

Abstract
Förster resonance energy transfer (FRET)-based biosensors for the quantitative analysis of intracellular signaling, including sensors for monitoring cyclic adenosine monophosphate (cAMP), are of increasing interest. The measurement of the donor/acceptor emission ratio in tandem biosensors excited at the donor excitation wavelength is a commonly used technique. A general problem, however, is that this ratio varies not only with the changes in cAMP concentration but also with the changes of the ionic environment or other factors affecting the folding probability of the fluorophores. Here, we use a spectral FRET analysis on the basis of two excitation wavelengths to obtain a reliable measure of the absolute cAMP concentrations with high temporal and spatial resolution by using an “exchange protein directly activated by cAMP”. In this approach, FRET analysis is simplified and does not require additional calibration routines. The change in FRET efficiency (E) of the biosensor caused by [cAMP] changes was determined as ΔE = 15%, whereas E varies between 35% at low and 20% at high [cAMP], allowing quantitative measurement of cAMP concentration in the range from 150 nM to 15 μM. The method described is also suitable for other FRET-based biosensors with a 1:1 donor/acceptor stoichiometry. As a proof of principle, we measured the specially resolved cAMP concentration within living cells and determined the dynamic changes of cAMP levels after stimulation of the Gs-coupled serotonin receptor subtype 7 (5-HT7).
INTRODUCTION
Förster resonance energy transfer (FRET)-based biosensors are frequently used for the analysis of molecular processes within living cells. However, using the fluorescence emission ratio in most cases allows only the analysis of qualitative changes. One important reason for that is the dependence of the emission properties of the donor and/or acceptor on their ionic environment (1).
Cyclic adenosine monophosphate (cAMP) is a ubiquitous second messenger that is regulated by G-protein coupled receptors (GPCRs) targeting the enzyme adenylyl cyclase (AC) to upregulate or downregulate the production of cAMP (2,3). These processes can be monitored online, with the “exchange protein directly activated by cAMP” (Epac) (4). Rich et al. (5) showed that cAMP signals differ spatially and temporally within cells. They also observed a stimulus-correlated rise of cAMP concentration that is limited to the inner side of the membrane. This finding promoted the concept of functional microdomains of cAMP production and action (6), in which metabotropic receptors, G-proteins, membrane-associated ACs, and cAMP targets are aggregated. The recent finding of a family of cytosolic ACs (7), however, suggests that cAMP may also act over long distances.
To analyze the spatiotemporal changes of cellular cAMP levels, several fluorescence biosensors of cAMP have been created using Epac as a backbone (8–10). Epacs have either one (Epac1) or two (Epac2) cAMP-binding domains, a guanine nucleotide exchange motif (GEF) for small GTPases Rap1 and Rap2, and a disheveled, EGL-10, pleckstrin domain, which determine their membrane localization (4). These biosensors consist either of the full-length Epac1 protein or a single domain, which includes only the cAMP-binding part of Epac1 or Epac2.
The function of such fluorescence biosensors for cAMP is based on FRET (11). Binding cAMP to the biosensor proteins leads to a conformational change of the FRET pair and, therefore, to a change in the FRET signal originating from a complementary change of donor and acceptor emission. Thus, the intensity ratio between donor and acceptor emission is typically used for the evaluation of FRET. All available biosensors suitable for quantitative cAMP measurements (8–10), however, are labeled with enhanced cyan fluorescence protein (eCFP) and enhanced yellow fluorescence protein (eYFP) (12). It is necessary to note that the fluorescence of eYFP is highly sensitive to changes in pH and other ion concentrations (e.g., [Cl−]) (1), which may vary in an activity-dependent manner (13–15). Therefore, physiological measurements of cAMP must be affected by such changes.
Here, we demonstrated that the eYFP/eCFP intensity ratio is not valid for quantitative cAMP measurements; it is only useful for determining relative cAMP level changes. As a solution, a simplified spectral FRET analysis method is presented to obtain a relative apparent FRET efficiency and to measure [cAMP] quantitatively. This method is applied to an Epac1 sensor CFP-Epac(δDEP-CD)-YFP (in the following, labeled “EPAC*”) developed by Ponsioen et al. (10). In this construct, the amino terminus of Epac1 is fused to eCFP, whereas the carboxy terminus is fused to eYFP. The catalytical property for Rap1 and the membrane-linking DEP domain are deleted, resulting in a cytosolic localization of the sensor. Binding cAMP to the Epac1 construct induces a conformational change of the protein, resulting in a distance and orientation change of eCFP between eYFP. By using this sensor, Ponsioen et al. have demonstrated that the reduction of intracellular cAMP leads to an increase in energy transfer between eCFP and eYFP, whereas it is diminished by a rise of cAMP.
Using this, to our knowledge, novel FRET analysis method, we applied FRET-based cAMP biosensor to obtain spatially resolved quantitative data on intracellular cAMP concentration by measuring the donor and acceptor fluorescence intensity signal at two excitation wavelengths. This approach was demonstrated for the quantitative detection of changes in [cAMP] in neuroblastoma cells expressing the serotonin receptor subtype 7 (5-HT7), which is known to be positively coupled to ACs.
MATERIALS AND METHODS
Plasmids
Cells were transfected with cDNA (complementary DNA) encoding for the following proteins: 1), eCFP (pECFP-N1, Clontech Laboratories, Mountain View, CA); 2), eYFP (pEYFP-N1, Clontech Laboratories); 3), empty vector (pcDNA3.1/CAT, Invitrogen, Karlsruhe, Germany); 4), the Epac1 construct eCFP-Epac(δDEP-CD)-eYFP in a pcDNA3 from Ponsioen et al. (10) (EPAC*); or 5), a cotransfection of the myc-tagged 5-HT7-receptor cloned into the pcDNA3.1 plasmid (16) together with EPAC*.
Adherent cell culture and transfection
Mouse N1E-115 neuroblastoma cells from the American Type Culture collection (LGC Promochem, Wesel, Germany) were grown in Dulbecco's modified Eagle's medium (Sigma-Aldrich, Munich, Germany) containing 10% fetal calf serum and 1% penicillin/streptomycin at 37°C under 5% CO2. Twenty-four hours before transient transfection, cells were seeded at low density (1 × 106 cells) either in 60 mm dishes (for fluorescence spectroscopy measurements) or in 10 mm dishes including glass coverslips on the bottom (for microscopic measurements). Cells were transfected with appropriate vectors using Lipofectamine2000 reagent (Invitrogen) according to the manufacturer's instruction. Three hours after transfection, cells were serum starved overnight before analysis. Continuative incubation led to a conglomeration of the EPAC* proteins.
Fluorescence spectroscopy
Sample preparation
Exposure of the in vivo used EPAC* proteins to a given solution was achieved by lysis of transfected N1E-115 cells. To remove phenol-red from the N1E-115 cells, cells were washed three times with intracellular solution (140 mM KCl, 5 mM NaCl, 1 mM MgCl2, and 10 mM HEPES at pH 7.2) and thereafter suspended in 2.3 mL intracellular solution and homogenized with an S30 homogenizer (Schütt Labortechnik, Goettingen, Germany) at 2500 rpm for 2 min. This homogenate was centrifuged for 1 min at 21,000 × g and at 4°C. Two milliliters of the supernatant was directly filled into quartz cuvettes equipped with a magnetic stirrer.
Fluorescence spectrometer
Spectroscopic measurements were performed using a Fluorolog-322 (Horiba Jobin Yvon, Munich, Germany) at 25°C. Measurements were performed in 1 nm wavelength steps with 2 nm spectral resolution for excitation and emission. Samples were placed in quartz cuvettes (10 × 10 mm2) and continuously mixed by a magnetic stirrer. To suppress scattering and reabsorption, spectra were measured in a front face arrangement.
