

Abstract
The ESCRT pathway mediates membrane remodeling during enveloped virus budding, cytokinesis, and intralumenal endosomal vesicle formation. Late in the pathway, a subset of membrane-associated ESCRT-III proteins display terminal amphipathic “MIM1” helices that bind and recruit VPS4 ATPases via their MIT domains. We now report that VPS4 MIT domains also bind a second, “MIM2” motif found in a different subset of ESCRT-III subunits. The solution structure of the VPS4 MIT-CHMP6 MIM2 complex revealed that MIM2 elements bind in extended conformations along the groove between the first and third helices of the MIT domain. Mutations that block VPS4 MIT-MIM2 interactions inhibit VPS4 recruitment, lysosomal protein targeting and HIV-1 budding. MIT-MIM2 interactions appear to be common throughout the ESCRT pathway and possibly elsewhere, and we suggest how these interactions could contribute to a mechanism in which VPS4 and ESCRT-III proteins function together to constrict the necks of budding vesicles.
The human ESCRT pathway helps facilitate membrane fission events required during enveloped virus budding, the abscission stage of cytokinesis, and intralumenal vesicle formation at the multivesicular body (MVB) (Bieniasz, 2006; Hurley, 2008; Saksena et al., 2007; Williams and Urbe, 2007). Most ESCRT pathway members function as subunits of one of four multiprotein complexes, termed ESCRT-0, -I, -II, and –III. The pathway also has a multiprotein ATPase complex, which is organized by the dodecameric VPS4 enzyme and is recruited to sites of action through direct interactions with ESCRT-III subunits (Babst et al., 1998; Obita et al., 2007; Stuchell-Brereton et al., 2007). Although mechanistic details are lacking, there is increasing evidence that the ESCRT-III and VPS4 complexes may act in concert to help to create and constrict the thin membrane “necks” that arise during virus budding, abscission, and MVB vesicle formation.
Human cells express a total of 11 ESCRT-III or CHMP (charged multivesicular body) proteins, that can be subdivided into seven different families (CHMP1−7). The CHMP1−6 families correspond to the six ESCRT-III-like proteins in S. cerevisiae, whereas CHMP7 has no yeast ortholog (Horii et al., 2006). ESCRT-III subunits co-polymerize at sites of action (Babst et al., 2002), and recent evidence indicates that ESCRT-III subunits of the CHMP4 family can form circular filaments around sites of membrane exvagination (Hanson et al., 2008). Crystallographic analyses have revealed that the core of another ESCRT-III subunit, CHMP3, is organized about an extended helical hairpin that is capped at one end by three smaller helices (Muziol et al., 2006). All CHMP proteins have related sequences and presumably adopt similar folds, but have evolved distinct activities, particularly in co-factor recruitment. For example, specific ESCRT-III subunits bind more tightly to the VPS4 ATPases (Stuchell-Brereton et al., 2007), the VPS4 activator Vta1p/LIP5 (Azmi et al., 2008; Shim et al., 2008; Xiao et al., 2008), the adaptor protein ALIX (CHMP4A-C) (McCullough et al., 2008), and the ubiquitin hydrolases AMSH (CHMP1A and 3) and UBPY (CHMP1A, 1B, 2A, 4C, and 7) (Row et al., 2007). In all well characterized cases, these cofactors are recruited by amphipathic helices located at or near the CHMP C-termini. VPS4A, VPS4B, AMSH, UPBY, and Vta1p/LIP5 (but not ALIX) all use MIT (microtubule interacting and transport) domains to bind these ESCRT-III recognition helices, which are termed MIM motifs (MIT Interaction Motifs) (Hurley and Yang, 2008). Remarkably, the VPS4 ATPase complex apparently contains 24 MIT domains, one from each of the 12 VPS4 subunits and two from each of the six associated Vta1p/LIP5 cofactors (Azmi et al., 2008; Xiao et al., 2008; Yu et al., 2008). Thus, it appears that ESCRT-III filaments present a polymeric array of MIM elements that recruit VPS4 and other MIT-domain containing proteins to complete the terminal events of the ESCRT pathway.
Recent structural studies have revealed that MIT domains are asymmetric three helix bundles that bind amphipathic MIM helices along the groove between MIT helices 2 and 3 (Obita et al., 2007; Stuchell-Brereton et al., 2007). Molecular recognition is mediated by three MIM leucine/hydrophobic residues and by flanking hydrophilic residues that make favorable interactions along the MIT groove. It was therefore surprising that our initial surveys of VPS4-ESCRT-III protein interactions revealed that VPS4A and VPS4B bind several human CHMP4−6 proteins ((von Schwedler et al., 2003) and data not shown), even though these ESCRT-III subunits lack C-terminal MIM consensus sequences. We therefore characterized the tightest of these complexes, between CHMP6 and VPS4A, and have discovered that MIT domains can bind their targets through a second type of interaction that is entirely distinct from the canonical MIM-MIT interaction. The three dimensional structure and functional importance of this new MIT interaction mode are reported.
RESULTS
Mapping the CHMP6-VPS4 MIT Domain Interaction
Biosensor binding assays showed that pure recombinant VPS4A MIT domain bound the immobilized GST-CHMP6 protein with a dissociation constant of KD=33±7μM (Fig. 1), which is similar to the affinities of other well characterized VPS4 MIT-ESCRT-III interactions (Obita et al., 2007; Stuchell-Brereton et al., 2007). As noted above, however, the CHMP6 terminus does not conform to the MIM consensus, and removing the final 20 CHMP6 residues actually increased VPS4A MIT binding affinity slightly (KD=23.2 ±0.2 μM), indicating that the VPS4A MIT-CHMP6 interaction must differ from all VPS4-ESCRT-III interactions characterized to date.
Figure 1. VPS4A MIT Binds a Unique Internal CHM6 MIT Interacting Motif (MIM2).

(A) Biosensorgrams showing concentration-dependent binding of purified VPS4A MIT (residues 1−84, 0−500 μM) to full length GST-CHMP61−201.
(B) Biosensor binding isotherms showing VPS4A MIT binding to a series of different GST-CHMP6 deletion mutants (see inset key). Binding to a GST control surface was negligible (not shown).
(C) Schematic illustration of the predicted CHMP6 structure, indicating the location of the CHMP6 MIM2 between helices α4 and α5. Dissociation constants for the different CHMP6 constructs are shown at right.
Deletion analyses were used to map the VPS4A MIT binding site to an internal sequence spanned by CHMP6 residues 170−181 (Figs. 1B and 1C, and see Supplemental Table 1 for all measured dissociation constants). Optimal binding was observed for the CHMP6165−181 fragment (KD=5.8±0.8 μM), and strong binding was also observed for the minimal CHMP170−181 fragment (KD=8±0.1 μM). Importantly, VPS4A MIT bound to these smaller CHMP6 fragments ∼5-fold more tightly than to the full length protein, which is consistent with the observation that full length ESCRT-III proteins fold into “autoinhibited” conformations that can mask protein binding sites (Lata et al., 2008; Lin et al., 2005). The VPS4A binding site on CHMP6 is conserved across eukaryotic CHMP6 proteins (Fig. 2A and see Supplemental Table 2), but the sequence is rich in prolines and does not exhibit significant helical propensity, again indicating a novel MIT binding mode.
Figure 2. Solution Structure of the VPS4A MIT-CHMP6 MIM2 Complex.
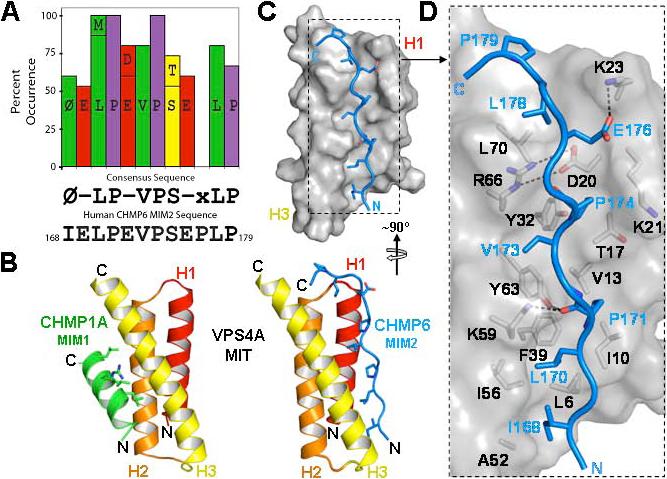
(A) Graphic summary showing MIM2 residues that are >50% conserved across eukaryotic CHMP6 proteins (and see Supplemental Table 2), the resulting MIM2 consensus sequence, and the CHMP6 MIM2 sequence. “Φ” denotes an aliphatic residue and “-“ denotes acidic residues.
