliquoted and mixed with either known DNA quantities (for a standard curve) or experimental unknown DNA quantities. The reaction was incubated for 10 min at 37°C, chilled on ice for 10 min before mixing with ENLITEN luciferase reagent. The light output was immediately measured in a luminometer.
Real-time PCR.
Input and IP samples were interrogated with gene-promoter-specific primers in triplicate reactions (supplemental Table 3, available at www.jneurosci.org as supplemental material) in real-time PCR analysis as described previously (Chen-Plotkin et al., 2006). Threshold amplification cycle numbers using iCycler software were used to calculate IP DNA quantities as percentages of corresponding inputs.
Ligation-mediated PCR amplification of ChIP products.
We used a ligation-mediated PCR (LM-PCR) procedure to amplify the very small amounts of immunoprecipitated DNA recovered through ChIP, and for subsequent ChIP-on-chip or ChIP-subcloning experiments. LM-PCR was performed as described previously (Lee et al., 2006).
ChIP-on-chip.
ChIP-on-chip was performed as described (Boyer et al., 2006; Lee et al., 2006), in collaboration with Agilent Technologies. Briefly, LM-PCR-amplified DNA was labeled and purified using Invitrogen Bioprime random primer labeling kits. Labeled DNA [immunoenriched DNA labeled with cyanine 5 (Cy5), input DNA labeled with Cy3] was combined (5 μg each of immunoenriched and input DNA) and cohybridized to mouse proximal promoter arrays (Whitehead format 13275 and 13276, with 4–5 60-mer oligonucleotide probes for 17,917 annotated transcription start sites) in Agilent hybridization chambers at 20 rpm for 40 h at 65°C. Arrays were washed and scanned on an Agilent scanner. The scanned image was analyzed using Feature Extraction software. Background and noise were removed from the data and the quality control generated by feature extraction was monitored. Fluorescent intensities of Cy5/Cy3 for each individual feature on the array represent the binding events of the amplified ChIP product enabling identification of Htt binding sites.
ChIP-on-chip data normalization and analysis.
Preprocessed data were normalized and analyzed using the ChIP Analytics software version 1.2. Replicates were analyzed together within the software (intra-array Lowess intensity-dependent normalization), and the results were averaged using the replicate support extended error model proposed by the software. Results of the combined analyses were used to generate reports of text files for the probes, segments, and gene-level results, University of California, Santa Cruz (UCSC) track files, Ensembl distributed annotated system files, and quality control reports.
Identifying Htt-enriched regions at known gene locations.
We selected probes with a weighted average log2 ratio (IP/input) >1 or less than −1 with a p value <0.01 for the STHdh7/7 and STHdh111/111 cell lines, identified the genes with at least 1 probe significantly enriched, and pulled down the results for all of the probes for each of these genes. Results were mapped on the chromosomes using the UCSC Genome Browser as custom tracks. Binding ratios for both cell lines were compared at each probe and gene location. To determine the amplitude of difference between Htt binding in the two cell lines, we calculated the log2 ratio of the STHdh111/111/STHdh7/7 binding ratios.
Comparing Htt-promoter binding and gene expression data.
We analyzed gene expression profiling data for genes identified through the ChIP-on-chip analysis. Probes that were absent (Affymetrix absent/presence call) in either STHdh7/7 or STHdh111/111 lines were removed as well as probes with no difference between the two lines [log2(STHdh111/111/STHdh7/7) <0.3 or more than −0.3]. Data from the remaining probes from the gene expression study and from the binding study were combined as a comparison of the log2 ratios (STHdh111/111/STHdh7/7) from the gene expression analysis compared with the binding ratios.
ChIP subcloning.
To isolate transcription factor targets in an unbiased manner, ChIP subcloning was performed using a modified method (Nelson et al., 2006). ChIP was performed as described with MAb2170 except that two sequential immunoprecipitations were performed. Annealed, phosphorylated oligonucleotides were ligated to the blunt-ended purified Htt ChIP product and amplified by PCR for 20 cycles of amplification as described by Nelson et al. (2006). DNA from the ligation-mediated PCR amplification of the Htt-ChIP product was cloned into pBluescript, transformed into XL1 Blue (MRF'; Stratagene) Escherichia coli, and positive clones were identified through blue/white colony selection. Initial analysis of bacterial clones was performed by picking each individual white bacterial colony into 200 μl of double-distilled H2O and vigorously vortexing. This preparation was used as a template in a PCR with Promega buffer, 2.5 mm MgCl2, 200 μm dNTPs, 10 ng/μl M13 forward and reverse primers, and 0.5 U/μl Taq polymerase (Promega). Cycling conditions were 90 s at 94°C; 35 times 30 s at 94°C, 30 s at 55°C, and 90 s at 72°C; and 10 min at 72°C. Clones with an insert larger than 180 bp were miniprepped (Qiagen) and submitted for sequencing at the Massachusetts General Hospital DNA core using the M13 forward primer.
DIP.
DNA immunoprecipitation is essentially a modified GST-pulldown technique. Equal amounts (5 μg) of each GST protein (on Sepharose beads) were incubated overnight with agitation at 4°C with 5 μg of DNA template in freshly made GST binding buffer (20 mm Tris pH 7.5, 150 mm NaCl, 0.1 mm EDTA, 1 mm dithiothreitol, 0.3% tert-octylphenoxy poly(oxyethlene)ethanol, and 20% glycerol). DNA templates include genomic DNA restriction digested to completion with Sau3AI or short double-stranded DNA molecules such as electrophoretic mobility shift assay probes (made from annealed oligonucleotides). Samples were cross-linked under UV light for 3 min before washing three times in 1 ml of GST binding buffer, and resuspended in 50 μl of PBS containing protease inhibitors.
Micrococcal nuclease assay coupled with Southern blotting.
Micrococcal nuclease (MNase) digestion was performed as described by Carey and Swale (2000), except that ∼106 cells were used. It was not possible to measure genomic DNA amounts before MNase digestion because of its high-molecular-weight state. After MNase digestion (0.5–50 U/ml), DNA was visualized via agarose gel electrophoresis. Southern blotting was performed as described previously (Mangiarini et al., 1996) except that probes were generated by PCR amplification and confirmed by sequencing of the relevant gene promoter from genomic DNA before radiolabeling (for primer information, see supplemental Table 3, available at www.jneurosci.org as supplemental material).
Conformation-dependent fluorescent quenching of YOYO-1/DNA complexes.
pMDR1-luc, a kind gift from Eric Stanbridge (University of California, Irvine CA), and pWWP1-luc, generously given by Bert Vogelstein (Johns Hopkins, Baltimore, MD), plasmid DNA were mixed at a ratio of 50 bp of double-stranded plasmid DNA to 1 dye molecule at a final concentration of 200 nm of YOYO-1 dye (Invitrogen). The DNA–YOYO-1 complex was incubated in a buffer containing a final concentration of 100 mm NaCl and 20 mm HEPES at pH 7.4 at room temperature for 5 h in the dark. As a control, poly-l-lysine (Sigma) was added to a final concentration of 0.0045%, which corresponds to an approximate ratio of two poly-l-lysine molecules to one phosphate on the DNA backbone. GST fusion proteins were added (0.5 μm) to the DNA–YOYO-1 complex and incubated for up to 120 min. Steady-state florescent measurements were performed on a Wallac Victor 1420 Multilabel Counter (PerkinElmer). The excitation wavelength was 485 nm and the emission was collected at 535 nm.
Results
Genes whose expression is dependent on the presence of wild-type or mutant Htt
Specific gene expression changes occur in the presence of mutant Htt in human HD patient brains, as well as in multiple cell and mouse models of HD (Luthi-Carter et al., 2000; Sipione et al., 2002; Sugars et al., 2004; Hodges et al., 2006). Htt can modulate reporter gene transcription in a polyglutamine-dependent manner (Steffan et al., 2000; Kegel et al., 2002; Yohrling et al., 2003; Obrietan and Hoyt, 2004). Furthermore, the presence of mutant Htt fragments restricted to the nucleus in vivo is sufficient to induce transcriptional dysregulation (Schilling et al., 2004; Benn et al., 2005). However, the extent to which exclusive expression of wild-type or mutant Htt can modulate gene expression is not clear. We therefore performed microarray gene expression profiling on immortalized striatal cell lines expressing only wild-type Htt (STHdh7/7), or homozygous for mutant Htt (STHdh111/111) (Trettel et al., 2000). The exclusive presence of wild-type or mutant Htt confers a transcriptional phenotype, with distinct effects on gene expression profiles (Fig. 1). We hypothesized that the most profound effect on gene expression would be presence versus absence of a mRNA species in one line compared with the other, and therefore generated lists of genes whose expression is dependent on or inhibited by either wild-type and mutant Htt (Fig. 1) (Sadri-Vakili et al., 2007). More genes were expressed exclusively in the presence of mutant Htt (2098 probes) compared with the number of genes expressed in the presence of only wild-type Htt (1382 probes). We also interrogated mRNA species that were present (14233 probes) or absent (23424 probes) in both STHdh7/7 and STHdh111/111 cell lines. For those mRNA species present in both cell lines, we determined whether the expression was upregulated or downregulated by mutant Htt. Genes expressed in both cell lines were more likely to be downregulated in the presence of mutant Htt (1105 probes, 85% of which had a p value <0.01) compared with genes that are upregulated in the presence of mutant Htt (964 probes, 79% of which had a p value <0.01). Therefore, we have identified four clusters of mRNA expression changes in response to the exclusive presence of wild-type or mutant Htt: (1) absent with wild-type Htt and present with mutant Htt, (2) present with wild-type Htt and absent with mutant Htt, (3) present in both wild-type and mutant but upregulated by mutant Htt, and (4) present in both wild-type and mutant but downregulated by mutant Htt. Of these, the two most prevalent are clusters 1 and 4.