For the estimate of spectral contributions due to light scattering and autofluorescence of the cells, reference spectra of transfected cells with an empty vector were recorded and considered an additional background component for the fitting procedure. Special care has been taken for scattering and reabsorption effects in respect to the cell lysate and the fluorophore expression level.
Fluorescence reference spectra of eCFP and eYFP
The cAMP-sensing EPAC* from Ponsioen et al. (10) is a biosensor based on a FRET pair of the fluorophores eCFP and eYFP. From a comprehensive analysis of the emission properties of EPAC*, its fluorescence spectrum was found as a linear combination of the fluorescence spectra of eCFP and eYFP in the complete cAMP concentration range (0 M – 10 mM) and for a pH range (pH 6.5–8). The reference emission spectra of eCFP and eYFP, 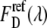 and
and 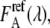 used for the unmixing procedure (see Fig. 2), were obtained at 420 nm excitation.
used for the unmixing procedure (see Fig. 2), were obtained at 420 nm excitation.
FIGURE 2.

The measured EPAC* emission spectrum (420 nm excitation) was fitted by a linear combination of reference spectra for eYFP, eCFP, Raman, and background. The reference spectra were obtained separately. Additionally, the residual of the fitted EPAC* spectrum is shown.
The fluorescence excitation and emission spectra of eCFP and eYFP were obtained separately. Thereby, the line shape of their emission spectrum did not change when the excitation wavelength was varied. Typical normalized spectra are shown in Fig. 1. The fluorescence excitation spectra were taken at 500 nm and 550 nm emission for eCFP and eYFP, respectively.
FIGURE 1.

Excitation (dashed line) and emission (solid line) spectra of eCFP (blue) and eYFP (green) measured in a fluorescence spectrometer. Spectra were obtained by measuring a diluted supernatant of homogenized and centrifuged N1E-115 cells transfected with eCFP and eYFP, respectively. The filter set used in the microscope is represented by the filled areas: excitation bandwidth for eCFP (violet) and for eYFP (green) as well as emission bandwidth for eCFP (light blue) and for eYFP (light yellow).
Choice of excitation wavelengths and emission channels
The optimal excitation wavelength for fluorescence spectrometry and emission channels in microscopic measurements have been determined from the fluorescence excitation and emission spectra of individual fluorescence properties of eCFP and eYFP in the diluted supernatant of homogenized and centrifuged N1E-115 cells that had been transfected with DNA for eCFP and eYFP. In addition to the reference spectra, the transmission spectra of the filter set used for microscopic measurements are shown in Fig. 1. The spectra illustrate the strong overlap of the emission spectrum of eCFP with the excitation spectrum of eYFP, which, on the one hand, is an essential condition for FRET but, on the other hand, complicates data analysis, especially in filter cube experiments. A donor excitation wavelength of 420/10 nm was chosen, which is a compromise between the excitation maximum of eCFP at 430 nm and the maximum of the eCFP/eYFP absorption ratio at ∼400 nm. Due to the small Stokes shift of eYFP of ∼20 nm, 500/10 nm was used as a second excitation wavelength instead of 515 nm, where eYFP excitation reaches its maximum. This allows the use of an emission bandpass filter, which includes the eYFP emission maximum at ∼525 nm and is well separated from excitation. In addition, eCFP is not excited at 500 nm excitation, which is a necessary condition for the derived formalism (see Data Analysis). Finally, the donor emission bandpass filter was chosen to separate from acceptor excitation and to detect as much eCFP signal as possible.
Fluorescence microscopy
Solution application
A coverslip with transfected N1E-115 cells was positioned underneath the microscope objective into a bath chamber equipped with a solution inflow and suction. Cells were kept in a solution of 150 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 10 mM natrium-D-glucose (pH 7.4). 5-HT7 agonist application was realized by exchanging the bath solution with solution containing 1 μM 5-carboxamidotryptamine ((5-CT), Tocris Bioscience, Bristol, UK).
Microscope
For microscopy, we used an upright epifluorescence microscope equipped with a water immersion objective (LUMFI, 40×, NA 1.1, Hamburg, Olympus, Germany). A 100 W xenon lamp attached to a monochromator (Optoscan, Cairn Research, Faversham, UK) was used as an excitation light source that was coupled to the microscope via fiber optics. A dichroic mirror (455 nm) was taken to separate emission from excitation light. According to its properties, the higher intensity of the second excitation wavelength was much less reflected, and, thus, similar intensities were obtained at the two excitation wavelengths, 420/10 nm and 500/10 nm. Using a DualView (Optical Insights, Tucson, AZ), the fluorescence emission signal was split by a dichroic mirror (515 nm) into 470/30 nm for the eCFP channel and 535/30 nm for the eYFP channel. With an iXon camera DV887DCS (Andor Technology, South Windsor, CT), three principal images were acquired: a) the donor image at eCFP excitation and eCFP emission wavelength, b) the FRET image at eCFP excitation and eYFP emission wavelength, and c) the acceptor image at eYFP excitation and eYFP emission wavelength. Camera gain and exposure times (∼3 s) were chosen according to the fluorescence intensity of the cells and were equal for all image series of an experiment. Special care was taken that bleaching was not significant.
Determination of the eYFP extinction ratio (α) and the eCFP “bleedthrough” (β)
The ratio of the eYFP extinction, 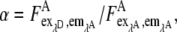 was obtained from the FRET and the eYFP images by measuring cells expressing only eYFP. Special care was taken to use similar experimental conditions as in the EPAC* experiments. For the fluorescence spectrometer, this value was determined to be α = 0.017. Because α depends on the excitation intensities I1 and I2, this value is highly device specific and must be obtained separately for every setup. For the microscope setup used in our experiments, α was measured to be α = 0.27. It is important to note that the images from microscopic measurement were corrected for inhomogeneous illumination.
was obtained from the FRET and the eYFP images by measuring cells expressing only eYFP. Special care was taken to use similar experimental conditions as in the EPAC* experiments. For the fluorescence spectrometer, this value was determined to be α = 0.017. Because α depends on the excitation intensities I1 and I2, this value is highly device specific and must be obtained separately for every setup. For the microscope setup used in our experiments, α was measured to be α = 0.27. It is important to note that the images from microscopic measurement were corrected for inhomogeneous illumination.
Due to the spectral overlap in the eCFP and eYFP emission, the eYFP emission filter did not block the bathochromic part of the eCFP emission (Fig. 1). Therefore, the FRET image was corrected for contribution of the eCFP emission in the eYFP channel. The eCFP bleedthrough, 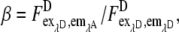 was obtained by measuring cells expressing only eCFP. The ratio of the eCFP emission obtained from the FRET image and the donor image was of about β = 1.20 for the used filter sets of our microscope setup. The eCFP bleedthrough is not an issue (compare Eqs. 8 and 11) in the fluorescence spectrometer calibration experiments, because the unmixing procedure separated eYFP from eCFP emission.
was obtained by measuring cells expressing only eCFP. The ratio of the eCFP emission obtained from the FRET image and the donor image was of about β = 1.20 for the used filter sets of our microscope setup. The eCFP bleedthrough is not an issue (compare Eqs. 8 and 11) in the fluorescence spectrometer calibration experiments, because the unmixing procedure separated eYFP from eCFP emission.
The same excitation wavelengths of 420 nm and 500 nm used in the microscopic measurements were used for the calibration experiments at the fluorescence spectrometer to keep measurements comparable.
Data analysis
The FRET signal
In donor/acceptor FRET experiments, the detected fluorescence signal F(λ) can be interpreted as a superposition of donor and acceptor emission quantities
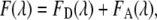 |
(1) |
where 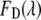 and
and 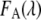 are the fluorescence signal with the characteristic of the donor or acceptor spectra. The contribution of the donor signal in
are the fluorescence signal with the characteristic of the donor or acceptor spectra. The contribution of the donor signal in 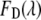 contains two components: I is the signal from free donors (D), and II is the unquenched fluorescence from donors within FRET complexes (DA). The contribution of the acceptor signal
contains two components: I is the signal from free donors (D), and II is the unquenched fluorescence from donors within FRET complexes (DA). The contribution of the acceptor signal 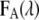 typically contains three parts (17,18) originating from III, the direct excitation of free acceptors (A), IV acceptors within FRET distance, and V acceptors excited via resonant energy transfer, often called sensitized emission.
typically contains three parts (17,18) originating from III, the direct excitation of free acceptors (A), IV acceptors within FRET distance, and V acceptors excited via resonant energy transfer, often called sensitized emission.