(B) Structures of human VPS4A MIT in complex with the MIM1 element from CHMP1A (left structure, MIM1 helix in green) and the CHMP6 MIM2 element (right, MIM2 strand in blue). The MIT domains are shown in the same orientation to emphasize that the MIM1 and MIM2 elements bind in different grooves of the MIT three helix bundle. Key MIM contact residues are shown explicitly in both structures.
(C, D) Overview (C) and detailed (D) views of the VPS4A MIT domain (gray and black, space filling representation) in complex with the CHMP6 MIM2 element (stick representation, blue). Key side chains on both sides of the interface are shown in (D), with selected intermolecular salt bridges and hydrogen bonds indicated by dashed lines.
Structure of the VPS4A-CHMP6 MIM2 Complex
To visualize how VPS4 recognizes CHMP6, we determined the solution structure of the VPS4A MIT domain in complex with its CHMP6166−181 binding site. NMR spectra of the 1:1 complex were of high quality, and backbone heavy atoms in the ordered core of the twenty lowest penalty complexes overlaid with low rmsd values (0.34±0.06 Å), indicating that the structure was well defined (see Supplemental Figs. 1 and 2, and Table 1). As shown in Fig. 2B (right panel), VPS4A MIT formed the expected three helix bundle and did not change structure significantly upon CHMP6 binding. The CHMP6166−181 element bound in an extended conformation along the groove between MIT helices 1 and 3, burying 1300 ± 150 Å2 of solvent-exposed surface. The CHMP6166−181 binding mode is therefore entirely different from previously characterized MIM-MIT interactions (compare right and left panels in Fig. 2B). We have designated this new binding element a type-2 MIT Interaction Motif (MIM2).
As shown in Fig. 2, the CHMP6166−181 MIM2 peptide is oriented parallel to MIT helices H1 and H3, and tracks in an extended but irregular conformation along the MIT helix 1/3 groove for most of its length until a kink at CHMP6 residue Pro177 orients the more mobile C-terminal residues (177−179) over top of the MIT helical bundle. The structure of a yeast Vps4p MIT-Vps2 complex also showed a proline-rich sequence binding in the helix 1/3 groove of the MIT domain (in addition to a canonical MIM1-MIT interaction made by the Vps2p terminus) (Obita et al., 2007). Although the VPS4 MIT-CHMP6166−181 and Vps4p MIT-VPS2183−232 structures share some similarities in binding proline sidechains in the MIT helix 1/3 groove, Obita et al. concluded that the proline interactions were crystal packing artifacts because the Vps4p MIT binding affinity was not reduced when the proline-rich Vps2p sequence was removed. We agree with this interpretation because the binding orientation of the Vps2 strand is opposite that seen here for the CHMP6166−181 MIM. Importantly, the orientation of the MIM2 element in our structure matches the orientation of an MIM1 element (i.e., both elements bind parallel to MIT helix 3), which enables MIT domains to bind different ESCRT-III subunits in the same relative orientation.
The VPS4A MIT-CHMP6166−181, structure, together with sequence and mutational analyses described below indicate that the central 170LP(E/D)VP174 CHMP6 sequence forms the core of the MIM2 element, with flanking residues also contributing binding affinity and specificity. The helix 1/3 groove is generally hydrophobic, but there is a salt bridge between MIT Arg66 and Glu20 at the top of the groove that remains intact in the complex and is partially buried by peptide binding. As shown in Fig. 2D, a series of CHMP6166−181 side chains project toward the MIT domain, and CHMP6 residues Ile168, Leu170, Pro171, Val173, Pro174, and Leu178 each make well defined hydrophobic interactions along the groove. All of these MIM2 residues are conserved in identity or hydrophobic character across metazoan CHMP6 MIM2 proteins (Fig. 2A and Supplemental Fig. 2), and the importance of the Leu170, Val173, and Leu178 interactions was confirmed by mutagenesis (see below). In addition, the MIT Lys59 and Tyr63 side chains form hydrogen bonds to the carbonyl oxygen of CHMP6 Pro171, which help stabilize the MIT-MIM2 core, and the CHMP6 Glu176 side chain also forms a salt bridge with MIT Lys23, which explains the conservation of this acidic MIM2 residue. In contrast, the hydrophilic CHMP6 Glu169, Glu172 and Ser175 residues all project into solution, and the conservation of the two acidic residues is not explained by the structure, except insofar as these residues contribute to complex solubility.
Mutational Analyses of the MIM2-MIT Interface
Mutational analyses were performed to assess the energetic contributions of the CHMP6 MIM2 Leu170, Val173, and Leu178 residues and to identify non-binding mutants for use in functional studies. As shown in Fig. 3A, the CHMP6 Leu170Asp and Val173Asp mutations eliminated all detectable VPS4A MIT binding, whereas the Leu178Asp mutation reduced binding affinity 8-fold. These data confirm the importance of the core MIM2 Leu170 and Val173 residues, and demonstrate that the flanking Leu178 residue, though not essential, also contributes to VPS4A MIT binding.
Figure 3. Mutational Analyses of the VPS4A MIT-CHMP6 Complex.

(A) Biosensor binding isotherms showing VPS4A MIT binding to wild type and mutant CHMP6 MIM2 peptides (labeled). Note that the CHMP6 L178D mutation reduces the binding affinity ∼8-fold (from KD=5.8±0.8 to KD=44.4±0.2 μM), whereas the L170D and Vl173D mutations block all detectable binding.
(B) Biosensor binding isotherms showing that the V13D mutation in VPS4A MIT helix 1 blocks CHMP6 MIM2 binding (triangles) but does not affect CHMP1B MIM1 binding (ovals).
(C) Biosensor binding isotherms showing that the L64D mutation in VPS4A MIT helix 3 blocks CHMP1B MIM1 binding (ovals), but has much less effect on CHMP6 MIM2 binding (triangles), altering CHMP6 MIM2 binding from KD = 5.8 ± 0.8 μM to KD = 26.1 ± 0.8 μM.
Point mutations in the two different VPS4A MIT binding surfaces were also examined to identify MIT mutations that could discriminate between the MIM1 and MIM2 binding modes. These experiments tested VPS4A MIT binding to the CHMP6 MIM2 element and to the well characterized MIM1 element at the C-terminus of human CHMP1B (Stuchell-Brereton et al., 2007). As shown in Fig. 3B, the VPS4A Val13Asp mutation, which sits in the center of the MIM2 binding groove, blocked all detectable CHMP6 binding (compare light and dark curves with triangular symbols). Importantly, this residue did not reduce CHMP1B binding affinity (compare light and dark curves with oval symbols). Conversely, a mutation within the VPS4A MIM1 binding site (Leu64Asp) blocked all detectable CHMP1B MIM1 binding, although this mutation also modestly reduced the affinity of CHMP6 MIM2 (4-fold reduction). This affinity reduction may arise because Leu64 is adjacent to Tyr63, which hydrogen bonds to the backbone carbonyl of CHMP6 Pro171. Nevertheless, these experiments identify VPS4A MIT mutations that can discriminate between the MIM1 and MIM2 binding modes and, in particular, demonstrate that the Val13Asp mutation blocks MIM2 binding without affecting the binding of a control MIM1 element.
MIM2 Binding Helps Recruit VPS4 to Class E Compartments
VPS4 ATPases are targeted to endosomal membranes via MIT domain interactions (Babst et al., 1998), and we therefore tested whether the MIM2 binding site on the MIT domain contributes to VPS4 recruitment. Endosomal localization is most easily visualized for VPS4 proteins that lack ATPase activity because these proteins are recruited but not released from the surfaces of aberrantly enlarged endosomes (termed “Class E” compartments). This phenomenon is illustrated in Fig. 4, where the wild type GFP-VPS4A protein was distributed diffusely in 91±2% of cells analyzed, whereas a GFP-VPS4AK173Q mutant, which is impaired in ATP binding, exhibited punctate Class E compartment localization in 80% of the cells (Fig. 4A, compare upper right and left panels, and Fig. 4B, compare lanes 1 and 5). As reported previously, the Leu64Asp MIT mutation, which primarily blocks MIM1 interactions, inhibited recruitment so that the VPS4AK173Q,L64D-GFP double mutant exhibited diffuse cytoplasmic distributions in 81±7% of cells (Fig. 4A, lower left panel and Fig. 4B, lane 6) (Stuchell-Brereton et al., 2007). Importantly, the Val13Asp mutation in the VPS4A MIM2 binding site also inhibited Class E compartment localization, resulting in a diffuse distribution of the GFP-VPS4AK173Q,V13D double mutant in 80±4 % of cells (Fig. 4A, lower right panel and Fig. 4B, lane 7). Not surprisingly, the GFP-VPS4K173Q,V13D,L64D triple mutant was recruited to Class E compartments even less efficiently, resulting in diffuse cytoplasmic distributions in 86±3% of cells (Fig. 4B, lane 8). These experiments indicate that both MIM1 and MIM2 interactions contribute in recruiting VPS4A to endosomal membranes.