Figure 1.
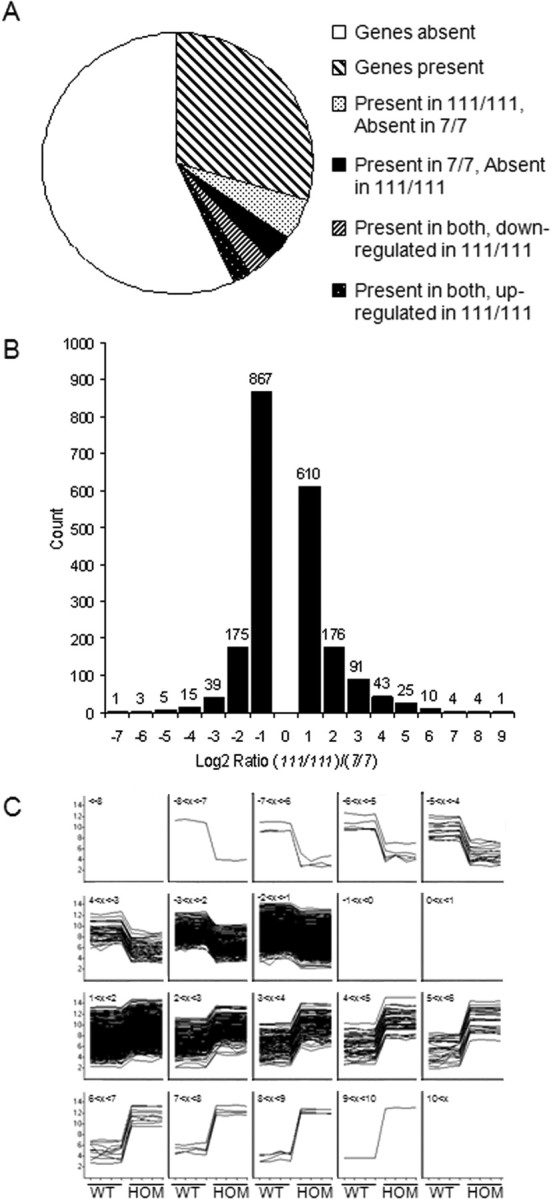
mRNA expression profiling reveals gene expression responses to wild-type (WT) or mutant Htt. Comparison of mRNA expression profiles yields numbers of genes present and absent in wild-type STHdh7/7 compared with mutant STHdh111/111 cell lines. A, Pie chart showing the distribution of the present and absent probes. Probes called present were present or marginal in at least three of the four replicates, probes called absent were absent in at least three of four replicates, and probes were called marginal if present and absent in two of the four replicates. More probes were present in STHdh111/111 and absent/marginal in STHdh7/7. B, Distribution of the binned log2(STHdh111/111/STHdh7/7) from the probes called present in both cell lines, with a log2 ratio >1 or less than −1 and a p value <0.05. More probes were downregulated in the STHdh111/111 versus the STHdh7/7, but the amplitude of change (i.e., the difference in expression) was larger for the probes upregulated in the STHdh111/111 versus the STHdh7/7 lines. C, A representation of the log2-normalized intensities for each replicate clustered by the log2 ratio bins determined from the probes called present in both cell lines, with a log2 ratio >1 or less than −1 and a p value <0.05, demonstrating the consistency of the results between the four replicates of each group and the amplitude of the differences between the two groups. The title of each box identifies which of the homozygote (HOM)/wild-type binned data are shown from −7 (top left) to +9 (bottom right). The y-axis shows the magnitude of change for each sample, whereas the x-axis labels identify the wild-type (first four, left) and homozygote (last four, right) samples for each box. Thus, within each bin, the traces represent the fold change in wild-type cells to homozygote cells.
PolyQ expansion of Htt modulates binding of many transcription factors
The control of gene expression depends on the function of transcription factors. Htt interacts directly with many transcription factors, and for some of these factors, the interaction is altered with polyglutamine expansion (Holbert et al., 2001; Dunah et al., 2002; Kegel et al., 2002; Li et al., 2002; Yohrling et al., 2003; Zuccato et al., 2003; Zhai et al., 2005). However, other than the few transcription factors studied in HD, we do not know the effect of mutant Htt on transcription factor binding activities. We therefore interrogated transcription factor binding activity using an unbiased transcription factor array in the full-length knock-in STHdh cell lines. There was an overall increase in transcription factor binding activities in STHdh111/111 compared with STHdh7/7 cells (Fig. 2A).
Figure 2.
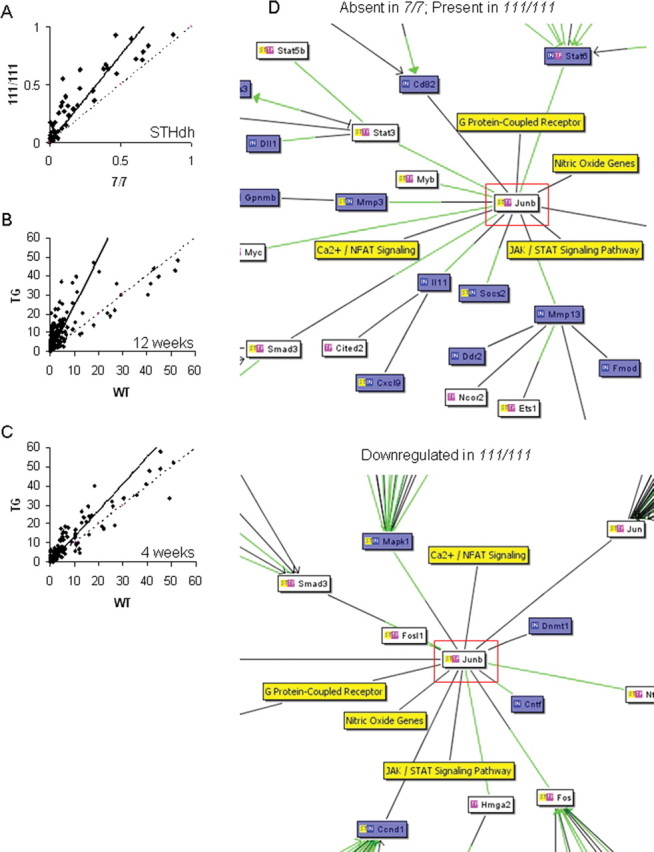
Mutant Htt alters the binding of many transcription factors. Densitometric analyses of transcription factor binding activity are scatter plotted, with each point representing the correlation of the binding activity of each transcription factor in two different conditions. Dashed line indicates the predicted correlation if there is no difference in binding activities between the two conditions, whereas the solid line indicates the actual correlation. A, Transcription factor binding activities are increased in the homozygote STHdh111/111 compared with STHdh7/7 cells (measured by film densitometry). B, C, The progressive increase in binding of many transcription factors at 12 weeks in the R6/2 transgenic mouse striatum (B) is evident at the presymptomatic age of 4 weeks (C). D, Increased transcription factor activity does not have straightforward effects on gene expression profiles (Table 1), but correlates well with gene expression changes. Interaction networks from cocitation literature mining generated by the Bibliosphere Pathway view (Genomatix) display the relationship of transcription factors (white boxes), mRNA expression levels (blue boxes), and expression pathways (yellow boxes). The green lines represent a binding site for the relevant transcription factor on the gene promoter. Shown are part of the interaction networks for JunB (highlighted in red), demonstrating different interactions in STHdh7/7 and STHdh111/111 cell lines. JunB exerts two types of gene expression effects (top, mRNA absent in STHdh7/7 and present in STHdh111/111 cells; bottom section shows part of an interaction network for mRNA present in both cell lines but downregulated in STHdh111/111 homozygotes). Full interaction networks generated by the Bibliosphere Pathway view (Genomatix) for each of the four lists of genes are shown in supplemental Figure 1 (available at www.jneurosci.org as supplemental material). JAK, Janus kinase; Fmod, fibromodulin; NFAT, nuclear factor of activated T-cells; STAT, signal transducer and activator of transcription.
For this finding to be relevant for HD pathogenesis, it needs to be confirmed in a separate model. R6/2 transgenic mice express only exon 1 of the human HD gene (Mangiarini et al., 1996). We therefore interrogated transcription factor binding activities at 12 weeks of age in wild-type and transgenic R6/2 mouse striatum. We observed an overall increase in transcription factor binding activity in 12-week-old R6/2 transgenic mice (Fig. 2B). The increased binding activities are an early event in R6/2 pathogenesis, as profiling of transcription factor activity at 4 weeks of age revealed the same trend toward increased activities at this presymptomatic time point (Fig. 2C). For some transcription factors, altered binding activity was further confirmed by electromobility shift assay (data not shown). Thus, mutant Htt increases the binding activities of many transcription factors, in multiple HD models.