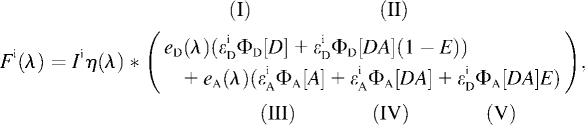 |
(2) |
where 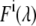 is the fluorescence signal at the excitation wavelength λi; Ii is the corresponding excitation intensity;
is the fluorescence signal at the excitation wavelength λi; Ii is the corresponding excitation intensity; 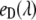 and
and 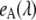 are the characteristic emission spectra of the donor and acceptor normalized to unit area,
are the characteristic emission spectra of the donor and acceptor normalized to unit area, 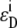 and
and 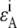 are extinction coefficients of donor and acceptor at the excitation wavelength
are extinction coefficients of donor and acceptor at the excitation wavelength  and
and 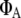 are fluorescence quantum yields of donor and acceptor; E is the characteristic FRET efficiency of the donor acceptor complex; and the function η(λ) is the wavelength-dependent detection efficiency of the instrument used.
are fluorescence quantum yields of donor and acceptor; E is the characteristic FRET efficiency of the donor acceptor complex; and the function η(λ) is the wavelength-dependent detection efficiency of the instrument used.
Fluorescence spectrometer measurements
Using a fluorescence spectrometer, the fractions of donor and acceptor emission signals can be deduced by fitting the linear combination of donor and acceptor reference spectra to the detected emission characteristic
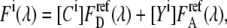 |
(3) |
where 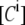 and
and 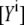 are the apparent concentrations of the donor and acceptor in the signal, and
are the apparent concentrations of the donor and acceptor in the signal, and 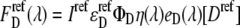 and
and 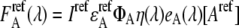 are the reference spectra, which must be obtained in a separate reference measurement of cells containing only donor and acceptor at the concentrations
are the reference spectra, which must be obtained in a separate reference measurement of cells containing only donor and acceptor at the concentrations  and
and 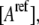 respectively.
respectively.
The apparent concentrations can then be derived from Eqs. 2 and 3 as
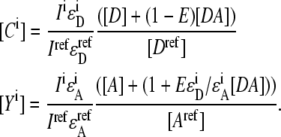 |
(4) |
The fractions of donor and acceptor in FRET complex is defined as
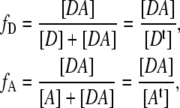 |
(5) |
where 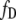 and
and 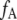 are the fractions and [Dt] and [At] are the total concentration of donor and acceptor, respectively, participating in complexes. Thus Eq. 4 can be rewritten as
are the fractions and [Dt] and [At] are the total concentration of donor and acceptor, respectively, participating in complexes. Thus Eq. 4 can be rewritten as
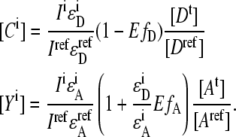 |
(6) |
In the defined situation of a one to one FRET construct like EPAC* with equal total donor and acceptor concentrations 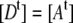
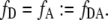 |
(7) |
EPAC* calibration
The cAMP dependence of EPAC* can be calibrated by the sensitized emission FRET signal using a fluorescence spectrometer. To achieve a calibration function that is independent from the EPAC* concentration, the apparent acceptor concentration 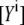 must be obtained at two excitation wavelengths: at donor excitation wavelength λ1, where mostly the donor is excited, and at the acceptor excitation wavelength λ2, where the donor must not be excited (19). Fitting both EPAC* fluorescence signals
must be obtained at two excitation wavelengths: at donor excitation wavelength λ1, where mostly the donor is excited, and at the acceptor excitation wavelength λ2, where the donor must not be excited (19). Fitting both EPAC* fluorescence signals 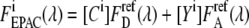 with the reference spectra obtained at one excitation wavelength using Eq. 6, the ratio of
with the reference spectra obtained at one excitation wavelength using Eq. 6, the ratio of 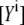 can be rewritten as
can be rewritten as
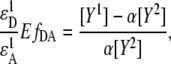 |
(8) |
where 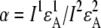 is the relative acceptor emission intensity for the two used excitations, which must be obtained in a separate experiment using an “acceptor only” sample. The fractions of acceptor emission
is the relative acceptor emission intensity for the two used excitations, which must be obtained in a separate experiment using an “acceptor only” sample. The fractions of acceptor emission 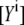 were obtained by unmixing the EPAC* signal (Fig. 2) with the reference emission spectra of eCFP
were obtained by unmixing the EPAC* signal (Fig. 2) with the reference emission spectra of eCFP 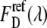 and eYFP
and eYFP 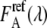 for both excitation wavelengths λi, according to
for both excitation wavelengths λi, according to 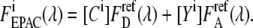 Note that to obtain
Note that to obtain 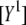 and
and 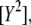 the spectra
the spectra  and
and  are fitted by the same reference spectra
are fitted by the same reference spectra 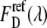 and
and 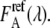
This determination of the apparent FRET efficiency, which was used to calibrate the cAMP-dependent EPAC* FRET signal, is similar to the one presented in Lakowicz (20), Hoppe et al. (19), and van Rheenen et al. (21). However, in contrast to the general FRET analysis, we do not need to determine the relative donor/acceptor extinction, which is labeled as 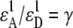 in Hoppe et al. (19) and is difficult to obtain (22,23). A universally valid derivation of the intensity-based FRET analysis can be found in Wlodarczyk et al. (18) from which Eq. 8 can be derived with the constraint that the donor must not be excited at the acceptor excitation wavelength λ2.
in Hoppe et al. (19) and is difficult to obtain (22,23). A universally valid derivation of the intensity-based FRET analysis can be found in Wlodarczyk et al. (18) from which Eq. 8 can be derived with the constraint that the donor must not be excited at the acceptor excitation wavelength λ2.
The equilibrium of the cAMP-binding reaction to the binding sites of the enzyme EPAC* can be expressed by a Hill equation. 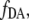 which is the fraction of EPAC* in bound state (Eqs. 5 and 7), describes the equilibrium of the cAMP-binding reaction. The cAMP dependency of
which is the fraction of EPAC* in bound state (Eqs. 5 and 7), describes the equilibrium of the cAMP-binding reaction. The cAMP dependency of 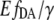 as a function of the Hill equation is
as a function of the Hill equation is
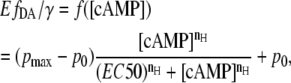 |
(9) |
where 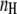 is the Hill coefficient indicating the amount of cAMP-binding places, p0 and
is the Hill coefficient indicating the amount of cAMP-binding places, p0 and 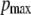 are offset and maximum amplitude parameters, respectively, and EC50 is the cAMP concentration when 50% of the cAMP-binding sites are occupied. The cAMP concentration can then be obtained by the reverse function
are offset and maximum amplitude parameters, respectively, and EC50 is the cAMP concentration when 50% of the cAMP-binding sites are occupied. The cAMP concentration can then be obtained by the reverse function
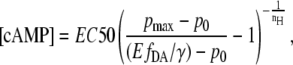 |
(10) |
where 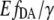 is obtained from the sensitized emission FRET experiment using Eq. 8.
is obtained from the sensitized emission FRET experiment using Eq. 8.