Figure 4. VPS4A MIT Mutations that Block MIM1 and MIM2 Binding also Inhibit Recruitment of VPS4AK173Q to Class E Endosomal Compartments.

A) Confocal fluorescence slices showing cellular distributions of wild type GFP–VPS4A (VPSAWT, upper left, negative control) and GFP-VPS4A proteins with the following mutations: K173Q (upper right, positive control, mutation blocks ATP binding and induces class E compartment formation), K173Q,L64D (lower left, K173Q mutation blocks ATP binding and L64D mutation blocks MIM1 binding), and K173Q,V13D (lower right, K173Q mutation blocks ATP binding and V13D mutation blocks MIM2 binding). Scale bar is 20 μm.
(B) Quantification of cells showing diffuse cytoplasmic localization of wild type and mutant GFP-VPS4A proteins (mutations are listed below). Two blinded sets of >100 cells of each type were scored, and error bars indicate standard deviations. Note that the VPS4A MIT L64D and V13D both mutations alleviate the Class E compartment recruitment induced by the K173Q mutation (compare lanes 6−8 to lane 5).
The CHMP6 MIM2 Binding Site is Required for Ferroportin Degradation
Several ESCRT functions, including HIV budding, are insensitive to CHMP6 depletion (Langelier et al., 2006). We have shown, however, that lysosomal degradation of ferroportin, a plasma membrane iron transporter, is impaired by CHMP6 depletion (De Domenico et al., 2007). This system was therefore used to test whether the CHMP6 MIM2 element was required for ferroportin trafficking through the MVB pathway. Treatment of wild type cells with the hormone hepcidin triggers ferroportin phosphorylation, endocytosis, ubiquitylation, endosomal trafficking, MVB sorting, and lysosomal degradation. In the absence of CHMP6, however, ferroportin can still be post-translationally modified and endocytosed, but not efficiently degraded because it is not incorporated into MVB vesicles. The protein therefore accumulates internally and can also be recycled back to the plasma membrane (De Domenico et al., 2007). As shown in Fig. 5, ferroportin-GFP was efficiently downregulated when endogenous CHMP6 was depleted and replaced by comparable levels of exogenously expressed, siRNA-resistant CHMP6-Myc. Efficient depletion of endogenous CHMP6 is shown in Fig. 5A (compare lanes 1 and 2, upper panel), and hepcidin-induced ferroportin downregulation in the presence of exogenous CHMP6-Myc was analyzed either by western blot detection of total cellular ferroportin-GFP (2.3-fold reduction in ferroportin upon hepcidin treatment, compare lanes 7 and 8 in Fig. 5B, panel 2), or by total fluorescence intensity (compare panels 7 and 8 in Fig. 5C and quantified as a 4.2-fold fluorescence intensity reduction in Fig. 5D). In contrast, hepcidin-induced ferroportin downregulation was inhibited by two different CHMP6 MIM2 point mutations that blocked VPS4A MIT binding (Leu170Asp and Val173Asp), as reflected by a lack of any observable ferroportin reduction in the western blot assays and less than 40% reductions in the fluorescence assays (Figs. 5B-D, compare lanes 9 and 11 to lanes 10 and 12). Control experiments behaved as expected, confirming that all CHMP6-Myc constructs were expressed at similar levels (Fig. 5B, panel 1, lanes 1−12), and that ferroportin downregulation occurred normally when the CHMP6-Myc proteins were expressed in the presence of endogenous CHMP6 (Figs. 5B-D, compare lanes 1, 3, and 5 to lanes 2, 4, and 6). Thus, mutations in the CHMP6 MIM2 element that block VPS4A MIT binding also block ferroportin downregulation, implying that the MIM2 element plays an essential role when CHMP6 functions in MVB protein sorting.
Figure 5. CHMP6 MIM2 Mutations Inhibit Hepcidin-induced Lysosomal Degradation of Ferroportin.

(A) Western blot analyses showing efficient siRNA depletion of endogenous CHMP6 protein (top blot, compare lanes 1 and 2), without changes in ferroportin–GFP (FPN) or α–Tubulin levels (bottom two blots).
(B) Western blot analyses of hepcidin–induced ferroportin–GFP downregulation in the presence of wild type and mutant CHMP6 proteins. Left panels (controls) show ferroportin levels (middle blot) in the absence (−) or presence (+) of hepcidin treatment of cells expressing wild type and mutant CHMP6-Myc proteins (top blot) in the presence of endogenous CHMP6 (control siRNA). Right panels show the same experiment, but with depletion of endogenous CHMP6, so that only the exogenous, siRNA-resistant CHMP6-Myc proteins were present. Integrated intensities were normalized to the untreated control and are shown below each band. Note that ferroportin–GFP levels decreased 2.3–fold upon hepcidin treatment in the presence of wild type CHMP6-Myc (compare lanes 7 and 8) but did not decrease in the presence of CHMP6-Myc proteins with inactivating MIM2 point mutations (L170D and V173D).
(C, D) Epifluorescence and differential interference contrast (DIC) images (C) and quantification (D) showing the effects of hepcidin treatment and CHMP6 mutations on ferroportin-GFP levels. Samples 1−12 correspond to the equivalent samples in part (B). Note that the data again show that wild type CHMP6-Myc supports efficient hepcidin-induced ferroportin-GFP downregulation (compare samples 7 and 8, 4.2-fold reduction), whereas the Myc-CHMP6 MIM2 mutants do not (compare samples 9 and 11 to 10 and 12). Scale bar is 20 μm, and DIC images of the hepcidin-treated cells are provided for reference in the lower panels of part (C). Quantifications presented in (D) show the average fluorescence/cell from two data sets (one blinded) of >100 cells each, and error bars indicate standard deviations.
The VPS4B MIM2 Binding Surface Is Required for HIV-1 Budding
As discussed below, MIM2 elements appear in several other proteins of the ESCRT pathway, suggesting that MIM2-MIT interactions may be a central, recurring theme in ESCRT pathway functions. To examine this idea, we tested the requirement for VPS4B MIT-MIM2 interactions in HIV-1 budding. HIV budding does not require CHMP6 (Langelier et al., 2006), and VPS4B binds only very weakly to CHMP6 (KD=786±6 μM, Supplemental Table 1). We therefore reasoned that if mutations in the MIM2 binding site of VPS4B MIT block virus budding, then additional MIM2-MIT interactions must also be required for this (and presumably other) ESCRT pathway functions.
These experiments again utilized an siRNA depletion/reconstitution system that allowed us to test whether VPS4B mutants could support HIV budding. siRNAs targeting both known human VPS4 isoforms (A and B) were co-transfected, and this procedure efficiently depleted both VPS4 proteins from 293T human cells (Fig. 6, top two panels, compare lanes 1 and 2). As expected, depletion of the two human VPS4 isoforms strongly inhibited HIV release as measured in a western blot assay (compare the release of virus-associated CA and MA proteins in lanes 1 and 2, panel 5 (Virus)) and also by a reduction in viral titer as measured in single cycle viral replication assays (graph at bottom, compare lanes 1 and 2, 18-fold reduction in titer and see (Fraile-Ramos et al., 2007)). Re-expression of an siRNA-resistant VPS4B construct rescued virus release and restored viral titers to 60% of the wild type control levels (compare lanes 2 and 3). In contrast, expression of a siRNA-resistant VPS4B construct that lacked ATPase activity failed to rescue virus budding (lane 4). Indeed, expression of the inactive VPS4BK180Q mutant resulted in an even lower viral titer than the siRNA depleted control alone (lane 2), presumably because the VPS4BK180Q mutant dominantly inhibited all residual VPS4 function (Martin-Serrano et al., 2003; Strack et al., 2003; von Schwedler et al., 2003). Thus, this new VPS4 depletion/reconstitution assay could be used to analyze the activity of mutant VPS4B proteins without confounding contributions from endogenous human VPS4 proteins, and the assay was therefore used to test the functional importance of the VPS4B MIT MIM1 and MIM2 binding surfaces in HIV budding.