We next sought to determine the relationship between increased transcription factor binding and gene expression abnormalities in the STHdh cell lines (Fig. 2D, Table 1, supplemental Fig. 1, available at www.jneurosci.org as supplemental material). To a large extent, altered transcription factor activity accounted for the differences in mRNA expression profiles. We found that there were groups of transcription factors with distinct gene expression effects, with the largest group of transcription factors potentially influencing all four types of gene expression changes (present in one cell line but absent in the other, or upregulated or downregulated in the homozygote cell lines) (Table 1). Other groups of transcription factors correlated with one, two, or three types of gene expression changes. Thus, increased transcription factor binding in vitro does not have unitary effects on gene expression, such as exclusive downregulation of specific genes.
Table 1.
Increased transcription factor activities correlate with gene expression profiles
| Group | TF | Binding activity | Number of genes |
|||
|---|---|---|---|---|---|---|
| Absent STHdh7/7, Present STHdh111/111 | Present STHdh7/7, Absent STHdh111/111 | Downregulated in STHdh111/111 | Upregulated in STHdh111/111 | |||
| 1 | + | + | + | + | ||
| Creb1 | 4.2 | 10 | 7 | 9 | 7 | |
| Egr1 | 1.2 | 9 | 4 | 12 | 13 | |
| Esr1 | 12.2 | 9 | 6 | 8 | 5 | |
| Ets1 | 11.4 | 6 | 4 | 7 | 4 | |
| Fos | 2.35 | 15 | 10 | 13 | 14 | |
| Jun | 2.41 | 28 | 13 | 24 | 29 | |
| Nfkb1 | 1.5 | 37 | 11 | 19 | 35 | |
| Myb | 1.3 | 5 | 2 | 4 | 4 | |
| Myc | 2.8 | 12 | 15 | 15 | 14 | |
| Pparg | 29.3 | 17 | 8 | 13 | 7 | |
| Rara | 2.8 | 7 | 3 | 3 | 7 | |
| Smad3 | 1.3 | 4 | 7 | 8 | 9 | |
| Smad4 | 1.3 | 4 | 2 | 7 | 5 | |
| Sp1 | 2.8 | 14 | 12 | 17 | 9 | |
| Stat3 | 2.9 | 10 | 5 | 11 | 8 | |
| 2 | + | + | ||||
| JunB | 2.41 | 6 | 5 | |||
| Hnf4a | 7.8 | 3 | 4 | |||
| Nfatc1 | 19.5 | 1 | 3 | |||
| Ppara | 29.3 | 7 | 11 | |||
| Stat6 | 3.7 | 5 | 4 | |||
| 3 | + | + | + | |||
| Crebbp | 4.2 | 7 | 7 | 5 | ||
| Stat1 | 1.7 | 15 | 8 | 12 | ||
| Irf1 | 3.9 | 12 | 4 | 6 | ||
| Stat5a | 3.4 | 6 | 6 | 6 | ||
| 4 | + | |||||
| Vdr | 1.4 | 5 | ||||
| Esr2 | 12.2 | 4 | ||||
| Pou2f1 | 2.9 | 6 | ||||
| Rarb | 2.8 | 5 | ||||
| 5 | + | + | + | |||
| E2f1 | 4.8 | 7 | 7 | 5 | ||
| Mycn | 2.8 | 4 | 10 | 3 | ||
| 6 | + | + | ||||
| Stat5b | 3.4 | 3 | 2 | |||
| Gata2 | 44.2 | 1 | 3 | |||
| 7 | + | |||||
| Hsf1 | 3.6 | 3 | ||||
| Gata1 | 44.2 | 4 | ||||
| 8 | Pgr | 3.1 | 5 | 4 | 4 | |
| Srf | 2.96 | 3 | 5 | |||
| Tbp | 4.2 | 3 | 5 | |||
| Gata4 | 44.2 | 3 | 4 | |||
| Gata3 | 44.2 | 4 | ||||
The Bibliosphere application was used to coanalyze the microarray and transcription factor (TF) array data to yield information about gene transcription factor relations and, hence, gene expression regulators. Using this information, we have grouped TFs according to the type of expression effect they have (first column). +, Type of gene expression effect present, shown along each row defining the group. Shown is the transcription factor binding activity in STHdh cell lines in the Panomics array (calculated as a ratio of binding activity, or spot intensity in STHdh111/111 vs STHdh7/7 cell lines) (third column), and the number of gene changes in each of four groups (fourth through seventh columns). As an example, group 2 contains the transcription factors involved in regulating two groups of genes: the group of genes that are not expressed (absent) in wild-type STHdh7/7 cell lines but expressed (present) in the homozygote STHdh111/111 cell lines, and the group of genes that are downregulated in STHdh111/111 cell lines (compared with levels in STHdh7/7 cell lines). Transcription factors in this group do not have any other type of gene expression effects, although some of the genes are regulated by other transcription factors. So, within group 2, JunB regulates six genes that are absent in STHdh7/7 and present in STHdh111/111, and five genes that are downregulated in STHdh111/111. Similarly, genes that are regulated by JunB can be regulated by other transcription factors, both in the same group and those in other groups. Specifically, one of the downregulated genes in STHdh111/111 cell lines, Tgfb1 is regulated by JunB, but also regulated by the transcription factors Creb1 (group 1), Stat6 (group 2), and Pgr (group 8), among others. However, within gene expression patterns, each gene is uniquely assigned to a type of gene expression (i.e., if it is downregulated in STHdh111/111, then it cannot belong to the group of genes that are upregulated in STHdh111/111); thus, each factor that is known to regulate a gene is correlated with the same type of expression effect. Crebbp, CREB binding protein; Pparg, peroxisome proliferator activated receptor gamma; Pgr, progesterone receptor; Rara, retinoic acid receptor A; Rarb, retinoic acid receptor B; Tbp, TATA-box binding protein; Srf, serum response factor; Vdr, Vitamin D receptor.
Huntingtin occupies gene promoters in vivo
The transcriptional machinery is clearly perturbed in HD, with altered mRNA gene expression profiles, abnormal transcription factor function, and altered occupancies of transcription factors at specific gene promoters in the presence of mutant Htt (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) (Kegel et al., 2002; Zuccato et al., 2003; Hu et al., 2004; Zhai et al., 2005; Chen-Plotkin et al., 2006; Cui et al., 2006). However, the mechanism by which mutant Htt alters the presence of transcription factors at specific gene promoters is not clear. A simple possibility is that mutant Htt directly occupies gene promoters, interfering with normal transcription factor access. To determine whether Htt is present at gene promoters in vivo, we used ChIP assays.
DNA quantitation confirmed that Htt antibodies immunoprecipitate significantly more DNA than IgG or the no-antibody mock control (Fig. 3A). The DNA pull-down was not attributable to an antibody artifact, because we consistently observed the same result with different antibodies to Htt (Fig. 3A) (data not shown). Either equivalent amounts or more DNA was pulled down in HD conditions compared with control conditions in cells and mouse models (Fig. 3B). Anti-Htt antibodies consistently immunoprecipitated DNA from human brain samples, although there was no difference in overall amounts of DNA immunoprecipitated between HD and control cortex (data not shown). We used the ChIP-Htt-immunoenriched DNA from wild-type and R6/2 striatum as a template in real-time PCR with gene-specific primers to determine the association of wild-type and mutant Htt with gene promoters. We surveyed genes whose expression is unaffected in HD (Actb, Grin1), genes that are downregulated in HD (Drd2, Cnr1, Penk1), and a gene that is not expressed in striatal cells [Bdnf (brain derived neurotrophic factor)]. Immunoprecipitation with the MAb2170 antibody, which immunostains the nucleus (Kegel et al., 2002), showed increased Htt-promoter occupancy in R6/2 transgenic mouse brains, regardless of the gene that was interrogated (Fig. 3C). The MAb2170 antibody does not recognize the exon 1 transprotein, suggesting an increased occupancy of the endogenous wild-type protein in the presence of the mutant transprotein. These data demonstrate that Htt is present at gene promoters in vivo. ChIP experiments with MAb2170 in STHdh cell lines and with S830 antibody in R6/2 mouse brains further confirmed an association of Htt with gene promoters, but failed to reveal clear differences in Htt association between wild-type and transgenic conditions that correlated with gene expression changes (data not shown). The possibility that Htt is present in transcriptional complexes in vivo is bolstered by the finding that addition of Htt antibodies supershifts several protein/probe complexes in an electromobility shift assay, indicating that Htt can assemble into transcriptional complexes in vitro (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 3.

Htt-gene-promoter occupancy is modulated by polyQ repeat length. A, DNA quantitation of ChIP using different Htt antibodies in wild-type brain and cell lines. MAb2166 (open bars) and MAb2170 (filled bars) compared with IgG or no antibody (hatched bars) shows that significant amounts of DNA are pulled down. Error bars indicate SEM (n = 3–4). B, ChIP with the MAb2170 Htt antibody in R6/2 mouse brain reveals nonselective increases in Htt-promoter occupancy in transgenic mice (filled bars) compared with wild-type mice (open bars), including those for genes whose expression is unchanged [Actb (β-actin) and Grin1 (NMDA receptor NR1 subunit)], and those for genes downregulated in HD [Drd2 (dopamine D2 receptor), Penk1 (preproenkephalin), Cnr1 (cannabinoid receptor 1), and Bdnf (brain derived neurotrophic factor), which is not expressed in striatal neurons]. Hatched bars represent negative control conditions. Error bars indicate SEM (n = 2–4).