Microscope measurements
In the microscopic analysis, the spectral information is obtained with bandpass filters. The experimental microscopic images were corrected for the background and for the inhomogeneous illumination according to the specific excitation wavelengths. The background image was acquired under identical settings as in the FRET experiments without applying excitation light. The inhomogeneous illumination was recorded by fluorescent slides (Chroma Technology, Rockingham, VT). The correction for inhomogeneous illumination was essential, because the intensity at the peripheral regions of the images dropped to ∼80%. The intensity drop showed an individual characteristic for the used excitation wavelengths and the specific emission channels. Slight pixel shifts between the donor and acceptor emission channel caused by imperfect alignment of the DualView were corrected with the help of a reference grid structure.
The three corrected images  of donor excitation and donor emission channel (donor image),
of donor excitation and donor emission channel (donor image),  donor excitation and acceptor emission channel (FRET image),
donor excitation and acceptor emission channel (FRET image), 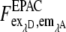 acceptor excitation and acceptor emission channel (acceptor image) were then used for the pixel-based relative apparent FRET efficiency calculation. In contrast to the fluorescence spectrometer measurement, the FRET signal must also be corrected for the emission signal of the donor in the acceptor emission channel. In the literature it is often denoted as “bleedthrough” or “cross talk” (24–26).
acceptor excitation and acceptor emission channel (acceptor image) were then used for the pixel-based relative apparent FRET efficiency calculation. In contrast to the fluorescence spectrometer measurement, the FRET signal must also be corrected for the emission signal of the donor in the acceptor emission channel. In the literature it is often denoted as “bleedthrough” or “cross talk” (24–26).
Thus Eq. 8 must be extended as
 |
(11) |
where the relative acceptor fluorescence signal 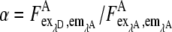 and the donor bleedthrough fraction
and the donor bleedthrough fraction 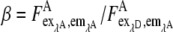 were obtained in acceptor and donor only measurements, respectively. Using the calibration measurement obtained by the fluorescence spectrometer, similar excitation conditions must be chosen due to the γ value. The pixel-based cAMP concentration was calculated by the inverse Hill equation (Eq. 10).
were obtained in acceptor and donor only measurements, respectively. Using the calibration measurement obtained by the fluorescence spectrometer, similar excitation conditions must be chosen due to the γ value. The pixel-based cAMP concentration was calculated by the inverse Hill equation (Eq. 10).
The calculated [cAMP] map was displayed in a two-dimensional RGB (red, green, blue color model) space with color coding of the concentration, whereas the accuracy of the [cAMP], calculated from the error of the three corrected images (see Appendix), was used for the brightness in the [cAMP] map. Thus thresholding the corrected images was not required.
All calculations were performed with MATLAB (MathWorks, Natick, MA) and the DIPimage Toolbox (image processing toolbox for Matlab, Delft University of Technology, The Netherlands). An error analysis is derived in the appendix.
RESULTS
cAMP dependence of EPAC*
The calibration of the EPAC* sensor was performed in lysates of EPAC*-transfected N1E-115 neuroblastoma cells under the same experimental conditions as the measurements for eCFP and eYFP references, which were used as references. Expectedly, there was no change in the reference spectra during changes of the cAMP concentration. The eCFP and eYFP reference spectra (Fig. 1) were used to unmix the [cAMP]-dependent spectra of EPAC* into linear fractions 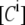 and
and 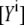 according to Eq. 3 (Fig. 2). Additionally, the cell background and the Raman peak of the excitation were taken into account during the linear unmixing procedure.
according to Eq. 3 (Fig. 2). Additionally, the cell background and the Raman peak of the excitation were taken into account during the linear unmixing procedure.
To calibrate the EPAC* using its FRET signal as a function of cAMP concentration, we exposed cell lysates from EPAC*-transfected cells to [cAMP] ranging from 0 M to 10 mM. As illustrated in Fig. 3 A, the background-corrected spectra of EPAC* revealed the characteristic FRET behavior. Moreover, the emission range of eCFP showed a higher intensity upon increase in [cAMP], whereas the intensity of the emission range of eYFP decreased. The contributions of the reference spectra are shown in Fig. 3 B. Thus, the emission of eCFP increased and the emission of eYFP decreased during a rise of [cAMP] as described by Ponsioen et al. (10). The reduced donor quenching induced an increased eCFP emission signal, whereas a reduced sensitized emission signal was responsible for the decrease in the eYFP emission signal. The [cAMP]-dependent changes in the spectra are illustrated by arrows in Fig. 3 B.
FIGURE 3.

(A) [cAMP]-dependent EPAC* emission spectrum. Spectra were obtained from a diluted supernatant of homogenized and centrifuged N1E-115 cells transfected with EPAC*. cAMP was directly applied into the supernatant solution. (B) EPAC* emission spectra were unmixed into linear fractions of eCFP and eYFP reference spectra. Curve shapes of these reference spectra were obtained by separate measurements (Fig. 1). The gray arrows indicate the intensity change with increasing [cAMP]. All presented spectra are corrected for background and autofluorescence.
At the excitation wavelength λ1 (420 nm) the eYFP is barely excited, resulting in a strong contribution (up to 40%) of the change in the eYFP to the EPAC* signal, whereas the change in the eCFP contribution was relatively low (∼15 %). Assuming that all EPAC* molecules are in the FRET state at low [cAMP] and in the non-FRET state at high [cAMP], the change in characteristic FRET efficiency E was calculated to be ΔE = 0.15 ± 0.01 (Eq. 2).
Calibration of EPAC*
Using the data from the unmixing calculations (Fig. 3 B), the ratios between the donor 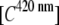 and acceptor concentrations
and acceptor concentrations 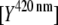 in the EPAC* emission signal (Eq. 3) were analyzed by varying [cAMP]. The eYFP/eCFP ratio is often taken as a relative value for the apparent FRET efficiency (e.g., Ponsioen et al. (10)). In Fig. 4 A this ratio is plotted versus [cAMP] for two characteristic calibration measurements. The curves were fitted by the Hill equation (Eq. 9). Even though the curve shapes were similar within different experiments, indicating
in the EPAC* emission signal (Eq. 3) were analyzed by varying [cAMP]. The eYFP/eCFP ratio is often taken as a relative value for the apparent FRET efficiency (e.g., Ponsioen et al. (10)). In Fig. 4 A this ratio is plotted versus [cAMP] for two characteristic calibration measurements. The curves were fitted by the Hill equation (Eq. 9). Even though the curve shapes were similar within different experiments, indicating 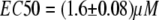 and
and 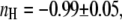 there was a clear offset between the curves.
there was a clear offset between the curves.
FIGURE 4.

[cAMP]-calibration curves for EPAC*. (A) eYFP/eCFP ratio (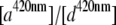 from Eq. 3) for [cAMP] calibration of EPAC* with (o) and without (x) autofluorescence correction. Data were obtained from two representive experiments (red and blue) with different cell batches that were similarly treated. The data are fitted using the Hill equation (Eq. 9). Resulting parameters are shown in the graph. (B) The relative apparent FRET efficiency
from Eq. 3) for [cAMP] calibration of EPAC* with (o) and without (x) autofluorescence correction. Data were obtained from two representive experiments (red and blue) with different cell batches that were similarly treated. The data are fitted using the Hill equation (Eq. 9). Resulting parameters are shown in the graph. (B) The relative apparent FRET efficiency 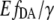 (Eq. 8) of the same data as in A is used for [cAMP] calibration. (C) pH dependence of the eYFP/eCFP ratio of the EPAC* emission signal is fitted with the Hill equation. (D) The pH dependency of
(Eq. 8) of the same data as in A is used for [cAMP] calibration. (C) pH dependence of the eYFP/eCFP ratio of the EPAC* emission signal is fitted with the Hill equation. (D) The pH dependency of 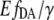 shows a constant behavior.
shows a constant behavior.