Figure 6. The VPS4B MIT MIM1 and MIM2 Binding Surfaces Both Contribute to Efficient HIV-1 Budding and Replication.

Western blots 1 and 2 show simultaneous siRNA depletion of endogenous VPS4A and VPS4B (Compare lanes 1 and 2) and re-expression of exogenous siRNA-resistant wild type and mutant VPS4B proteins (blot 1, Lanes 3−7). Western blot 3 shows an α–Tubulin loading control. Western blots 4 and 5 show levels of the viral Gag protein and its processed MA and CA products in the cytoplasm (Cell, panel 4, note that all cells were transfected with a proviral HIV-1 expression construct), and released as viral particles (Virus). Panel 6 shows viral titers, as assayed in single cycle replication assays of the culture supernatants. The experiment shows that simultaneous depletion of both human VPS4 isoforms blocks HIV-1 release and infectivity (panels 5 and 6, compare lanes 1 and 2), that reintroduction of a wild type, siRNA-resistant VPS4B protein construct significantly rescues virus release and titer (compare lanes 2 and 3), and that virus release and titer are inhibited by the VPS4B K180Q (lane 4, positive control), L66D (lane 5, inhibited MIM1 binding), A15D (lane 6, inhibited MIM2 binding) and L66D,A15D (lane 7, inhibited MIM1 and MIM2 binding).
An inactivating point mutation in the VPS4B MIM1 binding surface (Leu66Asp, which is equivalent to the VPS4A Leu64Asp mutation) reduced HIV release and titer ∼3-fold, as compared to the wild type VPS4B protein (compare lanes 3 and 5). Similarly, an inactivating point mutation in the VPS4B MIM2 binding surface (Ala15Asp, which is equivalent to the VPS4A Val13Asp mutation) resulted in an ∼2-fold reduction in HIV release and titer (compare lanes 3 and 6). Furthermore, simultaneous mutation of both MIM1 and MIM2 binding sites reduced levels of HIV-1 release and titer 10-fold (compare lanes 3 and 7), to nearly the same level as that seen in the absence of any exogenous VPS4B (compare lanes 2 and 7). Control experiments again showed that all of the different VPS4B proteins were expressed at similar levels, indicating that none of the mutations acted by destabilizing the protein (panel 1, compare lanes 3−7). We conclude that the VPS4B MIM1 and the MIM2 binding sites both contribute to the efficiency of HIV-1 budding, and that VPS4B is essentially inactive when both MIM binding sites are mutated.
DISCUSSION
Our studies reveal that in addition to their well characterized MIM1 binding interactions, MIT domains bind a second class of sequence motifs, which we term MIM2 elements. The structure of the VPS4A MIT-CHMP6166−181 complex reveals that MIM2 elements bind as extended strands in the groove between VPS4A MIT helices 1 and 3, and that conserved residues within the CHMP6 MIM2 element make a series of important contacts along the groove. Importantly, the CHMP6 MIM2 element is required for efficient lysosomal degradation of ferroportin in response to hepcidin treatment, demonstrating that this element is essential for the MVB sorting of specific membrane protein cargoes.
MIM2 Elements in Other Proteins
MIT domains are found in both ESCRT and non-ESCRT proteins, and MIT-MIM1 interactions continue to be documented in increasing numbers (Hurley and Yang, 2008; Tsang et al., 2006). Similarly, there are indications that MIT-MIM2 interactions may also be widespread. For example, VPS4B MIT-MIM2 interactions are required for efficient HIV budding, even though CHMP6 is not required for this process, indicating that the VPS4A MIT-CHMP6 MIM2 interaction is not the only biologically relevant MIT-MIM2 interaction. Conceivably, HIV-1 budding may instead involve the use of CHMP4 MIM2 elements. All three CHMP4 family members also have identifiable MIM2 elements, which are analogous to the CHMP6 MIM2 element in that: 1) the CHMP6 and CHMP4 MIM2 motifs reside at equivalent internal positions, 2) the CHMP4 MIM2 elements also bind VPS4A MIT, albeit with lower affinities than the CHMP6 MIM2 element (Supplemental Table 1), and 3) the isolated CHMP4 MIM2 elements bind more tightly than their full length proteins, again indicating that the full length proteins likely adopt autoinhibited conformations.
We have also identified several other ESCRT-associated proteins, including IST1, that contain functional MIM2 motifs (Bajorek et al., 2008; Morita et al., 2008). The VPS4A MIT-CHMP6166−181 structure, together with sequence analyses of the CHMP6, CHMP4A-C, and IST1 MIM2 elements, suggests a core consensus sequence of: (L/V)Px(V/L)P (where “x“ denotes a variable, but hydrophilic residue). Flanking CHMP6 residues, including Glu176 and Leu177, can also make favorable interactions and additional studies it will therefore be important to identify and analyze additional MIM2 elements to discern the absolute optimal MIT binding sequence(s).
The VPS4 adaptor IST1 is of particular interest because this protein contains MIM1 and MIM2 elements that can bind VPS4A MIT simultaneously, creating a particularly high affinity interaction (Bajorek et al., 2008). Yeast Ist1p inhibits Vps4p assembly in vitro (Dimaano et al., 2008), and we speculate that the protein may also serve to release CHMP protein substrates from VPS4 MIT domains, which could rationalize why IST1 needs to bind with high affinity and uses both types of MIM element. The VPS4 MIT-IST1 example also demonstrates that MIT domains can bind MIM1 and MIM2 elements simultaneously, and we have observed that this is true even when the MIM elements are presented by different proteins (not shown). Hence, it appears likely that the ability to engage both MIM1 and MIM2 elements is a general property of MIT domains. This multivalent binding capability provides a potential rationale for the unusual asymmetry of the MIT three helix bundle in which helices 1 and 3 are disposed at right angles to the central helix 2 (Scott et al., 2005b; Takasu et al., 2005). This geometry creates a shallow groove between helices 2 and 3 that can bind amphipathic MIM1 helices, and a deeper groove between helices 1 and 3 that is better suited to binding the extended strand of the MIM2 element. It will be of interest to learn whether the third, shallow groove between helices 1 and 2 can also be used to bind a third class of target proteins.
MIM2 Elements Contribute to VPS4 Recruitment
A specific point mutation in the VPS4A MIM2 binding site inhibited recruitment of VPS4AK173Q to endosomal Class E compartments, which indicates that MIT-MIM2 interactions help recruit VPS4 proteins to ESCRT-III assemblies. Although interactions between isolated VPS4 MIT domains and ESCRT-III subunits are relatively weak, we envision that four different phenomena likely contribute to creating high affinity VPS4 binding in vivo. Firstly, conformational changes in ESCRT-III subunits that accompany polymerization likely “unmask” VPS4 MIT binding sites that are autoinhibited in the monomeric proteins (Lata et al., 2008; Lin et al., 2005). This idea is supported by our observation that VPS4 MIT domains bind less tightly to the full length CHMP6 and CHMP4 proteins than to their isolated MIM2 elements. We note that a similar affinity enhancement is not observed for C-terminal MIM1 elements, which implies that ESCRT-III autoinhibition protects the internal MIM2 elements, but not the terminal MIM1 elements. Secondly, co-polymerization of ESCRT-III may bring MIM1 and MIM2 elements from different proteins into close juxtoposition, allowing individual MIT domains to engage both elements simultaneously, thereby increasing binding affinity (see Fig. 7). This idea is supported by our observations that MIT domains can bind MIM1 and MIM2 elements simultaneously ((Bajorek et al., 2008) and data not shown), and that both MIM binding surfaces are required to recruit VPS4 to class E compartments. Thirdly, co-polymerization of ESCRT-III subunits to create an array of immobilized MIM elements should dramatically enhance the binding avidity of the oligomeric VPS4 protein. Finally, the LIP5/Vta1 activator binds both VPS4 and ESCRT-III subunits and therefore likely helps tether the enzyme to the ESCRT-III lattice (Azmi et al., 2006; Azmi et al., 2008; Lottridge et al., 2006; Scott et al., 2005a; Shim et al., 2008; Xiao et al., 2008) (note that this interaction was omitted from Fig. 7 for clarity).
Figure 7. Model for VPS4 Recruitment and ESCRT-III Filament Constriction.

