

Abstract
Although the mechanism by which a kinesin-1 molecule moves individually along a microtubule is quite well-understood, the way that many kinesin-1 motor proteins bound to the same cargo move together along a microtubule is not. We identified a 60-amino-acid-long domain, termed Hinge 1, in kinesin-1 from Drosophila melanogaster that is located between the coiled coils of the neck and stalk domains. Its deletion reduces microtubule gliding speed in multiple-motor assays but not single-motor assays. Hinge 1 thus facilitates the cooperation of motors by preventing them from impeding each other. We addressed the structural basis for this phenomenon. Video-microscopy of single microtubule-bound full-length motors reveals the sporadic occurrence of high-compliance states alternating with longer-lived, low-compliance states. The deletion of Hinge 1 abolishes transitions to the high-compliance state. Based on Fourier transform infrared, circular dichroism, and fluorescence spectroscopy of Hinge 1 peptides, we propose that low-compliance states correspond to an unexpected structured organization of the central Hinge 1 region, whereas high-compliance states correspond to the loss of that structure. We hypothesize that strain accumulated during multiple-kinesin motility populates the high-compliance state by unfolding helical secondary structure in the central Hinge 1 domain flanked by unordered regions, thereby preventing the motors from interfering with each other in multiple-motor situations.
INTRODUCTION
Motor proteins in the myosin, dynein, and kinesin families mediate the transport of membrane-bounded organelles along actin filaments or microtubules. Several of these proteins, including kinesin-1 (1), kinesin-2 (2), cytoplasmic dynein (3), and myosin-V (4), are known to be processive, i.e., they are single-motor proteins capable of taking tens to hundreds of elementary steps along the corresponding filament before dissociating. Processivity is evidently an adaptation for the efficient transport of small organelles, such as vesicles, that can bind at most a small number of transport motors. Processivity implies that a single-motor protein must spend most of its time attached to the filament. Detachment for even a short period of time would result in the motor rapidly diffusing away from its binding site and toward dissociation from the filament (5). Such a high fraction of time bound is achieved by “two-headed” motors (i.e., those with two motor domains), of which all the abovementioned are examples, which move using a hand-over-hand mechanism (6–9), in which the unbinding of one head is contingent on the binding of the other (10), leading to an alternation of the binding of the two heads.
The high duty ratio of processive motors leads to a potential problem. If several processive motors are moving one organelle, as presumably occurs when kinesin-1 moves large organelles such as mitochondria (11) or kinesin-2 moves intraflagellar rafts (12,13), then the motion of one motor might be impeded by the other attached motors. However, such impedance occurs neither in vivo nor in vitro. For example, the speed of intraflagellar rafts is independent of the size of the raft (14), and therefore presumably the number of motors. Moreover, the speed of kinesin-1 in gliding assays, in which a microtubule is moved across a kinesin-coated surface, is independent of the length of the microtubule and the density of kinesin (1,15), and thus independent of the number of motors. These experiments show that motors are able to move in a coordinated manner to ensure a high speed, even when up to 100 motors are attached to and moving the microtubule at any one time.
A potential mechanism by which several motors may cooperate without impeding each other is suggested by the high torsional compliance (i.e., flexibility) of motor proteins. Observations of microtubules tethered to individual kinesin-1 molecules (1,16–18), and actin filaments tethered by individual myosin molecules (19,20), show that filaments undergo thermally induced rotations of 360° or more over several minutes. If a motor has high torsional compliance, then one would expect that the action of a motor would be independent of the angle between its base, through which it is anchored to the surface of a cargo, and its head, through which it is attached to the microtubule. Consistent with this expectation, Hunt and Howard (16) found that the speed of a kinesin-1 was independent of the orientation of the microtubule in gliding assays. Furthermore, high flexural compliance may indicate the presence of a disordered region in the motor domain or the cargo-binding site in the tail. Such a disordered region is also expected to have high flexibility under bending and stretching forces, in addition to torsional forces. Such flexibility would allow each motor in an ensemble to undergo its force-producing conformational change on its own, rather than stepping in synchrony with the other motors. Such asynchronous stepping is thought to underlie the fractional steps recently observed for kinesin-1, in which n motors moved at the same speed as one motor because they made n times as many steps that were only 1/nth the size of the elementary step (18). Thus several observations support the notion that motor compliance facilitates the cooperation of several motor proteins.
Hunt and Howard (16) suggested that the compliant region within kinesin-1 might correspond to a ∼45–65 amino-acid region near the motor domain. This region, termed Hinge 1, lies between the dimerization domain (also called the “neck,” amino acid (aa) 341–376 in Drosophila), a coiled coil just carboxy-terminal to the motor domain that is seen in the three-dimensional structure of the kinesin-1 dimer (21), and Coil 1 (aa 439–560), a coiled coil that forms part of the elongated kinesin tail domain (22) (Fig. 1). There are three reasons for thinking that Hinge 1 (aa 377–438) is flexible. First, it contains prolines not predicted to form a coiled coil (23,24) (Fig. 1 C), a protein domain that has high torsionally rigidity (25). Second, high torsional flexibility is still evident in kinesins truncated after aa 430 (18), and it is difficult to see where else the compliance might reside, because the motor and dimerization domains are expected to be torsionally rigid. A caveat is that the compliance is attributable to end effects: either the attachment to the cargo surface might be compliant (26), or the motor domain might transiently detach and rotate before reattaching. Third, removal of Hinge 1 from Neurospora crassa kinesin-1 drastically reduces the microtubule gliding speed at high motor densities (from 2.6 to 0.7 mm/s) (27). However, the activity was not tested in single-molecule assays, nor was the compliance of this region measured.