The offsets were attributed to different cell batches, indicating that, most likely during cell cultivation, an irreversible intensity change mainly of eYFP appeared; whereas its characteristic emission spectrum remained unchanged. Such intensity changes made it impossible to use the eYFP/eCFP ratio for a direct cAMP calibration of EPAC*. To clarify whether it is possible to refer to the 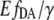 instead of the eYFP/eCFP ratio as a parameter for the calculation of cAMP concentration, we proved the independence of the
instead of the eYFP/eCFP ratio as a parameter for the calculation of cAMP concentration, we proved the independence of the 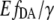 value from non-FRET related intensity changes in eYFP emission. For that, fluorescence spectra of EPAC* were taken at different pH conditions. As mentioned before, the pH value is known to interfere with the fluorescence properties of eYFP (1). Indeed, we observed pH-dependent changes in the intensity of the unmixed eYFP fluorescence signal (which, however, did not influence the characteristic emission spectrum
value from non-FRET related intensity changes in eYFP emission. For that, fluorescence spectra of EPAC* were taken at different pH conditions. As mentioned before, the pH value is known to interfere with the fluorescence properties of eYFP (1). Indeed, we observed pH-dependent changes in the intensity of the unmixed eYFP fluorescence signal (which, however, did not influence the characteristic emission spectrum 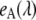 ). The fluorescence signal obtained for eCFP remained unchanged at a pH varying between 6.5 and 8.
). The fluorescence signal obtained for eCFP remained unchanged at a pH varying between 6.5 and 8.
The variations in eYFP intensity obtained in the above experiments resulted in a strong pH dependence of the eYFP/eCFP intensity ratio in the case of EPAC* (Fig. 4 C). A pH dependence, however, was not found for the 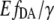 value (Fig. 4 D), which confirms that this value is independent from the non-FRET related variations in eYFP intensity. In Fig. 4 B,
value (Fig. 4 D), which confirms that this value is independent from the non-FRET related variations in eYFP intensity. In Fig. 4 B, 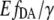 values are plotted as a function of the [cAMP]. By fitting this correlation with the Hill equation, there were no significant offset shifts between the
values are plotted as a function of the [cAMP]. By fitting this correlation with the Hill equation, there were no significant offset shifts between the 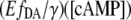 curves, indicating
curves, indicating 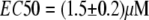 and
and 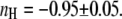 The comparison of Fig. 4, A and B, also shows that an
The comparison of Fig. 4, A and B, also shows that an 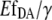 calibration curve can be applied to different experiments performed under similar conditions, whereas the calibration curves based on eYFP/eCFP intensity ratios were strongly dependent on the particular experiment. In addition, the Hill coefficient
calibration curve can be applied to different experiments performed under similar conditions, whereas the calibration curves based on eYFP/eCFP intensity ratios were strongly dependent on the particular experiment. In addition, the Hill coefficient 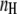 of about −1 (Fig. 4 B) corresponds with the assumption of a single cAMP-binding site described for Epac1 (4) and consequently for EPAC*.
of about −1 (Fig. 4 B) corresponds with the assumption of a single cAMP-binding site described for Epac1 (4) and consequently for EPAC*.
From the amplitudes of the fitted Hill equation, which was 6.5 ± 0.1 at low and 3.75 ± 0.1 at high [cAMP], the characteristic FRET efficiency was estimated to be 35% ± 2% and 20% ± 2%, respectively.
Spatially and time-resolved quantitative [cAMP] measurements
To analyze the spatial and temporal changes of [cAMP] in living cells, microscopic measurements were performed on N1E-115 neuroblastoma cells cotransfected with plasmids encoding for EPAC* and for 5-HT7 receptors. The 5-HT7 receptor is coupled to the stimulatory Gs-protein, and receptor stimulation with agonist results in the increase of the intracellular cAMP concentration (27,28). Receptor-mediated changes of the cAMP level were induced by bath application of the 5-HT7 receptor agonist, 5-CT (1 μM), which has a high affinity for 5-HT7 receptors (29). The bath was exchanged within 30 s, raising the local [5-CT] up to 90 % of its maximum within 10 s. The fluorescence was monitored over three periods of 117 s, corresponding to before, during, and after 5-CT application.
5-CT induced changes of [cAMP] were calculated quantitatively based on the changes in the apparent FRET efficiency (Fig. 5). The statistical analysis of 11 cotransfected cells showed that the receptor-mediated increase of [cAMP] ranged from a basal level of 0.32 ± 0.62 μM to reach a maximal concentration of 1.62 ± 1.83 μM upon 5-CT application. The large standard errors of means reflect major regional differences. This was also verified by examining specific subcellular regions of interest (Fig. 5, A–F; see also Supplementary Material, Movie S1 and Movie S2 for the whole time range). The basal [cAMP] was dispersed inhomogeneously, indicating the existence of microdomains with constitutive [cAMP] production. The lowest basal concentration was 0.1 ± 0.1 μM, which is at the lower resolution limit of EPAC*. Within the patchy microdomains, however, the basal [cAMP] ranged ∼0.4 ± 0.3 μM. Such preexisting microdomains with elevated basal [cAMP] showed a more pronounced response to the 5-CT application (“active” microdomains) (Fig. 5, B and E), and there the [cAMP] quickly increased up to 9 μM, whereas the increase of [cAMP] in surrounding “passive” regions remained below 2 μM.
FIGURE 5.

cAMP increase as a response to agonist activation of 5-HT7 receptor. (A, B, D, and E) [cAMP] map of two N1E-115 cells, which had been transfected with EPAC* and 5-HT7 receptor at basal level (A and D) and during stimulation (B and E) to 1 μM 5-CT. Quantitative [cAMP] are color coded in a range from 0 to 4.5 μM and 0 to 10 μM, respectively. The average [cAMP] time courses from selected microdomains (circles in concentration map) are shown in C and F. The region colors correspond to the line colors in C and F. The dotted line in white encircles the region of the nucleus.
The [cAMP] changes started at the plasma membrane, where it reached its highest levels and propagated in the form of a cAMP wave with a speed of ∼0.2 μm/s into the cytosol (Movie S2). It is noteworthy that [cAMP] did not increase synchronously within the various microdomains, and rather differently changed within individual microdomains that subsequently spilled over into neighboring microdomains (Fig. 5, D–F). These fluctuations in the spatial distribution of [cAMP] resulted in the highly dynamic movie. Although signals are seen in the area of the nucleus, the cell nucleus itself did not reveal any cAMP signals since EPAC* is expressed only in the cytosol. It is also notable that the agonist-mediated increase of the cAMP concentration was 5-HT7 receptor specific, because the agonist-mediated increase of cAMP concentration was completely blocked by parallel application of the receptor-specific antagonist SB269970 (data not shown).
DISCUSSION
The FRET technology becomes increasingly important for studying protein-protein interactions in various biological samples. For a quantitative analysis of intensity-based FRET measurements, however, a set of additional calibration measurements is required that includes fluorescence lifetime measurements performed under defined conditions. To overcome such extensive calibration steps, FRET-based biosensors with a fixed donor/acceptor stoichiometry of 1:1 have been designed that utilize the acceptor/donor fluorescence intensity ratio as a measure of the FRET signal. However, the fluorescence intensity ratio is often insufficient for calibrating and quantitative analysis because it depends on secondary, environmental factors, including pH and anion concentration (1).
In this study, we describe, to our knowledge, a novel method for the quantitative FRET analysis, which can be applied to the biosensor with a fixed donor/acceptor stoichiometry. The FRET-based cAMP biosensor EPAC* (10) was used as a model, The general spectroscopic properties of EPAC* as well as calibration experiments were achieved by fluorescence spectrometry allowing 1), a precise analysis of the spectral changes in the signal; 2), a higher sensitivity and accuracy due to a larger signal; and 3), averaging fluorescence signals from numerous cells simultaneously. We found that the spectrum of EPAC* can be fitted by superposition of the reference spectra of eCFP and eYFP (Fig. 3 B). EPAC* activation by cAMP increased the fluorescence intensity of eCFP, whereas the fluorescence intensity of eYFP decreased, as was expected according to a decrease in the FRET signal. The profile of the fluorescence spectra of eCFP and eYFP remained unchanged even when cAMP concentrations varied. It is notable that the calibration curves obtained by using the eYFP/eCFP intensity ratio plotted against cAMP concentration varied between different cell batches (Fig. 2 A). However, only variations in the ratio (po and pmax − vertical shift)—but neither in the EC50 values of 1.6 ± 0.1 μM nor in the Hill coefficient of nH = −1 ± 0.5—were observed. Variations in the cAMP concentration before lysing cells cannot explain such behavior.