Schematic model showing how different ESCRT-III subunits (blue and green) could co-assemble into concentric rings that display C-terminal MIM1 (green) and internal MIM2 elements (blue), thereby creating a high affinity VPS4 binding surface (orange, with the three helices of the MIT domains in red, orange, and yellow, respectively). Recruited VPS4 ATPases could then remove individual ESCRT-III subunits, constricting the rings about cargoes (red) and thereby helping drive vesicle extrusion and neck closure (see text for full details). We note that six molecules of the Vps4 activator, LIP5/Vta1p, also bind the VPS4 beta domains and make additional MIT-ESCRT-III interactions, but for clarity these additional interactions are not shown.
A Possible Mechanism for VPS4 Action
Like VPS4, several other AAA ATPases also initially recognize their substrates by binding small C-terminal epitope “tags” on their target proteins. Prominent examples include the recognition of tubulin tails by the microtubule severing enzyme, spastin (Roll-Mecak and Vale, 2008; White et al., 2007), and the recognition of terminal ssrA (and other) tags by bacterial translocators of the Clp family. Extensive structural and biochemical analyses of the translocators have established that substrates are initially bound by a substrate recognition domain (or separate protein), transferred to a binding site within the central pore of the hexameric AAA ATPase rings, and then “pulled” through the central pore by the ATP-dependent motions of central loops, which change conformation in response to ATP binding and hydrolysis (reviewed in (Licht and Lee, 2008). We previously suggested that Vps4 might remove subunits from the assembled ESCRT-III lattice by functioning in an analogous fashion: first binding the MIM tags on individual ESCRT-III subunits, then transferring them into the central pore, and then using the energy of ATP hydrolysis to pull the subunits out of the lattice. Although this idea is still speculative, it is supported by our observation that mutations in the central Pore 1 motif of VPS4 inhibit HIV budding (Scott et al., 2005a), and by the recent report of a second, conserved internal ESCRT-III sequence element that may represent the VPS4 pore binding site (Shim et al., 2008).
As noted earlier, a recent study showed that overexpressed CHMP4 proteins can form concentric rings that surround the bases of membrane tubules that extrude out from the plasma membrane (Hanson et al., 2008). This observation suggests that native ESCRT-III lattices may also consist of concentric circular filaments that surround sites of vesicle extrusion/virus budding. Combining these models for VPS4 and ESCRT-III function leads to a plausible scenario in which VPS4 and ESCRT-III might work together to produce the constriction necessary for vesicle extrusion and/or membrane fission (Fig. 7). Specifically, we envision that co-polymerization of different classes of ESCRT subunits into concentric circular filaments would expose arrays of MIM1 and MIM2 sites and create high affinity VPS4 binding sites. VPS4 could then act as a disassembly chaperone by removing individual ESCRT-III subunits from the filaments. Subsequent ring reclosure would reduce the diameter of the circular filament(s), creating a constricting force on the encircled cargoes and helping to drive vesicle extrusion and neck closure. Future studies will be necessary to distinguish this attractive model from alternatives in which the ESCRT-III lattice could help create vesicles by other means, for example by functioning as a membrane bending machine or by constricting the neck as a single continuously sliding helical filament.
EXPERIMENTAL PROCEDURES
Expression Constructs and Plasmids
Expression constructs are summarized in Supplemental Table 3 and footnotes.
Protein Expression and Purification
VPS4A MIT Expression and Purification
Recombinant VPS4A MIT (VPS4A residues 1−84) and VPS4A MITL64D were purified as described previously (Scott et al., 2005b; Stuchell-Brereton et al., 2007). GST-VPS4A MITV13D was expressed in BL21 (DE3) codon+ (RIPL) E. coli cells in auto-induction media, ZYP-5052 (Studier, 2005) at 37°C for 5 h, then transferred to 23°C for 16 h. Subsequent steps were at 4°C except where noted. Cells were lysed by sonication and incubation in lysis buffer (50 mM Tris HCl pH 8.0, 150 mM NaCl and 2 mM β-ME) containing lysozyme (1.0 mg/ml), deoxycholate (0.125%), and protease inhibitors (Sigma). Clarified lysate was bound to a GSTPrep column (GE Healthcare), washed with lysis buffer and elulted with lysis buffer supplemented with 20mM reduced glutathione. The GST domain was removed with TEV protease (50μg/ml) during dialysis against 4 liters of lysis buffer (100 mM NaCl, 16 hours at 23°C). The protein was concentrated and purified further by gel filtration on a Superdex 75 column (GE Healthcare). Protein fractions were pooled, concentrated, and passed over a glutathione column to remove residual GST. TEV cleavage left two nonnative residues at the N-terminus of VPS4A MIT (Gly-His) not included in our numbering scheme. The expected mass was confirmed by mass spectrometry.
Synthesis and Purification of CHMP6166−181 Peptides
Unlabeled CHMP6166−181 peptide was prepared by solid phase synthesis and purified by reversed phase HPLC. 13C/15N CHMP6165−181 was expressed as a (HIS)10TrpΔLE peptide fusion in isotopic 13C- and 15N-labeled M9 minimal media, purified from inclusion bodies under denaturing conditions, cleaved with cyanogen bromide to remove the TrpΔLE fusion, and repurified by reversed phase HPLC (Stuchell-Brereton et al., 2007). Expected peptide masses were confirmed by mass spectrometry and final peptide concentrations were determined by amino acid analyses.
Biosensor Binding Assays
Biosensor binding experiments were performed as previously described using BIACORE 2000 and 3000 instruments (Stuchell-Brereton et al., 2007). Briefly, GST-CHMP6, -CHMP4A, -CHMP5, and -CHMP1B proteins and mutants were expressed and captured directly from BL21 (DE3) codon+ (RIPL) E. coli extracts onto anti-GST antibody derivatized CM5 sensor chips. Pure VPS4 MIT proteins were diluted at the designated concentrations in binding buffer, injected in triplicate (50 μL/min, 20°C) and binding data were collected at 2 Hz during the 30 second association and dissociation phases (see Fig. 1A). All interactions reached equilibrium rapidly and dissociated within seconds during the dissociation phase, and binding constants were obtained by fitting the equilibrium responses to 1:1 binding models (e.g., see Fig. 1B).
NMR Sample Preparation
Samples used for NMR structure determination were prepared in a buffer containing 25 mM sodium phosphate pH 5.75, 50 mM NaCl, 1.0 mM EDTA, 40 μM sodium azide, 90% H2O, and 10% D2O. The following NMR samples were prepared: 1) 1.4 mM uniformly 15N-/13C-labeled VPS4A MIT + 1.8 mM unlabeled CHMP6166−181 (NMR structure of bound VPS4A MIT and identification of intermolecular NOEs, KD=5.8 μM, 98.5% occupancy), 2) 1.55 mM unlabeled VPS4A MIT + 1.22 mM uniformly 15N-/13C-labeled CHMP6166−181 (NMR structure of bound CHMP6166−181 and identification of intermolecular NOEs, 98.5% occupancy) 3) 1.0 mM uniformly 15N-labeled VPS4A MIT + 2.0 mM unlabeled CHMP6166−181 (isotropic 1JNH of bound VPS4A MIT, KD=5.8 μM, 99.5% occupancy) and 4) sample 3 + 3% (w/v) C12E5 with C12E5:n-hexanol molar ratio of 1:0.85 (1JNH + 1DNH for bound VPS4A MIT, KD=5.8 μM, 99.5% occupancy)(Rückert and Otting, 2000). All complexes were in fast exchange.
Data Collection and Resonance Assignment
Chemical shift assignments were made using a standard suite of triple resonance NMR experiments (Cavanagh et al., 1996) recorded at 25 °C on Varian Inova 600 or 800 MHz NMR spectrometers equipped with triple-resonance 1H,13C,15N cryogenic (600 MHz) or room temperature (800 MHz) probes. Data were processed with FELIX 2004 (Felix NMR, Inc), and resonances assigned using standard approaches within the SPARKY program (T.D. Goddard and D.G. Kneller, University of California, San Francisco).
Nearly complete resonance assignments were obtained for VPS4A MIT-CHMP6166−181 (98% of the 1H and directly attached 13C/15N resonances) and include 140 stereospecific assignments of prochiral methylene and methyl signals. All prolines were in trans conformations. 140 backbone phi and psi torsion angles were estimated from 13Cα chemical shifts using TALOS (Cornilescu et al., 1999). Unlabeled CHMP6166−181 (sample 1) and unlabeled VPS4A MIT (sample 2) resonance assignments were critical for interpretation of intermolecular NOEs. These assignments were obtained from a combination of 2D F1/F2-13C15N filtered [1H,1H] NOESY (60 ms mix time), 2D F1/F2-13C15N filtered [1H,1H] TOCSY (35, 70 ms mix times), and analysis of 2D [15N,1H] HSQC (Supplemental Figure 1) and 2D [13C, 1H] HSQC during the titration. Double edited NOESY experiments were unnecessary owing to the small size of the system.