Wild-type and mutant Htt have different genomic binding sites
Chromatin immunoprecipitation with antibodies against Htt shows that wild-type and mutant Htt differentially occupy gene promoters, with mutant Htt present to greater extents at all gene promoters tested. A limitation of interrogating ChIP-immunoenriched DNA with gene-specific primers is that only a small number of genes can be interrogated. A broader and more unbiased approach is to survey the genome for all Htt binding sites. To determine the genomic targets of wild-type and mutant Htt, we used a ChIP-on-chip approach, hybridizing ChIP products to high-resolution genomic DNA microarrays probing proximal promoter regions. We used the full-length STHdh7/7 and STHdh111/111 cell lines, facilitating equivalent wild-type and mutant Htt pull-down in ChIP with MAb2170. Analysis of Htt ChIP product binding at the individual gene level revealed that the level and pattern of binding to each probe is similar between the STHdh7/7 and STHdh111/111 cell lines, with relatively few probes showing significantly different binding patterns (Fig. 4A–C). Htt was significantly immunoenriched at 419 (2.3%) of the promoter regions for known protein-coding genes in the wild-type (STHdh7/7) cell line (Fig. 4A), and at 338 (1.9%) of promoter regions in the homozygote (STHdh111/111) cell lines (Fig. 4B). This percentage of promoters bound is in the same range of magnitude as transcripts with altered expression as assessed by microarray analysis, typically 2–4% of transcripts (Luthi-Carter et al., 2000). Of the genes that were bound, only 62 genes (0.3%) were bound by Htt in both wild-type and homozygote cell lines, suggesting that the majority of gene targets of wild-type and mutant Htt may be distinct (Fig. 4D,E, supplemental Table 1, available at www.jneurosci.org as supplemental material). We used sequence information from the probes most significantly bound by wild-type and mutant Htt using the binding-site estimation suite of tools (BEST) (Che et al., 2005) to determine whether a sequence-specific preference existed in this paradigm. BEST analyses failed to reveal any distinct sequence preferences by either wild-type or mutant Htt (data not shown).
Figure 4.

Genomic binding sites of wild-type and mutant Htt. A–C, Representative examples of binding events for more wild-type Htt (open circles) bound genes [Slc8a1 (solute carrier family 8, member 1), Sox11 (SRY-box containing gene 11), Rpn1 (ribophorin I)] (A), more mutant Htt (filled squares) bound genes (Tlr8 [Toll-like receptor 8], Pcoln3 [procollagen (type III) N-endopeptidase], Jam3 [junction adhesion molecule 3]) (B), and genes showing no difference between wild-type and mutant Htt [Actb (cytoplasmic β-actin), Foxo6 (forkhead box O6), Abca2 (ATP-binding cassette, subfamily A, member 2)] (C). For A–C, binding ratios (y-axis) are plotted against chromosomal location (x-axis) for STHdh7/7 (open circles) and STHdh111/111 (filled squares), although the precise chromosomal location on the x-axis is omitted for simplicity. D, Genes on chromosome 2 for which significant binding events have been identified include those bound more by wild-type Htt (open bars), and those with increased binding by mutant Htt (filled bars). Shown are relative binding levels for each probe and the ratio of mutant/wild-type Htt binding (y-axis), plotted as a function of chromosomal probe localization (x-axis), with the genes indicated (top). E, Genes bound in STHdh7/7 (419) and STHdh111/111 (338) cell lines show little overlap (62), suggesting that wild-type and mutant Htt may have distinct targets. F, ChIP subcloning analysis using control and HD patient brain as a source material shows that the majority of clones bind within intronic or intergenic regions as opposed to proximal promoters.
Although ChIP-on-chip analysis yields insight into the binding of Htt at gene proximal promoters, many transcription factor binding sites are not limited to the proximal promoter region of a gene (Nelson et al., 2006). To determine whether Htt binds genomic regions other than proximal promoters, we also used a ChIP subcloning approach. We performed ChIP with Htt antibody MAb2170 on human HD patient brains and normal controls, and subcloned and sequenced the Htt-immunoenriched DNA. Sixty percent of clones from the HD patient brains, but only 40% of clones from control patient brains, were successfully and significantly aligned with a genomic region. Of the clones that were aligned, the majority were localized to intronic regions, followed by intergenic regions (Fig. 4F, supplemental Table 2, available at www.jneurosci.org as supplemental material). More than one-third of clones from both control and HD sequences contained repetitive elements such as microsatellites that aligned to multiple genomic loci. Other clones did not significantly align with defined genomic regions and were thus unidentifiable. These results suggest that Htt binding sites are not limited to proximal promoters, but also include intronic or intergenic regions. More clones from the control source material were unidentifiable, whereas clones derived from HD patient brains appeared to derive from an interaction with DNA (Fig. 4F).
We identified genes whose expression is modulated differentially according to the polyglutamine repeat length in Htt, and additionally have identified some genes bound by wild-type and mutant Htt. To determine whether wild-type and mutant Htt binding events correlated with expression of nearby genes, we interrogated STHdh7/7 and STHdh111/111 microarray expression profiles (Fig. 1) (Sadri-Vakili et al., 2007) for genes with significant ChIP-on-chip binding events for wild-type and mutant Htt, and filtered the genes on the basis of whether they were expressed in the cell lines. We found no correlation between Htt ChIP-on-chip binding events and mRNA expression profiles at either a gene-by-gene level, or at a global level (Fig. 5).
Figure 5.

No correlation between ChIP-on-chip data and mRNA expression profiling. A, ChIP-on-chip binding ratios (dashed line) for STHdh7/7 (open bars) and STHdh111/111 (filled bars) do not show a straightforward correlation with mRNA expression level changes (solid line) on a gene-by-gene basis: shown are representative combined data from ChIP-on-chip and gene expression studies for significant genes on chromosomes 14, 15, and 16. B, ChIP-on-chip binding ratios (open triangles, ranked in order) and gene expression profiles (crosses) show no correlation at a global level. Also plotted are STHdh7/7 (open circles) and STHdh111/111 (filled squares) binding ratios.
Exon 1 huntingtin binds directly to DNA in the absence of other proteins
ChIP-based experiments demonstrate that Htt is present at genomic loci, but these experiments cannot discriminate between direct binding of Htt to DNA and indirect binding via an interacting protein. To determine whether Htt can bind DNA directly, we have established and validated a DIP technique using positive controls (known DNA-binding proteins) and negative controls (proteins known not to interact with DNA) (Fig. 6A). We found that GST-exon 1 Htt fusion proteins interact with genomic DNA at significant levels. We then asked the question of whether it was possible to assess sequence specificity for DNA binding using double-stranded DNA fragments or plasmids containing transcription factor consensus sequences (Fig. 6B) (data not shown). Interestingly, GST-exon 1 Htt fusion proteins were able to interact with the p53 consensus sequence by this technique (Fig. 6B). Furthermore, when GST-exon 1 Htt fusion proteins and GST-p53 fusion proteins were incubated together, DNA binding was inhibited rather than potentiated, suggesting competition for binding sites (Fig. 6C). Thus, exon 1 Htt can bind DNA directly in vitro, in a manner that potentially interferes with the activities of other transcription factors.
Figure 6.

Exon 1 Htt binds nonspecifically to DNA directly in the absence of other proteins in a polyQ-dependent manner. A, Validation of the DIP technique with known DNA interactors [DNA-binding domains of CBP (GST-CBP4, GST-CBP1), GST-p53] and proteins known not to interact with DNA [GST-only, non-DNA-binding domains of CBP (GST-CBP2, GST-CBP5)]. Exon 1 Htt proteins bind significant amounts of genomic DNA, with expanded repeats (GST-HD32Q and GST-HD53Q) typically binding equivalent or more DNA than wild type (HD20Q). Error bars are SEM (n = 4). B, Exon 1 Htt proteins bind a p53 consensus sequence, as does the nonspecific DNA-binding domain of GST-CBP4. C, Exon 1 Htt proteins interfere with GST-p53 DNA binding. The dashed line indicates the predicted amount of genomic DNA binding (estimated by adding the level of binding of GST-p53 and the exon 1 Htt protein species). D, DNA binding by GST fusion proteins on a transcription factor array shows appropriate binding by GST-p53 to its recognition element (boxed area) in addition to some ectopic binding, some of which is explained through GST-only. The GST-HD20Q binding pattern resembles that GST-p53; however, the polyQ repeat expansion in GST-HD53Q potentiates DNA binding. Binding levels were not normalized, because we assayed the presence/absence of binding only. E, Comparing GST–protein binding ratios (HD53Q/GST-HD20Q) with transcription factor activity in cell lines (STHdh111/111/STHdh7/7) shows that transcription factor recognition elements with more binding in HD conditions are those bound by pure mutant exon 1 Htt in vitro (Table 2).
In an attempt to determine DNA-binding sequences for Htt, we incubated GST-Htt with labeled probes consisting of known DNA consensus sequences and hybridized the bound probes to a transcription factor array (Fig. 6D). As expected, GST-p53 bound to the p53 consensus sequence in addition to some ectopic binding, possibly because of the excess of pure protein. GST alone bound to some sequences, which were excluded from additional analysis. Both wild-type and mutant exon 1 Htt proteins bound to the same motifs as GST-p53, but binding was not limited to these motifs. Expanded polyQ repeat protein binds more DNA sequences than wild-type protein, including response elements for transcription factors whose activity is modulated by Htt such as p53 and the thyroid hormone receptor (Steffan et al., 2000; Yohrling et al., 2003). Interestingly, the transcription factor recognition elements that are bound by mutant Htt are remarkably similar to the transcription factors with increased activity in HD models (Fig. 6E, Table 2).
Table 2.