It has been reported that the fluorescence intensity of the eYFP depends on the pH and other anion concentrations (1), whereas the profile of its fluorescence spectra remains unchanged. To evaluate the role of pH for the obtained variations in the eYFP/eCFP ratio, we measured emission spectra of eCFP and eYFP upon different pH conditions. The intensity of eCFP remained stable (±4%) as the pH ranged from 6.5 to 8, whereas the eYFP intensity was dramatically quenched (compare Fig. 2 C). Therefore differences in the eYFP/eCFP intensity ratio obtained for EPAC* seem to originate from pH-dependent changes in the intensity of eYFP rather than of eCFP. Consequently, the eYFP/eCFP ratio cannot be used as a direct quantitative measure of the cAMP concentration. To overcome this limitation, we developed a simplified FRET-based analysis method. This method is based on two-wavelength excitation in which the sensitized emission signal is scaled by the eYFP intensity, resulting in the 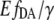 value (Eq. 9). The exposure of the cell lysates containing EPAC* to different pH values revealed that the value
value (Eq. 9). The exposure of the cell lysates containing EPAC* to different pH values revealed that the value 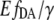 does not significantly change at different pH values (Fig. 4 D), whereas the eYFP/eCFP ratio showed a significant change with pH (Fig. 4 C). The finding demonstrates that the
does not significantly change at different pH values (Fig. 4 D), whereas the eYFP/eCFP ratio showed a significant change with pH (Fig. 4 C). The finding demonstrates that the 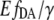 value is independent from non-FRET related variations of eYFP signals, whereas the eYFP reference spectrum remains constant. The calibration of EPAC* by using the
value is independent from non-FRET related variations of eYFP signals, whereas the eYFP reference spectrum remains constant. The calibration of EPAC* by using the 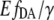 value instead of the eYFP/eCFP ratio resulted in similar curves of different cell batches. The extreme pmax and po varied within an error of <±1%, whereas the ratio exhibited a variation of >±7%.
value instead of the eYFP/eCFP ratio resulted in similar curves of different cell batches. The extreme pmax and po varied within an error of <±1%, whereas the ratio exhibited a variation of >±7%.
Fitting the 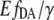 data by the Hill equation provided additional important information about EPAC* properties: the Hill coefficient of nH = −1.05 ± 0.05 is in line with the assumption of a single binding cAMP site suggested for Epac1 and Epac2, the EC50 value for EPAC* was 1.5 ± 0.2 μM, which is in disagreement with that obtained by Ponsioen et al. (10), who proposed an EC50 value of 14 μM. Two things may lead to such a discrepancy. First, we observed significant degradation of cAMP in a frozen stock solution after a few weeks by a shift of the EC50 value. Thus we use only a solution of freshly dissolved cAMP for our experiments. Second, we could prove in our study here that the ratiometric analysis could be afflicted with an error of non-FRET related intensity changes of the acceptor.
data by the Hill equation provided additional important information about EPAC* properties: the Hill coefficient of nH = −1.05 ± 0.05 is in line with the assumption of a single binding cAMP site suggested for Epac1 and Epac2, the EC50 value for EPAC* was 1.5 ± 0.2 μM, which is in disagreement with that obtained by Ponsioen et al. (10), who proposed an EC50 value of 14 μM. Two things may lead to such a discrepancy. First, we observed significant degradation of cAMP in a frozen stock solution after a few weeks by a shift of the EC50 value. Thus we use only a solution of freshly dissolved cAMP for our experiments. Second, we could prove in our study here that the ratiometric analysis could be afflicted with an error of non-FRET related intensity changes of the acceptor.
Besides the EC50 value, the characteristic FRET efficiency E must also be taken into account, because it affects the signal/noise ratio. From the donor quenching (ΔE = 15% ± 1%) and the cAMP concentration dependence of 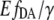 value, a characteristic FRET efficiency E of EPAC* was estimated to be 35% ± 2% at low [cAMP], as E was reduced to 20% ± 2% at high [cAMP]. In the “FRET-positive” conformation state of EPAC*, E was found to be only slightly lower than the E of 37% obtained for eCFP-eYFP tandem constructs (18). In the so-called “FRET-negative” state, the apparent FRET efficiency was reduced to ∼57% of its maximal value. Thus, the presumed conformational changes between the EPAC molecule at cAMP bound and nonbound states are not very pronounced in the case of EPAC*. It is more likely that only slight changes in distance and/or orientation of the EPAC-bound fluorophores are responsible for the moderate differences in the apparent FRET efficiency. The accuracy of the biosensor could be optimized by exploiting the complete dynamic range of the eCFP-eYFP FRET pair. For further studies, this method can also be employed to obtain the characteristic FRET efficiencies of other cAMP biosensors working on the base of Epac1 and Epac2.
value, a characteristic FRET efficiency E of EPAC* was estimated to be 35% ± 2% at low [cAMP], as E was reduced to 20% ± 2% at high [cAMP]. In the “FRET-positive” conformation state of EPAC*, E was found to be only slightly lower than the E of 37% obtained for eCFP-eYFP tandem constructs (18). In the so-called “FRET-negative” state, the apparent FRET efficiency was reduced to ∼57% of its maximal value. Thus, the presumed conformational changes between the EPAC molecule at cAMP bound and nonbound states are not very pronounced in the case of EPAC*. It is more likely that only slight changes in distance and/or orientation of the EPAC-bound fluorophores are responsible for the moderate differences in the apparent FRET efficiency. The accuracy of the biosensor could be optimized by exploiting the complete dynamic range of the eCFP-eYFP FRET pair. For further studies, this method can also be employed to obtain the characteristic FRET efficiencies of other cAMP biosensors working on the base of Epac1 and Epac2.
After the FRET-based biosensor was characterized and a calibration curve had been acquired, the optimized FRET-based analysis method was applied to the microscopic studies at the single-cell level. Only two intrinsic constants, α and β, of the microscope needed to be determined. However, it was recognized that there are additional factors that may create significant artifacts. Slight variations in the emission intensity profile were found for different excitation wavelengths. Thus, besides the standard background correction, the intensity shading (up to 20%) also has to be corrected to achieve the required accuracy. Also, the pixel-based analysis required an optimized beam pathway. Remaining slight pixel shifts in the image were corrected during image processing. Shifts and rotation of the images were more pronounced when an image splitter was used. To diminish systematic errors while applying the calibration curve obtained by the fluorescence spectrometer to the data from microscopy, we suggest obtaining at least one characteristic point of the calibration curve (i.e., p0 for low [cAMP] or pmax for high [cAMP]) at the microscope setup. In contrast, the EC50 and the nH values can be taken over from the calibration measurements and must not be redetermined in a microscope measurement since they depend only on the binding affinity of cAMP to EPAC*.
To prove the 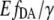 -based FRET method, we performed microscopic measurements of [cAMP] in neuroblastoma cells coexpressing EPAC* and 5-HT7 receptor. The [cAMP] increase was pronounced in all measurements but never surpassed the upper concentration limit of the biosensor, which is ∼15 μM. The agonist-mediated [cAMP] increase was localized in several subcellular microdomains. The spatial and temporal resolved analyses of individual cells revealed [cAMP] gradients of 0.5 μM/μm. At basal conditions, microdomains with three times higher [cAMP] coexist and are surrounded by areas that remained inactive at low [cAMP] (<0.3 μM). Stimulation of the 5-HT7 receptor with an agonist-induced [cAMP] increased in a range of several μM within specific microdomains. This finding corresponds to the two-compartment model proposed by Rich et al. (30) assuming a diffusion restriction between microdomains and the cytosol. The microdomains in neuroblastoma cells had dimensions that are similar to the cAMP microdomains analyzed in neonatal cardiac myocytes (31).