Amide residual dipolar couplings, 1DNH, were obtained from 2D [15N,1H] IPAP-HSQC (Ottiger et al., 1998) or analysis of 2D [15N,1H] HSQC and TROSY of isotropic and aligned sample 3 and 4, respectively. Only those amides in regular α-helical regions were included in the final structure calculation and included in Table 1.
Structure Calculations
Intra and intermolecular NOEs were obtained using 3D [1H,13C/15N,1H] NOESY and 3D F1-13C/15N-filtered [1H,13C/15N,1H] NOESY experiments (60 msec mixing times). Intermolecular NOEs were obtained from the F1-13C15N-filtered [1H,13C/15N,1H] NOESY while intramolecular NOEs were obtained from difference of 3D [1H,13C/15N,1H] NOESY and 3D F1-13C15N-filtered [1H,13C/15N,1H] NOESY. NOE coordinates (1H,15N/13C,1H) and intensities were obtained by using the tools in SPARKY, and exported into CYANA (Guntert, 2004) for structure calculations. NOEs were assigned and structures calculated using the automated NOE assignment module implemented in CYANA (Version 2.1). Intermolecular NOEs were manually assigned and incorporated into the complex structure calculation. Final CYANA calculations included α-helical hydrogen bond restraints, derived from the preliminary structures.
The lowest energy CYANA structures were further refined in XPLOR-NIH using a simulated annealing protocol that implements amide residual dipolar coupling constraints. Structure statistics for the VPS4A MIT-CHMP6166−181 complex are summarized in Table 1. Structures were visualized, superimposed, and figures made using PYMOL (DeLano Scientific).
Confocal Fluorescence Microscopy
Transfection and confocal fluorescence imaging of COS7 cells expressing wild type and mutant GFP-VPS4A mammalian expression constructs were performed as described (Stuchell-Brereton et al., 2007).
Ferroportin Downregulation Assays
CHMP6 Depletion and Reconstitution
HEK293T cells were transfected with non-specific or CHMP6 siRNA oligonucleotides at final concentrations of 50−100 nM using OligofectAMINE (Invitrogen) as previously described (De Domenico et al., 2007; Langelier et al., 2006). 24 to 48 h post-transfection, cells were trypsinized and plated onto 60-mm plates without or with coverslips (for Western or fluorescence analyses, respectively). 24 h later, the cells were co-transfected with 5 μg pEGFP-N1-FPN and 5 μg wild-type or mutant siRNA-resistant CHMP6 expression constructs using Nucleofector technology (Amaxa) according to the manufacturer's directions. After an additional 18 h, cells were incubated in the presence of cyclohexamide (75 μg/ml) for 2 hours followed by incubation ± 1 μg/ml hepcidin for 1 h prior to epifluorescence analysis. Fluorescent images were captured on an Olympus BX51 microscope using an Olympus U-CMAD-Z camera and a 60x/1.30 NA oil-immersion objective lens. Images were acquired using Picture Frame 2.5 software (Olympus America)
Western Blot Analyses
Western analyses were conducted in the same way, but with incubation ± hepcidin for 4 hours. Cell lysates were prepared as described previously (De Domenico et al., 2007), and samples were normalized for the total protein concentration using the bicinchoninic acid assay (Pierce) prior to loading. Western detection used either anti-GFP (1:10,000, ab6556; Abcam); mouse anti-tubulin (1:1000; GeneTex); rabbit anti-CHMP6 (1:1000, Covance), or mouse anti-Myc, (1:1000, Abcam) followed by either peroxidase-conjugated goat anti-mouse IgG (1:10,000; Jackson ImmunoResearch Laboratories) or peroxidase-conjugated goat anti-rabbit IgG (1:10,000; Jackson ImmunoResearch Labs). Blots were scanned and quantified using Quanti One software (BioRad).
Quantification of Average Ferroportin-GFP Fluorescence
Average fluorescence per cell was calculated with ImageJ (rsbweb.nih.gov) software. Individual cells were outlined in freehand mode and the background corrected average fluorescence was calculated for >100 cells for each condition analyzed in two separate experiments (one blinded).
siRNA Depletion of VPS4 and HIV-1 Release Assays
HEK293T cells were seeded at 2.5×105 cells per well in a 6-well plate and incubated for 24 h (37°C, 5%CO2). siRNAs specific for VPS4A (sense: 5’ CCGAGAAGCUGAAGGAUUAtt; lower case denotes deoxynucleotides) and VPS4B (sense 5’-CCAAAGAAGCACUGAAAGAtt) were transfected using Lipofectamine RNAi MAX (Invitrogen) at a concentration of 20 nM according to manufacturers instructions. Transfected cells were incubated for 24 hours and then fresh media was added. Cells were co-transfected a second time with the same VPS4 siRNAs (20 nM each), together with the siRNA-resistant VPS4B mammalian expression vectors and with the HIV-1 R9 proviral expression vector (0.5 μg/well each), and incubated for 48 h. Cytoplasmic extracts and virus-containing supernatants were harvested and used for infectivity assays or Western blotting as described previously (Garrus et al., 2001). Briefly, titers were determined in triplicate using single cycle MAGIC assays in HeLa P4 cells. Gag-derived proteins in cytoplasmic and viral lysates were detected by Western blots using rabbit anti-CA and MA primary antibodies at 1:3,000; VPS4 protein levels in cell lysates were detected by Western blotting with rabbit anti-VPS4A and VPS4B polyclonal antibodies at 1:1,000 (von Schwedler et al., 2003); and α-tubulin controls were detected with a mouse anti-α-tubulin antibody (Sigma, DM1A, 1:1000). Bound primary antibodies were detected using donkey anti-rabbit IgG IR Dye 800 (Rockland) and goat anti-mouse IgG Alex 680 conjugates (Invitrogen) at 1:10,000 and visualized on an Odyssey scanner (software version 3.0, LI-COR).
Accession Codes
Protein Data Bank: Coordinates and chemical shift assignments have been deposited with the PDB, accession code: 2k3w (to be released at time of publication).
Supplementary Material
Supplemental Figure 1. NMR Spectra of the VPS4A MIT-CHMP6166−181 Complex
A) 2D [15N, 1H] HSQC spectrum of 15N-labeled VPS4A MIT in 1:1.25 complex with the CHMP6166−181 MIM2 peptide. Black crosspeaks are main chain amides and red crosspeaks are aliased arginine side chain NεHεs.
B) Selected regions from the 2D [15N,1H] HSQC spectrum of 15N-labeled VPS4A MIT titrated with different ratios of unlabeled CHMP6166−181 peptide. Spectra are colored according to the number of molar equivalents CHMP6166−181 added: blue, 0 equivalents; cyan, 0.25 equivalents; orange, 0.5 equivalents; maroon, 0.75 equivalents; magenta, 1.0 equivalents and black, 1.25 equivalents. Equivalent aliased arginine NεHε resonances are shown in green, gray, cyan, coral, beige, and red, respectively. Solid green arrows track VPS4A MIT resonances from free to bound and dashed arrows show connections where one of the two resonances is outside of the expanded region. The largest Δω values are observed for T17-NH and R66-NεH, both of which are situated within the complex interface. The downfield shifted VPS4A MIT R66-NεH signal and its large Δω upon titration with CHMP6161−181 are consistent with retention of the R66-D20 salt bridge in both the free and complex structures.