GST fusion protein binding to transcription factor recognition sequences
| Transcription Factor | GST only | GST-p53 | GST-HD20Q | GST-HD53Q | 53Q/20Q ratio | 111/111 to 7/7 ratio |
|---|---|---|---|---|---|---|
| AP-1(1) (Jun) | + | 5.1 | 5.9 | |||
| AP-2(2) (Jun) | ++ | ++ | ++++ | 1.9 | 2.6 | |
| Brn-3 | + | 8.9 | 2.0 | |||
| Cebp | ++ | 21.1 | ||||
| Cebpz | + | + | ++ | 1.9 | 6.2 | |
| Cutl1 | 21.6 | |||||
| c-Myb | ++ | +++ | +++ | 1.2 | 5.8 | |
| AP-1(2) (Jun) | + | ++ | 1.6 | 5.3 | ||
| Creb | + | + | ++ | ++++ | 3.7 | 3.9 |
| E2f1 | + | ++ | +++ | ++++ | 1.6 | 3.9 |
| Egr1 | ++++ | ++++ | ++++ | ++++ | 1.0 | 1.5 |
| ERE | +++ | 10.7 | 5.5 | |||
| Ets | ++ | 5.5 | 7.1 | |||
| Etv4 | ++ | 7.5 | 1.6 | |||
| GAS/ISRE | 2.9 | |||||
| AP-2(2) (Jun) | ++ | ++ | +++ | 1.2 | 1.1 | |
| GATA | 0.9 | 2.9 | ||||
| GRE | ++ | 36.9 | 2.4 | |||
| Hnf4 | + | + | ++ | 30.2 | 0.7 | |
| Irf1 | 2.7 | |||||
| Mef1 | + | ++++ | 4.0 | 3.0 | ||
| Mef2 | 3.8 | |||||
| Myc-Max | + | ++ | 3.9 | 3.4 | ||
| NF-1 | + | 4.5 | 1.5 | |||
| Nfatc | 4.6 | |||||
| Yy1 | ++ | +++ | 2.5 | 1.7 | ||
| Nfe2 | + | ++++ | 13.7 | 1.4 | ||
| NF-κ B | ++++ | 30.1 | 1.7 | |||
| Oct–1 | + | 3.8 | 3.0 | |||
| p53 | ++ | ++ | ++++ | 1.7 | 1.2 | |
| Pax-5 | + | ++ | 15.0 | 1.1 | ||
| Pbx1 | + | 4.6 | 1.4 | |||
| Pit1 | 8.6 | |||||
| PPAR | 0.5 | |||||
| PRE | ++ | 31.3 | 1.2 | |||
| RAR(DR-5) | + | +++ | 6.3 | 1.2 | ||
| RXR(DR-1) | + | 5.3 | 1.1 | |||
| SIE | + | +++ | 4.5 | 1.5 | ||
| Smad SBE | 1.8 | |||||
| Smad 3/4 | + | ++ | 3.7 | 1.2 | ||
| Sp1 | ++ | ++ | ++++ | 3.0 | 3.7 | |
| Stat1 | ++ | 5.7 | 1.7 | |||
| Stat3 | ++ | 6.5 | 1.3 | |||
| Stat4 | + | ++ | 3.8 | 1.2 | ||
| Stat5 | 0.9 | |||||
| Stat6 | 0.6 | |||||
| TFIID | + | 1.2 | 2.1 | |||
| TR | + | ++ | +++ | 3.5 | 6.8 | |
| TR(DR-4) | +++ | 4.3 | 1.7 | |||
| Usf1 | ++ | + | ++ | 3.0 | 1.5 | |
| VDR (DR-3) | + | ++ | 2.2 | 1.5 | ||
| HSE | 4.2 | |||||
| MRE | ++ | +++ | 1.8 | 1.3 |
Shown is the level of binding to a given transcription factor recognition sequence for each GST protein. The ratio of binding for GST-HD53Q/GST-HD20Q was calculated to compare with the ratio of binding of these transcription factors in STHdh cell lines. +, Some binding; ++, binding; +++, some binding at 1:10 dilution; ++++, binding at 1:10 dilution. ERE, Estrogen response element; GRE, glucocorticoid response element; HSE, heat shock element; ISRE, interferon-stimulated response element; MRE, MEF2 response element; NF-κ B, nuclear factor κ B; PPAR, peroxisome proliferator-activated receptor; RAR, retinoic acid receptor; RXR, retinoid X receptor; SIE, sis-inducible element; TFIID, mouse and human TATA-factors; USF-1, upstream stimulatory factor 1; VDR, vitamin D receptor. Ets1, E26 avian leukemia oncogene 1; GATA, GATA binding protein 1 (globin transcription factor 1); GAS, interferon-γ activation factor (GAF) binding site; CBF, CCAAT/enhancer binding protein zeta; CDP, Cut-like 1; C/EBP, CCAAT/enhancer binding protein; EGR, early growth response 1; TR, thyroid hormone receptor; Smad SBE, MAD homologue 1 binding element; MRE, myocyte enhancing factor 2 response element; SIE, serum inducing element.
Htt alters DNA conformation
ChIP-based and direct-DNA interaction assays suggest that there is no sequence-specific basis for the Htt–DNA interaction. Mutant exon 1 Htt can differentially modulate DNA structure in vitro, as suggested by DNase footprinting assays (data not shown). To further assess whether DNA structure is affected by mutant Htt in vivo, we performed micrococcal nuclease assays on extracts prepared from STHdh7/7 and STHdh111/111 cell lines (Fig. 7A). Titrating the micrococcal nuclease enzyme on equivalent amounts of cell extract lead to differential patterns of digestion, with more digestion occurring in the mutant 111/111 cell lines compared with the 7/7 cell lines for a given amount of enzyme, suggesting a more open DNA structure that is more accessible to the digesting enzyme. To further confirm that DNA structure is altered around gene promoters, we performed Southern hybridization with various gene-specific probes. We observed radiolabeled probe hybridization to both high- and low-molecular-weight species of DNA in the MNase-treated 7/7 cells, but only to low-molecular-weight DNA species in MNase-treated 111/111 cells, consistent with the notion that the mutant 111/111 cell lines comprise a more open DNA structure that is more accessible to digestion by MNase (Fig. 7B) (data not shown).
Figure 7.
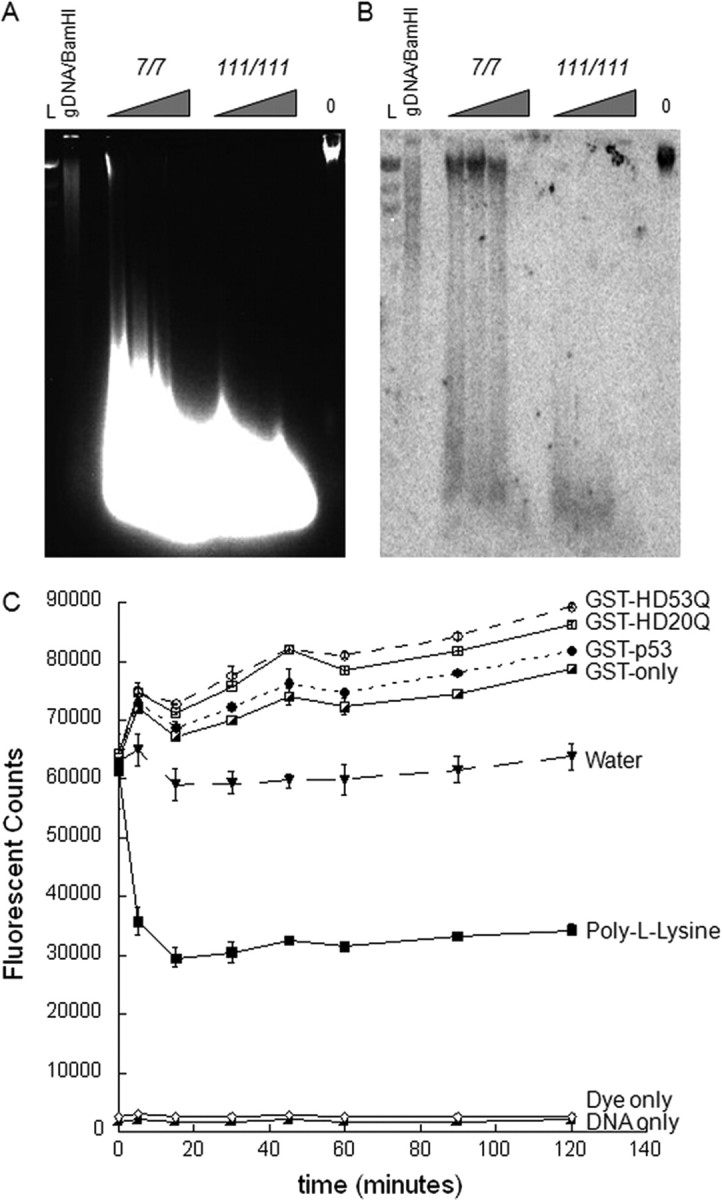
Mutant Htt may contribute to a more open DNA conformation. A, Micrococcal nuclease assay on STHdh7/7 and STHdh111/111 cell lines show more digestion in the mutant Htt cell lines than the wild-type cell lines, which suggests a more open conformation of DNA, facilitating access of the enzyme. Triangles represent amounts of titrated MNase enzyme (from 2.5 to 12.5 units). Also shown is control genomic DNA from STHdh111/111 cell lines with 0 units of MNase enzyme. L is the λHindIII DNA ladder. B, Southern hybridization of a cathepsin H (Ctsh)-specific probe to the MNase-digested chromosomal DNA revealed the presence of high-molecular-weight fragments in the STHdh7/7 cell lines only, with levels of radiolabeled probe binding to high-molecular-weight fragment much higher than levels of high-molecular-weight DNA in the native gel. In contrast, the Ctsh probe hybridized only to low-molecular-weight fragments in the homozygote STHdh111/111 cell lines, suggesting a more open chromatin conformation accessible to digestion by MNase. Probes specific for other genes have demonstrated a similar pattern (data not shown). C, Exon 1 Htt exerts distinct polyQ-dependent effects on DNA conformation, as assayed by DNA–YOYO-1 fluorescence. Addition of 0.5 μm GST fusion proteins increases the YOYO-1 fluorescent signal, which corresponds to a more open DNA conformation. We observed more fluorescence with addition of GST-p53 than GST-only, as expected. Furthermore, addition of the mutant Htt protein GST-HD53Q induces more fluorescence than the wild-type GST-HD20Q. Quenching of the fluorescence by poly-l-lysine, a DNA condensing agent, confirms the assay. Data shown in C are a representative experiment from four independent trials. Error bars indicate SEM.