-based FRET method, we performed microscopic measurements of [cAMP] in neuroblastoma cells coexpressing EPAC* and 5-HT7 receptor. The [cAMP] increase was pronounced in all measurements but never surpassed the upper concentration limit of the biosensor, which is ∼15 μM. The agonist-mediated [cAMP] increase was localized in several subcellular microdomains. The spatial and temporal resolved analyses of individual cells revealed [cAMP] gradients of 0.5 μM/μm. At basal conditions, microdomains with three times higher [cAMP] coexist and are surrounded by areas that remained inactive at low [cAMP] (<0.3 μM). Stimulation of the 5-HT7 receptor with an agonist-induced [cAMP] increased in a range of several μM within specific microdomains. This finding corresponds to the two-compartment model proposed by Rich et al. (30) assuming a diffusion restriction between microdomains and the cytosol. The microdomains in neuroblastoma cells had dimensions that are similar to the cAMP microdomains analyzed in neonatal cardiac myocytes (31).
The high basal [cAMP] levels within distinct cell microdomains seem to be caused by a constitutive activity of 5-HT7 receptors (32), which are known to be clustered within membrane microdomains (E. Ponimaskin, D. W. Richter, unpublished results). Moreover, an agonist-induced increase in [cAMP] was observed mainly in these microdomains that were endogenously active and produced spatial gradients of up to 3 μM/μm. This suggests a higher density of 5-HT7 receptors and/or its effectors (including Gs-protein and AC) within microdomains than within surrounding regions with low activity. An additional reason for the existence of cAMP microdomains may be a compartmentalization of ACs or phosphodiesterase (6,33). During 5-HT7 receptor activation, the [cAMP] increase started from the plasma membrane as a wave of the cAMP signal that propagates with a speed of ∼0.2 μm/s. This speed is lower than the free diffusion of cAMP (300 μm2/s) in cytoplasm (34,35). Although we obtained a diffusion constant of 6–7 μm2/s for cAMP-free EPAC*, it cannot be excluded that the observed propagation of the cAMP signal is masked by an EPAC* diffusion. Our data are in line with the findings of Rich et al. (30), who showed that the microdomains need ∼150 s to be filled up by a process that is not related to unrestricted diffusion (2 ms). We conclude that cAMP waves start at clustered receptors and continue with directed cAMP fluxes along connected microdomains toward regions surrounding the nucleus. The functional significance of such directed cAMP traffic needs further clarification.
So far, we have assumed that the buffer capacity of EPAC* is negligible, because this was not the focus of our study. However the buffer capacity needs to be considered for further single-cell analysis of quantitative [cAMP], which is a principal aspect of all biosensors.
In conclusion, the presented FRET-based approach can be applied to different FRET-based biosensors for a quantitative analysis of [cAMP], the characteristic FRET efficiency, the dynamic range, and its specificity. This approach is independent from the ionic environment and can be applied to microscopy at a subcellular level with a high spatial and temporal resolution. The change in the apparent FRET efficiency of EPAC* caused by the conformational change is often far from the theoretical optimum, which diminishes the signal/noise ratio of the response and thus its sensitivity. Such a technique for quantitative analysis of FRET-based biosensors is also a requirement for the high throughput screening. The prospect of (to our knowledge) the novel FRET-based approach for quantitative measurements of [cAMP] is to analyze signaling processes quantitatively, including the influence of basal concentrations and the option to verify the significance of microdomains and their dynamics.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft through the Research Center Molecular Physiology of the Brain (FZT 103 and EXC 171).
We thank Prof. Dr. Erwin Neher (MPI for Biophysical Chemistry, Goettingen), Prof. Dr. Fred. S. Wouters, and Andrew Woehler (both in Center for Physiology and Pathophysiology, Goettingen) for helpful comments and for critically reading the manuscript. We thank Dr. Kees Jalink from the Department of Cellular Biophysics, The Netherlands Cancer Institute, who kindly provided us with cDNA encoding for the eCFP-Epac(δDEP-CD)-eYFP fusion construct.
APPENDIX
Error analysis
The error of 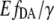 from Eq. 11 follows according to the exact differential method:
from Eq. 11 follows according to the exact differential method:
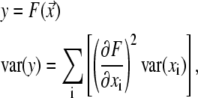 |
where var(y) is the variance of y. The standard error of 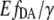 is then
is then 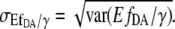
According to the total derivative
 |
var (α) and var (β) are significantly smaller than var (F) because α and β are obtained from large regions of interest (nROI ≫ 1) in reference measurements with var (α)∼ 1/nROI,α and var(β)∼ 1/nROI,β. Thus the error of EfDA/γ is determined mainly by the terms containing 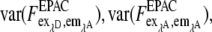 and
and 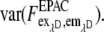
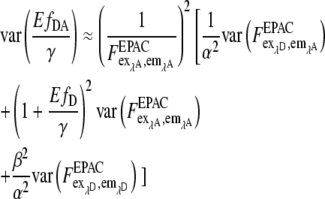 |
assuming that the error of a pixel is dominated by Poisson noise of the background-corrected image 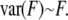 Thus,
Thus,
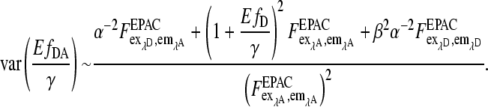 |
(12) |
Note that as a reasonable approximation, 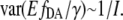 Thus to take the inverse of the 1-norm
Thus to take the inverse of the 1-norm 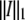 of the three obtained images with
of the three obtained images with 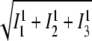 is a reasonable approach to estimate the reliability of the measurement rather than the two-norm
is a reasonable approach to estimate the reliability of the measurement rather than the two-norm 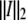 with
with 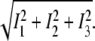 To better calculate a measure for the accuracy, Eq. 12 can be used:
To better calculate a measure for the accuracy, Eq. 12 can be used:
 |
(13) |
Because we used Eq. 13 only for the brightness information of our pseudocolor images, no significant difference was found between it and the simpler one-norm approach.
Error estimation of [cAMP]
The error of [cAMP] can now be estimated from the total derivative of Eq. 10.
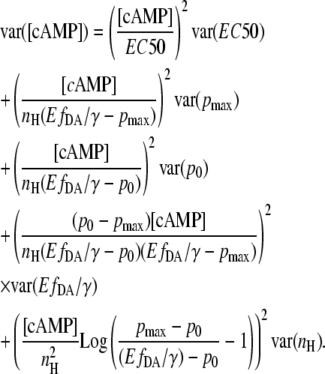 |
Assuming the parameters EC50, p0, pmax, and nH can be obtained precisely, the equation simplifies to
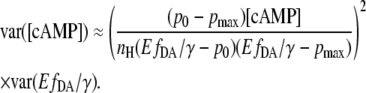 |
The relative error for [cAMP] is than
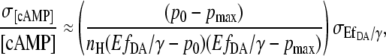 |
(14) |
where 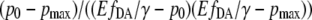 represents the relative “position” at the slope of the calibration curve.
represents the relative “position” at the slope of the calibration curve.
In contrast to the image brightness correction (Eq. 13), where only a factor proportional to 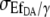 was needed for the relative error of [cAMP], at least the proportionally constant (i.e., detector gain) must be obtained. This is usually done by the pixel intensity fluctuation analysis of a homogeneous fluorescent sample. Taking into account the simplifications introduced to receive Eq. 14, obtaining the relative error of [cAMP] directly from a homogeneous [cAMP] region of an image or from an image sequence is recommended.
was needed for the relative error of [cAMP], at least the proportionally constant (i.e., detector gain) must be obtained. This is usually done by the pixel intensity fluctuation analysis of a homogeneous fluorescent sample. Taking into account the simplifications introduced to receive Eq. 14, obtaining the relative error of [cAMP] directly from a homogeneous [cAMP] region of an image or from an image sequence is recommended.