C. Strip plots from 3D [1H,13C/15N,1H] NOESY (blue) and 3D F1-13C,15N-filtered [1H,13C/15N,1H] NOESY (black) for five different CH strips of residues within the VPS4A MIT–CHMP6161−181 interface (Pascal S.M., Muhandiram D.R., Yamazaki T., Forman-Kay J. D. and Kay L. E. (1994) Simultaneous Acquisition of 15N- and 13C-Edited NOE Spectra of Proteins Dissolved in H2O, J. Magn. Reson. 103B, 197−201, Zwahlen C., Legault, P., Vincent, S.J.F., Greenblatt, J., Konrat, R., and Kay, L.E. (1997) Methods for Measurement of Intermolecular NOEs by Multinuclear NMR Spectroscopy: Application to a Bacteriophage λ N-Peptide/boxB RNA Complex. J. Am. Chem. Soc. 119, 6711−6721). The left two pairs of strips show NOEs detected on VPS4A MIT and the right three pairs on CHMP6. Horizontal lines denote inter-molecular NOEs and their assignments follow: 1, L170-QD1; 2, P171-HB2; 3, P171-QG; 4, P171-HB3; 5, P171-HD2; 6, P171-HD3; 7, L170-QD1; 8, V172-QG2; 9, P171-HB2; 10, P171-QG; 11, V171-HA; 12, I56-QG1/2; 13, L6-QD2; 14, K59-HG2; 15, K59-HB2; 16, K59-HD2; 17, K59-HB3; 18, I56-HG13; 19, K59-QE; 20, I56-HA; 21, K59-HA; 22, S55-HB2; 23, S55-HB3; 24, Y63-QE; 25, F39-QE; 26, R66-QG; 27, R66-HB3; 28, Q62-HB2; 29, R66-HD2; 30, R66-HD3; 31, R66-HA; 32, Y63-HA; 33, Y63-QE; 34, Y32-QE; 35, Y63-QD; 36, R66-HN; 37, L70-QD1; 38, L70-QD2; 39, K69-HB3; 40, K69-HB2; 41, L70-HG; 42, Y73-HB2; 43, Y73-HB3; 44, L70-HA; 45, Y73-QD.
Supplemental Figure 2. Stereoview of Superimposed VPS4A MIT-CHMP6166−181 Structures
Twenty structures are overlaid and shown in the same orientation as in Figure 2C and with the same backbone color scheme as Figure 2B. Interface side chains are shown for VPS4A (black, labeled subset; I10, V13, Y63, and L70) and CHMP6161−181 (cyan, I168, L170, P171, V173, P174, L178, and P179). Side atoms within the R66-D20 and K23-E176 salt bridges are shown in maroon (carbon), red (oxygen), and blue (nitrogen).
Supplemental Table 1. Measured Dissociation Constants for VPS4 MIT Domain-CHMP Protein Interactions
Supplemental Table 2. Alignment of CHMP6 MIM2 Sequences from 14 Eukaryotic Model Organisms
Supplemental Table 3. Expression Vectors
Acknowledgments
We thank the following scientists and University of Utah Core facilities for valuable assistance and advice: David Myszka and Rebecca Rich (Protein Interactions Core), Scott Endicott and Bob Schackmann (DNA/Peptide Core), Chris Rodesh (Fluorescence Imaging Core), and also thank Sanaz Ghaffarian and Melissa Stuchell-Brereton for initial studies on CHMP6-VPS4 interactions. This work was supported by NIH Grants GM082545, AI51174, HL26922, and DK070947.
REFERENCES
- Azmi I, Davies B, Dimaano C, Payne J, Eckert D, Babst M, Katzmann DJ. Recycling of ESCRTs by the AAA-ATPase Vps4 is regulated by a conserved VSL region in Vta1. J Cell Biol. 2006;172:705–717. doi: 10.1083/jcb.200508166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azmi IF, Davies BA, Xiao J, Babst M, Xu Z, Katzmann DJ. ESCRT-III Family Members Stimulate Vps4 ATPase Activity Directly or via Vta1. Dev Cell. 2008;14:50–61. doi: 10.1016/j.devcel.2007.10.021. [DOI] [PubMed] [Google Scholar]
- Babst M, Katzmann D, Estepa-Sabal E, Meerloo T, Emr S. Escrt-III. An endosome-associated heterooligomeric protein complex required for mvb sorting. Dev Cell. 2002;3:271–282. doi: 10.1016/s1534-5807(02)00220-4. [DOI] [PubMed] [Google Scholar]
- Babst M, Wendland B, Estepa EJ, Emr SD. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 1998;17:2982–2993. doi: 10.1093/emboj/17.11.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajorek M, Morita E, Skalicky JJ, Morham SG, Babst M, Sundquist WI. Biochemical Analyses of Human IST1 and Its Function in Cytokinesis. 2008 doi: 10.1091/mbc.E08-05-0475. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniasz PD. Late budding domains and host proteins in enveloped virus release. Virology. 2006;344:55–63. doi: 10.1016/j.virol.2005.09.044. [DOI] [PubMed] [Google Scholar]
- Cavanagh J, Fairbrother WJ, Palmer AG, 3rd, Skelton NJ. Protein NMR Spectroscopy, Principles and Practice. Academic Press; San Diego: 1996. [Google Scholar]
- Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- De Domenico I, Ward DM, Langelier C, Vaughn MB, Nemeth E, Sundquist WI, Ganz T, Musci G, Kaplan J. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol Biol Cell. 2007;18:2569–2578. doi: 10.1091/mbc.E07-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimaano C, Jones CB, Hanono A, Curtiss M, Babst M. Ist1 regulates vps4 localization and assembly. Mol Biol Cell. 2008;19:465–474. doi: 10.1091/mbc.E07-08-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraile-Ramos A, Pelchen-Matthews A, Risco C, Rejas MT, Emery VC, Hassan-Walker AF, Esteban M, Marsh M. The ESCRT machinery is not required for human cytomegalovirus envelopment. Cellular microbiology. 2007;9:2955–2967. doi: 10.1111/j.1462-5822.2007.01024.x. [DOI] [PubMed] [Google Scholar]
- Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- Guntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Roth R, Lin Y, Heuser JE. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J Cell Biol. 2008;180:389–402. doi: 10.1083/jcb.200707031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horii M, Shibata H, Kobayashi R, Katoh K, Yorikawa C, Yasuda J, Maki M. CHMP7, a novel ESCRT-III-related protein, associates with CHMP4b and functions in the endosomal sorting pathway. Biochem J. 2006;400:23–32. doi: 10.1042/BJ20060897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH. ESCRT complexes and the biogenesis of multivesicular bodies. Curr Opin Cell Biol. 2008;20:4–11. doi: 10.1016/j.ceb.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Yang D. MIT domainia. Dev Cell. 2008;14:6–8. doi: 10.1016/j.devcel.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Langelier C, von Schwedler UK, Fisher RD, De Domenico I, White PL, Hill CP, Kaplan J, Ward D, Sundquist WI. Human ESCRT-II complex and its role in human immunodeficiency virus type 1 release. J Virol. 2006;80:9465–9480. doi: 10.1128/JVI.01049-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata S, Roessle M, Solomons J, Jamin M, Gottlinger HG, Svergun DI, Weissenhorn W. Structural basis for auto-inhibition of ESCRT-III CHMP3. J Mol Biol. 2008 doi: 10.1016/j.jmb.2008.03.030. epublication ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht S, Lee I. Resolving Individual Steps in the Operation of ATP-Dependent Proteolytic Molecular Machines: From Conformational Changes to Substrate Translocation and Processivity. Biochemistry. 2008;47:3595–3605. doi: 10.1021/bi800025g. [DOI] [PubMed] [Google Scholar]
- Lin Y, Kimpler LA, Naismith TV, Lauer JM, Hanson PI. Interaction of the mammalian endosomal sorting complex required for transport (ESCRT) III protein hSnf7−1 with itself, membranes, and the AAA+ ATPase SKD1. J Biol Chem. 2005;280:12799–12809. doi: 10.1074/jbc.M413968200. [DOI] [PubMed] [Google Scholar]
- Lottridge JM, Flannery AR, Vincelli JL, Stevens TH. Vta1p and Vps46p regulate the membrane association and ATPase activity of Vps4p at the yeast multivesicular body. Proc Natl Acad USA. 2006;103:6202–6207. doi: 10.1073/pnas.0601712103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J, Yaravoy A, Perez-Caballero D, Bieniasz PD. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad USA. 2003;100:12414–12419. doi: 10.1073/pnas.2133846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough J, Fisher R, Whitby FG, Sundquist WI, Hill CP. Proceedings of the National Academy of Science. USA: 2008. CHMP4-ALIX interactions in the human ESCRT pathway. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, McCullough J, Gygi SP, Morham SG, Sundquist WI. TOM1 Family Members Deliver Membrane Proteins into the MVB Pathway. 2008. in preparation.