To test the hypothesis that wild-type and mutant Htt alter DNA conformation directly, we chose to use a DNA condensation assay to measure the direct effect of Htt protein on DNA structure. YOYO-1, a dimeric oxazole yellow cyanine dye that asymmetrically bis-intercalates into the DNA strand, has been used to assess changes in DNA structure (Rye et al., 1992; Wong et al., 2001; Krishnamoorthy et al., 2002, 2003). Validation of this assay with a known DNA-condensing agent (poly-l-lysine) (Wong et al., 2001) confirmed quenching of the YOYO-1 fluorescent signal (Fig. 7C). In contrast, addition of GST fusion proteins increases the YOYO-1 fluorescent signal, which is suggestive of a more open DNA conformation. We observed more fluorescence after addition of GST-p53 than GST-only as expected. (Fig. 7C). Furthermore, addition of the mutant Htt protein GST-HD53Q induces more fluorescence than the wild-type GST-HD20Q (Fig. 7C). This raises the possibility that direct Htt binding can modulate genomic DNA structure.
Discussion
We set out to identify the molecular mechanism underlying huntingtin-mediated transcriptional dysregulation in HD. We have identified genes whose expression is modulated differentially according to the polyglutamine repeat length in Htt. Moreover, mutant Htt modulates the binding activities of many transcription factors, which in part induces the aberrant mRNA expression profiles. In addition, Htt-ChIP product analyses by gene-specific PCR shows Htt occupation of gene promoters in vivo. Htt ChIP analyses were extended to ChIP-on-chip and ChIP subcloning, thus enabling us to identify genomic regions bound by wild-type and mutant Htt in an unbiased manner. We found distinct localization patterns of wild-type and mutant Htt at proximal promoter regions within the genome. Furthermore, proximal promoter Htt binding sites may represent a subset of total Htt binding sites, because the majority of Htt binding sites appear to be within intronic and intergenic regions. Additionally, exon 1 Htt binds directly to multiple DNA sequences, and binding is enhanced with longer polyQ repeat lengths. Interestingly, transcription factors whose recognition elements are bound directly by Htt have altered binding activity in HD conditions. Finally, exon 1 Htt binding directly alters DNA conformation in vitro. The data presented in this manuscript, together with previous observations that nuclear mutant Htt fragments are sufficient for transcriptional dysregulation in vivo (Benn et al., 2005), suggest a model whereby mutant Htt interacts directly with DNA, altering DNA conformation and transcription factor binding, and thus ultimately leads to transcriptional dysregulation (Fig. 8).
Figure 8.
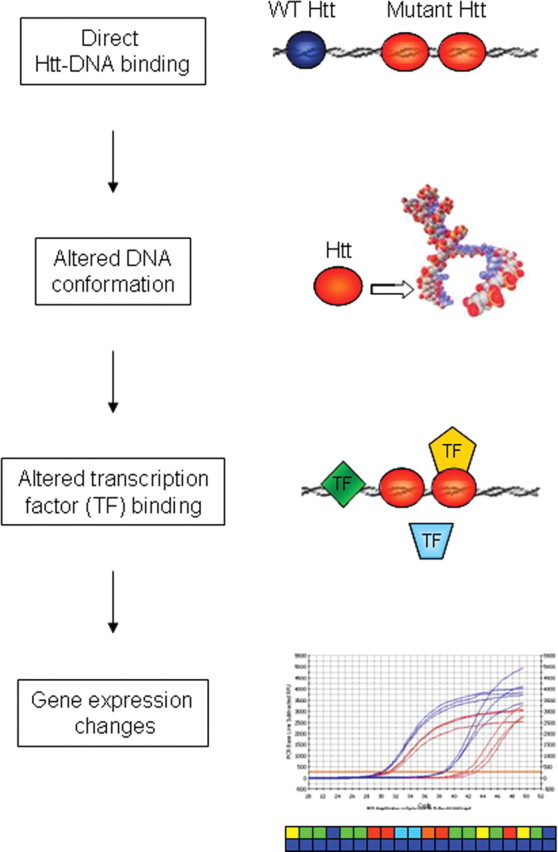
Schematic of wild-type (WT) and mutant Htt mediated effects on genomic DNA. Nuclear-localized mutant Htt facilitates direct Htt–DNA binding, which in turn leads to altered DNA conformation and altered transcription factor (TF) binding, ultimately resulting in gene expression dysregulation. Altered DNA conformation could be reflected in chromatin structure alterations, as suggested by histone modification profiles (Sadri-Vakili et al., 2007).
Microarray gene expression profiling reveals selective mRNA alterations in the STHdh111/111 compared with STHdh7/7 cell lines. The mRNA expression profile does not appear to match that of global transcriptional suppression (Hoshino et al., 2006), suggesting that the molecular basis of mutant-Htt-mediated transcriptional dysregulation is more selective. Although the pattern of mRNA alterations did not correlate with published gene expression profiles from mouse models and human HD patient brain, gene ontology analyses suggest that the differences are attributable to profiling cell lines as opposed to intact brain tissues (Sadri-Vakili et al., 2007).
Transcriptional regulation requires an extensive network of proteins, with many proteins functioning at several levels and the integration of multiple signaling pathways. Htt is implicated in a wide range of cellular functions including synaptic transmission, signal transduction and normal development (Bates et al., 2002; Woda et al., 2005). Furthermore, Htt interacts with a number of transcription factors, including but not limited to NCoR (nuclear receptor corepressor), p53, CREB, TBP, CtBP (C-terminal binding protein), Sp1 and its coactivator TAFII130, as well as proteins identified through their interaction with Htt such as the DNA-binding protein p231 HYPB (Huntingtin yeast partner B), recently identified as a histone methyltransferase (Steffan et al., 2000; Holbert et al., 2001; Rega et al., 2001; Dunah et al., 2002; Kegel et al., 2002; Li et al., 2002; Yohrling et al., 2003; Sun et al., 2005; Zhai et al., 2005). We confirm that wild-type and mutant Htt affect transcription factor binding activity, with increased binding of many transcription factors in mutant conditions at an early time point. These in vitro data support the hypothesis that transcriptional dysregulation does not arise simply from depletion of transcription factors in Htt inclusions (Yu et al., 2002). Indeed, we have previously found that the presence of inclusions has no effect on the levels of specific mRNA molecules in neurons (Sadri-Vakili et al., 2006). Together, mutant Htt does not merely reduce transcription factor levels, implicating a more direct role for mutant Htt in transcriptional dysregulation.
Many transcription factors had increased binding activity in the STHdh111/111 compared with the STHdh7/7 cell lines, and the dysregulated genes are governed by the transcription factors with altered activities. The sphere of effects for transcription factors is larger than expected, with many transcription factors correlating with varying effects on gene expression. The recognition elements bound directly by mutant exon 1 Htt and those transcription factors with increased binding activities are similar, suggesting that mutant Htt may block the access of these transcription factors to gene promoters. In this context, the observation that mutant Htt may alter DNA conformation is significant, as transcription factor access to DNA is dependent on the structural properties of DNA (Roeder, 2005). Indeed, given the larger number of transcription factors with altered activity, altered transcription factor binding as a consequence of direct Htt binding to DNA is a more parsimonious explanation than the less likely scenario in which Htt interacts with each transcription factor in individual protein–protein interaction.
We observed that both wild-type and mutant Htt occupy gene promoters in vivo, regardless of whether these genes have altered expression in HD. In addition, the ChIP data demonstrates the presence of Htt at genomic loci in vivo. Cui et al. (2006) showed increased occupancy of mutant Htt at two gene promoters in a knock-in mouse model of HD, but did not interrogate genes whose expression was not dysregulated. Surveying genome-wide Htt binding in an unbiased manner unmasked Htt binding at proximal promoter regions within the mouse genome for wild-type and mutant knock-in Htt cell lines. Binding levels of wild-type or mutant Htt were remarkably similar at many genomic loci, with a few distinct regions for which only wild-type or only mutant Htt were bound, thus suggesting a potential role for wild-type Htt in the nucleus. However, quality-control data (gene-specific amplication after LM-PCR amplification) indicate that there may be compression of gene enrichment by LM-PCR, which suggests that we did not observe a full representation of Htt–DNA binding events in the ChIP-on-chip experiments. Furthermore, ChIP-on-chip using current technology is biased toward surveying proximal promoter regions. Genomic Htt–DNA binding sites identified through ChIP subcloning were predominantly localized to intronic or intergenic regions, which would be consistent with a widely distributed localization of Htt within the genome. Indeed, given that the majority of Htt binding sites do not reside within proximal promoter regions, attempting to correlate the ChIP-on-chip data with expression profiling data may not reveal the full picture of Htt-induced transcriptional events. Furthermore, restricting ChIP analyses to proximal promoter regions is a partial representation of all Htt binding sites. Given that the presence of wild-type and mutant Htt directly perturbs gene expression as assayed by reporter construct assays (Steffan et al., 2000; Yohrling et al., 2003; Cui et al., 2006), more investigation is clearly required to understand the precise nature of the role of Htt in transcriptional regulation.