Petrus S. Salonikidis and André Zeug contributed equally to this work.
Andre Zeug's and Evgeni Ponimaskin's present address is Dept. of Cellular Neurophysiology, Medical School of Hannover, Germany.
Editor: Enrico Gratton.
References
- 1.Nagai, T., K. Ibata, E. S. Park, M. Kubota, K. Mikoshiba, and A. Miyawaki. 2002. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 20:87–90. [DOI] [PubMed] [Google Scholar]
- 2.Taskén, K., and E. M. Aandahl. 2004. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol. Rev. 84:137–167. [DOI] [PubMed] [Google Scholar]
- 3.Taylor, S. S., J. A. Buechler, and W. Yonemoto. 1990. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu. Rev. Biochem. 59:971–1005. [DOI] [PubMed] [Google Scholar]
- 4.Bos, J. L. 2003. Epac: a new cAMP target and new avenues in cAMP research. Nat. Rev. Mol. Cell Biol. 4:733–738. [DOI] [PubMed] [Google Scholar]
- 5.Rich, T. C., K. A. Fagan, T. E. Tse, J. Schaack, D. M. Cooper, and J. W. Karpen. 2001. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc. Natl. Acad. Sci. USA. 98:13049–13054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper, D. M. 2003. Regulation and organization of adenylyl cyclases and cAMP. Biochem. J. 375:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu, K., J. Zippin, D. Huron, M. Kamenetsky, U. Hengst, J. Buck, L. Levin, and S. Jaffrey. 2006. Soluble adenylyl cyclase is required for netrin-1 signaling in nerve growth cones. Nat. Neurosci. 9:1257–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiPilato, L. M., X. Cheng, and J. Zhang. 2004. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc. Natl. Acad. Sci. USA. 101:16513–16518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nikolaev, V. O., M. Bünemann, L. Hein, A. Hannawacker, and M. J. Lohse. 2004. Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 279:37215–37218. [DOI] [PubMed] [Google Scholar]
- 10.Ponsioen, B., J. Zhao, J. Riedl, F. Zwartkruis, G. van der Krogt, M. Zaccolo, W. Moolenaar, J. Bos, and K. Jalink. 2004. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 5:1176–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ponimaskin, E., M. Heine, A. Zeug, T. Voyno-Yasenetskaya, and P. Salonikidis. 2007. Monitoring receptor-mediated changes of intracellular cAMP level by using ion channels and fluorescent proteins as biosensors. In Serotonin Receptors in Neurobiology (Frontiers in Neuroscience). A. Chattopadhyay, editor. CRC Press, Boca Raton, FL. 19–40. [PubMed]
- 12.Patterson, G., R. Day, and D. Piston. 2001. Fluorescent protein spectra. J. Cell Sci. 114:837–838. [DOI] [PubMed] [Google Scholar]
- 13.Hübner, C. A., V. Stein, I. Hermans-Borgmeyer, T. Meyer, K. Ballanyi, and T. J. Jentsch. 2001. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 30:515–524. [DOI] [PubMed] [Google Scholar]
- 14.Tyzio, R., R. Cossart, I. Khalilov, M. Minlebaev, C. A. Hübner, A. Represa, Y. Ben Ari, and R. Khazipov. 2006. Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science. 314:1788–1792. [DOI] [PubMed] [Google Scholar]
- 15.Xu, J. Y., and B. R. Sastry. 2007. θ-bursts induce a shift in reversal potentials for GABA-A receptor-mediated postsynaptic currents in rat hippocampal CA1 neurons. Exp. Neurol. 204:836–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kvachnina, E., G. Liu, A. Dityatev, U. Renner, A. Dumuis, D. W. Richter, G. Dityateva, M. Schachner, T. A. Voyno-Yasenetskaya, and E. G. Ponimaskin. 2005. 5–HT7 receptor is coupled to G α-subunits of heterotrimeric G12-protein to regulate gene transcription and neuronal morphology. J. Neurosci. 25:7821–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clegg, R. M. 1992. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 211:353–388. [DOI] [PubMed] [Google Scholar]
- 18.Wlodarczyk, J., A. Woehler, F. Kobe, E. Ponimaskin, A. Zeug, and E. Neher. 2008. Analysis of FRET-signals in the presence of free donors and acceptors. Biophys. J. 94:986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoppe, A., K. Christensen, and J. A. Swanson. 2002. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys. J. 83:3652–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lakowicz, J. 2006. Principles of Fluorescence Spectroscopy. Springer, New York.
- 21.van Rheenen, J., M. Langeslag, and K. Jalink. 2004. Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys. J. 86:2517–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen, H., H. L. Puhl, S. V. Koushik, S. S. Vogel, and S. R. Ikeda. 2006. Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophys. J. 91:L39–L41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagy, P., L. Bene, W. Hyun, G. Vereb, M. Braun, C. Antz, J. Paysan, S. Damjanovich, J. Park, and J. Szöllsi. 2005. Novel calibration method for flow cytometric fluorescence resonance energy transfer measurements between visible fluorescent proteins. Cytometry A. 67A:86–96. [DOI] [PubMed] [Google Scholar]
- 24.Erickson, M. G., B. A. Alseikhan, B. Z. Peterson, and D. T. Yue. 2001. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron. 31:973–985. [DOI] [PubMed] [Google Scholar]
- 25.Gordon, G. W., G. Berry, X. H. Liang, B. Levine, and B. Herman. 1998. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 74:2702–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia, Z., and Y. Liu. 2001. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 81:2395–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin, S. L., N. N. Johnson-Farley, D. R. Lubinsky, and D. S. Cowen. 2003. Coupling of neuronal 5-HT7 receptors to activation of extracellular-regulated kinase through a protein kinase A-independent pathway that can utilize Epac. J. Neurochem. 87:1076–1085. [DOI] [PubMed] [Google Scholar]
- 28.Ruat, M., E. Traiffort, J. M. Arrang, J. Tardivel-Lacombe, J. Diaz, R. Leurs, and J. C. Schwartz. 1993. A novel rat serotonin (5-HT6) receptor: molecular cloning, localization and stimulation of cAMP accumulation. Biochem. Biophys. Res. Commun. 193:268–276. [DOI] [PubMed] [Google Scholar]
- 29.Wood, M., M. Chaubey, P. Atkinson, and D. R. Thomas. 2000. Antagonist activity of meta-chlorophenylpiperazine and partial agonist activity of 8-OH-DPAT at the 5-HT(7) receptor. Eur. J. Pharmacol. 396:1–8. [DOI] [PubMed] [Google Scholar]
- 30.Rich, T. C., K. A. Fagan, H. Nakata, J. Schaack, D. M. Cooper, and J. W. Karpen. 2000. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J. Gen. Physiol. 116:147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaccolo, M., and T. Pozzan. 2002. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 295:1711–1715. [DOI] [PubMed] [Google Scholar]
- 32.Krobert, K. A., and F. O. Levy. 2002. The human 5-HT7 serotonin receptor splice variants: constitutive activity and inverse agonist effects. Br. J. Pharmacol. 135:1563–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaccolo, M., G. Di Benedetto, V. Lissandron, L. Mancuso, A. Terrin, and I. Zamparo. 2006. Restricted diffusion of a freely diffusible second messenger: mechanisms underlying compartmentalized cAMP signalling. Biochem. Soc. Trans. 34:495–497. [DOI] [PubMed] [Google Scholar]
- 34.Chen, C., T. Nakamura, and Y. Koutalos. 1999. Cyclic AMP diffusion coefficient in frog olfactory cilia. Biophys. J. 76:2861–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lowe, G., and G. H. Gold. 1993. Contribution of the ciliary cyclic nucleotide-gated conductance to olfactory transduction in the salamander. J. Physiol. 462:175–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.