- Muziol T, Pineda-Molina E, Ravelli RB, Zamborlini A, Usami Y, Gottlinger H, Weissenhorn W. Structural basis for budding by the ESCRT-III factor CHMP3. Dev Cell. 2006;10:821–830. doi: 10.1016/j.devcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Obita T, Saksena S, Ghazi-Tabatabai S, Gill DJ, Perisic O, Emr SD, Williams RL. Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature. 2007;449:735–739. doi: 10.1038/nature06171. [DOI] [PubMed] [Google Scholar]
- Ottiger M, Delaglio F, Bax A. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J Magn Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- Roll-Mecak A, Vale RD. Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature. 2008;451:363–367. doi: 10.1038/nature06482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Row PE, Liu H, Hayes S, Welchman R, Charalabous P, Hofmann K, Clague MJ, Sanderson CM, Urbe S. The MIT domain of UBPY constitutes a CHMP binding and endosomal localization signal required for efficient epidermal growth factor receptor degradation. J Biol Chem. 2007;282:30929–30937. doi: 10.1074/jbc.M704009200. [DOI] [PubMed] [Google Scholar]
- Rückert M, Otting G. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J Am Chem Soc. 2000;122:7793–7797. [Google Scholar]
- Saksena S, Sun J, Chu T, Emr SD. ESCRTing proteins in the endocytic pathway. Trends Biochem Sci. 2007;32:561–573. doi: 10.1016/j.tibs.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Scott A, Chung HY, Gonciarz-Swiatek M, Hill GC, Whitby FG, Gaspar J, Holton JM, Viswanathan R, Ghaffarian S, Hill CP, et al. Structural and mechanistic studies of VPS4 proteins. EMBO J. 2005a;24:3658–3669. doi: 10.1038/sj.emboj.7600818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott A, Gaspar J, Stuchell-Brereton MD, Alam SL, Skalicky JJ, Sundquist WI. Structure and ESCRT-III protein interactions of the MIT domain of human VPS4A. Proc Natl Acad USA. 2005b;102:13813–13818. doi: 10.1073/pnas.0502165102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim S, Merrill SA, Hanson PI. Novel Interactions of ESCRT-III with LIP5 and VPS4 and their Implications for ESCRT-III Disassembly. Mol Biol Cell. 2008 doi: 10.1091/mbc.E07-12-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack B, Calistri A, Craig S, Popova E, Gottlinger HG. AIP1/ALIX Is a Binding Partner for HIV-1 p6 and EIAV p9 Functioning in Virus Budding. Cell. 2003;114:689–699. doi: 10.1016/s0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- Stuchell-Brereton MD, Skalicky JJ, Kieffer C, Karren MA, Ghaffarian S, Sundquist WI. ESCRT-III recognition by VPS4 ATPases. Nature. 2007;449:740–744. doi: 10.1038/nature06172. [DOI] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Takasu H, Jee JG, Ohno A, Goda N, Fujiwara K, Tochio H, Shirakawa M, Hiroaki H. Structural characterization of the MIT domain from human Vps4b. Biochem Biophys Res Commun. 2005;334:460–465. doi: 10.1016/j.bbrc.2005.06.110. [DOI] [PubMed] [Google Scholar]
- Tsang HT, Connell JW, Brown SE, Thompson A, Reid E, Sanderson CM. A systematic analysis of human CHMP protein interactions: Additional MIT domain-containing proteins bind to multiple components of the human ESCRT III complex. Genomics. 2006;88:333–346. doi: 10.1016/j.ygeno.2006.04.003. [DOI] [PubMed] [Google Scholar]
- von Schwedler UK, Stuchell M, Muller B, Ward DM, Chung HY, Morita E, Wang HE, Davis T, He GP, Cimbora DM, et al. The protein network of HIV budding. Cell. 2003;114:701–713. doi: 10.1016/s0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- White SR, Evans KJ, Lary J, Cole JL, Lauring B. Recognition of C-terminal amino acids in tubulin by pore loops in Spastin is important for microtubule severing. J Cell Biol. 2007;176:995–1005. doi: 10.1083/jcb.200610072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RL, Urbe S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol. 2007;8:355–368. doi: 10.1038/nrm2162. [DOI] [PubMed] [Google Scholar]
- Xiao J, Xia H, Zhou J, Azmi IF, Davies BA, Katzmann DJ, Xu Z. Structural basis of Vta1 function in the multivesicular body sorting pathway. Dev Cell. 2008;14:37–49. doi: 10.1016/j.devcel.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Gonciarz MD, Sundquist WI, Hill CP, Jensen GJ. Cryo-EM structure of dodecameric Vps4p and its 2:1 complex with Vta1p. J Mol Biol. 2008;377:364–377. doi: 10.1016/j.jmb.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. NMR Spectra of the VPS4A MIT-CHMP6166−181 Complex
A) 2D [15N, 1H] HSQC spectrum of 15N-labeled VPS4A MIT in 1:1.25 complex with the CHMP6166−181 MIM2 peptide. Black crosspeaks are main chain amides and red crosspeaks are aliased arginine side chain NεHεs.
B) Selected regions from the 2D [15N,1H] HSQC spectrum of 15N-labeled VPS4A MIT titrated with different ratios of unlabeled CHMP6166−181 peptide. Spectra are colored according to the number of molar equivalents CHMP6166−181 added: blue, 0 equivalents; cyan, 0.25 equivalents; orange, 0.5 equivalents; maroon, 0.75 equivalents; magenta, 1.0 equivalents and black, 1.25 equivalents. Equivalent aliased arginine NεHε resonances are shown in green, gray, cyan, coral, beige, and red, respectively. Solid green arrows track VPS4A MIT resonances from free to bound and dashed arrows show connections where one of the two resonances is outside of the expanded region. The largest Δω values are observed for T17-NH and R66-NεH, both of which are situated within the complex interface. The downfield shifted VPS4A MIT R66-NεH signal and its large Δω upon titration with CHMP6161−181 are consistent with retention of the R66-D20 salt bridge in both the free and complex structures.
C. Strip plots from 3D [1H,13C/15N,1H] NOESY (blue) and 3D F1-13C,15N-filtered [1H,13C/15N,1H] NOESY (black) for five different CH strips of residues within the VPS4A MIT–CHMP6161−181 interface (Pascal S.M., Muhandiram D.R., Yamazaki T., Forman-Kay J. D. and Kay L. E. (1994) Simultaneous Acquisition of 15N- and 13C-Edited NOE Spectra of Proteins Dissolved in H2O, J. Magn. Reson. 103B, 197−201, Zwahlen C., Legault, P., Vincent, S.J.F., Greenblatt, J., Konrat, R., and Kay, L.E. (1997) Methods for Measurement of Intermolecular NOEs by Multinuclear NMR Spectroscopy: Application to a Bacteriophage λ N-Peptide/boxB RNA Complex. J. Am. Chem. Soc. 119, 6711−6721). The left two pairs of strips show NOEs detected on VPS4A MIT and the right three pairs on CHMP6. Horizontal lines denote inter-molecular NOEs and their assignments follow: 1, L170-QD1; 2, P171-HB2; 3, P171-QG; 4, P171-HB3; 5, P171-HD2; 6, P171-HD3; 7, L170-QD1; 8, V172-QG2; 9, P171-HB2; 10, P171-QG; 11, V171-HA; 12, I56-QG1/2; 13, L6-QD2; 14, K59-HG2; 15, K59-HB2; 16, K59-HD2; 17, K59-HB3; 18, I56-HG13; 19, K59-QE; 20, I56-HA; 21, K59-HA; 22, S55-HB2; 23, S55-HB3; 24, Y63-QE; 25, F39-QE; 26, R66-QG; 27, R66-HB3; 28, Q62-HB2; 29, R66-HD2; 30, R66-HD3; 31, R66-HA; 32, Y63-HA; 33, Y63-QE; 34, Y32-QE; 35, Y63-QD; 36, R66-HN; 37, L70-QD1; 38, L70-QD2; 39, K69-HB3; 40, K69-HB2; 41, L70-HG; 42, Y73-HB2; 43, Y73-HB3; 44, L70-HA; 45, Y73-QD.
Supplemental Figure 2. Stereoview of Superimposed VPS4A MIT-CHMP6166−181 Structures
Twenty structures are overlaid and shown in the same orientation as in Figure 2C and with the same backbone color scheme as Figure 2B. Interface side chains are shown for VPS4A (black, labeled subset; I10, V13, Y63, and L70) and CHMP6161−181 (cyan, I168, L170, P171, V173, P174, L178, and P179). Side atoms within the R66-D20 and K23-E176 salt bridges are shown in maroon (carbon), red (oxygen), and blue (nitrogen).
Supplemental Table 1. Measured Dissociation Constants for VPS4 MIT Domain-CHMP Protein Interactions
Supplemental Table 2. Alignment of CHMP6 MIM2 Sequences from 14 Eukaryotic Model Organisms
Supplemental Table 3. Expression Vectors