Htt does not appear to function as a typical transcription factor because we have been unable to identify a DNA consensus sequence, although the transcription factor arrays suggest that exon 1 Htt does not bind indiscriminately to every DNA consensus sequence. Although exon 1 Htt appears to be required for DNA binding, it remains possible that other domains within Htt may confer specificity, as evidenced by the distinct binding patterns of wild-type and mutant Htt at proximal promoter regions. An alternative possibility is that specificity may be bestowed through protein–protein interactions, a mechanism suggested by recent studies on other polyglutamine repeat disorders (Friedman et al., 2007; Goold et al., 2007; Lim et al., 2008). Yet another possibility is that the apparent lack of DNA sequence specificity exhibited by exon 1 Htt may implicate a role at a structural, higher-order level of chromatin organization. This would be consistent with data showing clusters of deregulated transcripts at a genomic level, supporting a hypothesis of a genome-wide mechanism of disrupted transcription (Anderson et al., 2008). Interestingly, Htt has been shown to interact tightly with the genomic matrix (Kegel et al., 2002).
The striking finding that exon 1 Htt binds directly to DNA and alters DNA conformation in a polyQ-dependent manner in the absence of other proteins at least in vitro points to a novel and, thus far, unknown function of Htt. Increased DNA binding by mutant Htt in itself is intriguing, given that many transcription factors possess a polyQ repeat domain, and the transactivation properties are altered with polyQ repeat expansion or abolition (Callewaert et al., 2003; Chen et al., 2004; Miyashita et al., 2005). Htt may thus belong to a growing number of polyQ repeat disease proteins with a transcriptional function. Of particular interest is the TATA-binding protein, TBP, a general transcription factor and a core component of the transcriptional initiation complex. PolyQ expansion of TBP leads to SCA17, and although the underlying molecular mechanism is unknown, this disorder highlights the paradox of how mutation of a general and heavily involved transcription factor can lead to a late-onset neurological disorder affecting only specific neurons (Riley and Orr, 2006).
Together, we propose a molecular mechanism whereby mutant Htt perturbs normal transcriptional regulation: enhanced genomic DNA binding by mutant Htt alters the DNA conformation and subsequent binding of transcription factors, thus disrupting the normal control of mRNA expression at an early time point in HD pathogenesis (Fig. 8). This mechanism suggests that pharmacological modulation of aberrant Htt–DNA binding could be a novel, relevant, and potentially effective therapeutic target in HD.
Footnotes
This work was supported by the Huntington's Disease Society of America Coalition for the Cure Program, the Glendorn Foundation, National Institutes of Health Grants NS38106 and NS45242, and the Massachusetts General Hospital Fund for Medical Discovery (C.L.B.). We would like to acknowledge the expert and generous guidance of Erik Nelson and David Frank (Dana Farber Cancer Institute, Boston, MA) with ChIP subcloning. Human brain samples were generously provided by the Alzheimer's Research Disease Center at Massachusetts General Hospital. We thank Marcy E. MacDonald (Massachusetts General Hospital, Boston, MA) for generously sharing the STHdh striatal cell lines. We are grateful to Eileen M. Denovan-Wright and Geraldine Gomez (Dalhousie University, Halifax, Canada) for help with DNase footprinting assays and helpful discussions. We thank Prianka Chawla, Kelly E. Glajch, Ryan P. Overland, Arjun Vasan, and Samuel Bitoun for technical assistance. We are indebted to Weiguo Zhai and Robert Tjian for helpful discussions regarding the RAP30 and RAP74 chromatin immunoprecipitations. Finally, we especially thank Gillian P. Bates for her insightful comments, constructive discussions, and help with Southern blotting.
The authors declare no conflict of interest.
References
- Anderson AN, Roncaroli F, Hodges A, Deprez M, Turkheimer FE. Chromosomal profiles of gene expression in Huntington's disease. Brain. 2008;131:381–388. doi: 10.1093/brain/awm312. [DOI] [PubMed] [Google Scholar]
- Bates GP, Harper PS, Jones AL. In: Huntington's Disease. Ed 3. Bates GP, Harper PS, Jones AL, editors. Oxford: Oxford UP; 2002. [Google Scholar]
- Benn CL, Landles C, Li H, Strand AD, Woodman B, Sathasivam K, Li SH, Ghazi-Noori S, Hockly E, Faruque SM, Cha JH, Sharpe PT, Olson JM, Li XJ, Bates GP. Contribution of nuclear and extranuclear polyQ to neurological phenotypes in mouse models of Huntington's disease. Hum Mol Genet. 2005;14:3065–3078. doi: 10.1093/hmg/ddi340. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- Callewaert L, Christiaens V, Haelens A, Verrijdt G, Verhoeven G, Claessens F. Implications of a polyglutamine tract in the function of the human androgen receptor. Biochem Biophys Res Commun. 2003;306:46–52. doi: 10.1016/s0006-291x(03)00902-1. [DOI] [PubMed] [Google Scholar]
- Carey M, Swale S. Transcriptional regulation in eukaryotes: concepts, strategies and techniques. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2000. Electromobility shift assays; pp. 338–346. [Google Scholar]
- Che D, Jensen S, Cai L, Liu JS. BEST: binding-site estimation suite of tools. Bioinformatics. 2005;21:2909–2911. doi: 10.1093/bioinformatics/bti425. [DOI] [PubMed] [Google Scholar]
- Chen S, Peng GH, Wang X, Smith AC, Grote SK, Sopher BL, La Spada AR. Interference of Crx-dependent transcription by ataxin-7 involves interaction between the glutamine regions and requires the ataxin-7 carboxy-terminal region for nuclear localization. Hum Mol Genet. 2004;13:53–67. doi: 10.1093/hmg/ddh005. [DOI] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Sadri-Vakili G, Yohrling GJ, Braveman MW, Benn CL, Glajch KE, DiRocco DP, Farrell LA, Krainc D, Gines S, MacDonald ME, Cha JH. Decreased association of the transcription factor Sp1 with genes downregulated in Huntington's disease. Neurobiol Dis. 2006;22:233–241. doi: 10.1016/j.nbd.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Cornett J, Cao F, Wang CE, Ross CA, Bates GP, Li SH, Li XJ. Polyglutamine expansion of huntingtin impairs its nuclear export. Nat Genet. 2005;37:198–204. doi: 10.1038/ng1503. [DOI] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Dorsman JC, Smoor MA, Maat-Schieman ML, Bout M, Siesling S, van Duinen SG, Verschuuren JJ, den Dunnen JT, Roos RA, van Ommen GJ. Analysis of the subcellular localization of huntingtin with a set of rabbit polyclonal antibodies in cultured mammalian cells of neuronal origin: comparison with the distribution of huntingtin in Huntington's disease autopsy brain. Philos Trans R Soc Lond B Biol Sci. 1999;354:1061–1067. doi: 10.1098/rstb.1999.0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- Friedman MJ, Shah AG, Fang ZH, Ward EG, Warren ST, Li S, Li XJ. Polyglutamine domain modulates the TBP-TFIIB interaction: implications for its normal function and neurodegeneration. Nat Neurosci. 2007;10:1519–1528. doi: 10.1038/nn2011. [DOI] [PubMed] [Google Scholar]
- Goold R, Hubank M, Hunt A, Holton J, Menon RP, Revesz T, Pandolfo M, Matilla-Dueñas A. Down-regulation of the dopamine receptor D2 in mice lacking ataxin 1. Hum Mol Genet. 2007;16:2122–2134. doi: 10.1093/hmg/ddm162. [DOI] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, Hollingsworth ZR, Collin F, Synek B, Holmans PA, Young AB, Wexler NS, Delorenzi M, Kooperberg C, Augood SJ, Faull RL, et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- Holbert S, Denghien I, Kiechle T, Rosenblatt A, Wellington C, Hayden MR, Margolis RL, Ross CA, Dausset J, Ferrante RJ, Néri C. The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: neuropathologic and genetic evidence for a role in Huntington's disease pathogenesis. Proc Natl Acad Sci U S A. 2001;98:1811–1816. doi: 10.1073/pnas.041566798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino M, Qi ML, Yoshimura N, Miyashita T, Tagawa K, Wada Y, Enokido Y, Marubuchi S, Harjes P, Arai N, Oyanagi K, Blandino G, Sudol M, Rich T, Kanazawa I, Wanker EE, Saitoe M, Okazawa H. Transcriptional repression induces a slowly progressive atypical neuronal death associated with changes of YAP isoforms and p73. J Cell Biol. 2006;172:589–604. doi: 10.1083/jcb.200509132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, McCaw EA, Hebb AL, Gomez GT, Denovan-Wright EM, et al. Mutant huntingtin affects the rate of transcription of striatum-specific isoforms of phosphodiesterase 10A. Eur J Neurosci. 2004;20:3351–3363. doi: 10.1111/j.1460-9568.2004.03796.x. [DOI] [PubMed] [Google Scholar]
- Jackson WS, Tallaksen-Greene SJ, Albin RL, Detloff PJ. Nucleocytoplasmic transport signals affect the age at onset of abnormalities in knock-in mice expressing polyglutamine within an ectopic protein context. Hum Mol Genet. 2003;12:1621–1629. doi: 10.1093/hmg/ddg163. [DOI] [PubMed] [Google Scholar]
- Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, Sapp E, Wang Y, Qin ZH, Chen JD, Nevins JR, Aronin N, DiFiglia M. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem. 2002;277:7466–7476. doi: 10.1074/jbc.M103946200. [DOI] [PubMed] [Google Scholar]
- Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice [see comments] Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy G, Duportail G, Mély Y. Structure and dynamics of condensed DNA probed by 1,1′-(4,4,8,8-tetramethyl-4,8-diazaundecamethylene)bis[4-[[3-methylbenz-1,3-oxazol-2-yl]methylidine]-1,4-dihydroquinolinium] tetraiodide fluorescence. Biochemistry. 2002;41:15277–15287. doi: 10.1021/bi020440y. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy G, Roques B, Darlix JL, Mély Y. DNA condensation by the nucleocapsid protein of HIV-1: a mechanism ensuring DNA protection. Nucleic Acids Res. 2003;31:5425–5432. doi: 10.1093/nar/gkg738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, Becanovic K, Pouladi MA, Sathasivam K, Cha JH, Hannan AJ, Hayden MR, Leavitt BR, Dunnett SB, Ferrante RJ, Albin R, Shelbourne P, Delorenzi M, Augood SJ, Faull RL. Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet. 2007;16:1845–1861. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- Kurokawa R, Kalafus D, Ogliastro MH, Kioussi C, Xu L, Torchia J, Rosenfeld MG, Glass CK. Differential use of CREB binding protein-coactivator complexes. Science. 1998;279:700–703. doi: 10.1126/science.279.5351.700. [DOI] [PubMed] [Google Scholar]
- Lee TI, Johnstone SE, Young RA. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc. 2006;1:729–748. doi: 10.1038/nprot.2006.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, Li XJ. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol. 2002;22:1277–1287. doi: 10.1128/mcb.22.5.1277-1287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Crespo-Barreto J, Jafar-Nejad P, Bowman AB, Richman R, Hill DE, Orr HT, Zoghbi HY. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature. 2008;452:713–718. doi: 10.1038/nature06731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi-Carter R, Cha JH. Mechanisms of transcriptional dysregulation in Huntington's disease. Clin Neurosci Res. 2003;3:165–177. [Google Scholar]
- Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- McCaw EA, Hu H, Gomez GT, Hebb AL, Kelly ME, Denovan-Wright EM. Structure, expression and regulation of the cannabinoid receptor gene (CB1) in Huntington's disease transgenic mice. Eur J Biochem. 2004;271:4909–4920. doi: 10.1111/j.1432-1033.2004.04460.x. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Tabuchi A, Fukuchi M, Hara D, Kisukeda T, Shimohata T, Tsuji S, Tsuda M. Interference with activity-dependent transcriptional activation of BDNF gene depending upon the expanded polyglutamines in neurons. Biochem Biophys Res Commun. 2005;333:1241–1248. doi: 10.1016/j.bbrc.2005.06.032. [DOI] [PubMed] [Google Scholar]
- Nelson EA, Walker SR, Li W, Liu XS, Frank DA. Identification of human STAT5-dependent gene regulatory elements based on interspecies homology. J Biol Chem. 2006;281:26216–26224. doi: 10.1074/jbc.M605001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrietan K, Hoyt KR. CRE-mediated transcription is increased in Huntington's disease transgenic mice. J Neurosci. 2004;24:791–796. doi: 10.1523/JNEUROSCI.3493-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rega S, Stiewe T, Chang DI, Pollmeier B, Esche H, Bardenheuer W, Marquitan G, Pützer BM. Identification of the full-length huntingtin-interacting protein p231HBP/HYPB as a DNA-binding factor. Mol Cell Neurosci. 2001;18:68–79. doi: 10.1006/mcne.2001.1004. [DOI] [PubMed] [Google Scholar]
- Riley BE, Orr HT. Polyglutamine neurodegenerative diseases and regulation of transcription: assembling the puzzle. Genes Dev. 2006;20:2183–2192. doi: 10.1101/gad.1436506. [DOI] [PubMed] [Google Scholar]
- Roeder RG. Transcriptional regulation and the role of diverse coactivators in animal cells. FEBS Lett. 2005;579:909–915. doi: 10.1016/j.febslet.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Rye HS, Yue S, Wemmer DE, Quesada MA, Haugland RP, Mathies RA, Glazer AN. Stable fluorescent complexes of double-stranded DNA with bis-intercalating asymmetric cyanine dyes: properties and applications. Nucleic Acids Res. 1992;20:2803–2812. doi: 10.1093/nar/20.11.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadri-Vakili G, Menon AS, Farrell LA, Keller-McGandy CE, Cantuti-Castelvetri I, Standaert DG, Augood SJ, Yohrling GJ, Cha JH. Huntingtin inclusions do not down-regulate specific genes in the R6/2 Huntington's disease mouse. Eur J Neurosci. 2006;23:3171–3175. doi: 10.1111/j.1460-9568.2006.04871.x. [DOI] [PubMed] [Google Scholar]
- Sadri-Vakili G, Bouzou B, Benn CL, Kim MO, Chawla P, Overland RP, Glajch KE, Xia E, Qiu Z, Hersch SM, Clark TW, Yohrling GJ, Cha JH. Histones associated with downregulated genes are hypo-acetylated in Huntington's disease models. Hum Mol Genet. 2007;16:1293–1306. doi: 10.1093/hmg/ddm078. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Schilling G, Savonenko AV, Klevytska A, Morton JL, Tucker SM, Poirier M, Gale A, Chan N, Gonzales V, Slunt HH, Coonfield ML, Jenkins NA, Copeland NG, Ross CA, Borchelt DR. Nuclear-targeting of mutant huntingtin fragments produces Huntington's disease-like phenotypes in transgenic mice. Hum Mol Genet. 2004;13:1599–1610. doi: 10.1093/hmg/ddh175. [DOI] [PubMed] [Google Scholar]
- Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J, Kooperberg C, Olson JM, Cattaneo E. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum Mol Genet. 2002;11:1953–1965. doi: 10.1093/hmg/11.17.1953. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL. SUMO modification of Huntingtin and Huntington's disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Brown R, Cook LJ, Swartz J, Rubinsztein DC. Decreased cAMP response element-mediated transcription: an early event in exon 1 and full-length cell models of Huntington's disease that contributes to polyglutamine pathogenesis. J Biol Chem. 2004;279:4988–4999. doi: 10.1074/jbc.M310226200. [DOI] [PubMed] [Google Scholar]
- Sun XJ, Wei J, Wu XY, Hu M, Wang L, Wang HH, Zhang QH, Chen SJ, Huang QH, Chen Z. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J Biol Chem. 2005;280:35261–35271. doi: 10.1074/jbc.M504012200. [DOI] [PubMed] [Google Scholar]
- Takano H, Gusella JF. The predominantly HEAT-like motif structure of huntingtin and its association and coincident nuclear entry with dorsal, an NF-kB/Rel/dorsal family transcription factor. BMC Neurosci. 2002;3:15. doi: 10.1186/1471-2202-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Woda JM, Calzonetti T, Hilditch-Maguire P, Duyao MP, Conlon RA, MacDonald ME. Inactivation of the Huntington's disease gene (Hdh) impairs anterior streak formation and early patterning of the mouse embryo. BMC Dev Biol. 2005;5:17. doi: 10.1186/1471-213X-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Kong S, Dragowska WH, Bally MB. Oxazole yellow homodimer YOYO-1-labeled DNA: a fluorescent complex that can be used to assess structural changes in DNA following formation and cellular delivery of cationic lipid DNA complexes. Biochim Biophys Acta. 2001;1527:61–72. doi: 10.1016/s0304-4165(01)00149-0. [DOI] [PubMed] [Google Scholar]
- Xia J, Lee DH, Taylor J, Vandelft M, Truant R. Huntingtin contains a highly conserved nuclear export signal. Hum Mol Genet. 2003;12:1393–1403. doi: 10.1093/hmg/ddg156. [DOI] [PubMed] [Google Scholar]
- Yohrling GJ, Farrell LA, Hollenberg AN, Cha JH. Mutant huntingtin increases nuclear corepressor function and enhances ligand-dependent nuclear hormone receptor activation. Mol Cell Neurosci. 2003;23:28–38. doi: 10.1016/s1044-7431(03)00032-0. [DOI] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Nguyen HP, Li XJ. Huntingtin inclusions do not deplete polyglutamine-containing transcription factors in HD mice. Hum Mol Genet. 2002;11:905–914. doi: 10.1093/hmg/11.8.905. [DOI] [PubMed] [Google Scholar]
- Zhai W, Jeong H, Cui L, Krainc D, Tjian R. In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell. 2005;123:1241–1253. doi: 10.1016/j.cell.2005.10.030. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]