FIGURE 1.

Domain organization of Hinge 1 region of Drosophila kinesin-1. (A) Amino-acid sequence alignment made with ClustalW of wild-type kinesin-1 (Dm, Drosophila melanogaster; Hs, Homo sapiens; Mm, Mus musculus; Nc, Neurospora crassa). Letters “a” and “d” denote mainly hydrophobic positions in heptad repeats. Horizontal lines represent sequences sampled by model peptides 1–5 (left to right). (B) Structures of corresponding regions from x-ray crystallography (21), circular dichroism (44), and NMR (45). (C) Coiled coil likelihood calculated from PairCoil (46) and Coils (23) programs, using a 21-aa window. The x axis represents amino-acid position. (D) Predicted probability of secondary-structure formation given by PSIPRED (47) and PredictProtein (PP) (48). Predicted secondary structure is presented below each plot; the subset (from PP) with probability higher than 80% is shown. (E) Secondary-structural organization of Hinge 1, based on this study. Interstrand interactions of central part of Hinge 1 are hypothetical (see Discussion for details).
To test the structural and functional importance of Hinge 1, we measured the motility and torsional compliance of kinesin-1 in which this putative flexible region was removed. We demonstrate that Hinge 1 is necessary for high speed at high kinesin density, but is not required for high speed at low kinesin density, consistent with the necessity of Hinge 1 for efficient many-motor activity. However, we find that the compliance of this region is not high. To understand this unexpected behavior, we synthesized five 30-aa polypeptides that cover Hinge 1, and studied their secondary structures and thermal stability. This peptide approach is similar to that used by Tripet et al. (28) to study the role of the neck region in processivity. We found evidence for secondary structure that leads us to hypothesize that the Hinge 1 region is sufficiently structured to resist thermal forces, but that it is disrupted by the forces generated during motility that prevent it from hindering motion in many-motor conditions.
MATERIALS AND METHODS
Kinesin constructs
The domain organization of Drosophila melanogaster kinesin heavy chain (kinesin-1) is shown in Fig. 1. In the first-deletion construct, denoted ΔT, the tail domain was truncated after aa 559. The second-deletion construct, denoted ΔTΔH, was identical to ΔT, except that aa 380–442 of Hinge 1 were removed. All proteins were expressed in Escherichia coli as in Coy et al. (29), i.e., carrying a C-terminal 6xHis-tag. The deletion construct plasmids for ΔT and ΔTΔH were prepared as in Coy et al. (29). Tubulin was purified and microtubules were polymerized as in Schief et al. (30). All reagents were purchased from Sigma-Aldrich (St. Louis, MO), except as noted. All enzymes were purchased from New England Biolabs (Ipswich, MA), except for T4 DNA ligase, which was from GIBCO BRL (Division of Invitrogen, Paisley, United Kingdom). All DNA propagation was performed in DH5α except as noted. All oligonucleotides were purchased from Operon (Köln, Germany).
Motility assays
Flow cells were made as previously described (1), with the exception of using double-sided tape as spacer. The flow cell volume was ∼5 μL (30). Glass coverslips (Corning, Corning, NY) for flow cells were washed as follows: 1 h in acetone, 10 min in ethanol, rinsed in ddH2O, 1 h at 60°C in 70% H2SO4 (Merck, Whitehouse Station, NJ) plus 30% H2O2, rinsed three times with ddH2O, 15 min in 0.1 M KOH, rinsed twice in ddH2O, dried with N2, silanized with dichlorodimethylsilane (0.05% DDS in Trichloroethylene (Merck), excess silane removed by sequential 5-min, 15-min, and 30-min washes in methanol (Merck), dried with N2, and stored in layers of lens paper). The chamber was first incubated with 50 μg/mL Antipenta-Histidine IgG (Qiagen, Hilden, Germany). The remaining hydrophobic surfaces were blocked with 1% (w/v in ddH2O) F-127 (mol wt 12,600). Subsequently, kinesin was incubated for 5 min, followed by motility solution with microtubules (320 nM polymerized tubulin microtubules 2–6 μm long) and the stated ATP concentration. Motility assays at high and medium kinesin density correspond to 200–2000 μm−2 and 20–100 μm−2 for ΔT and ΔTΔH, respectively, assuming 100% binding of protein to the two antibody-coated surfaces. Single-molecule assays were performed with 0.01–0.1 nM kinesin construct. The temperatures of the flow cell and the working space were maintained at 23 ± 1°C.
Image acquisition
Microtubules containing a 3:1 ratio of unlabeled/tetramethylrhodamine-labeled tubulin were visualized with an Axioplan 2 epi-fluorescence microscope (Carl Zeiss AG, Oberkochen, Germany). Representative fields of high and low motor density motility assays were digitally captured for 40–60 s with IP Labs software (BD BioSciences, San Jose, CA), and TIFF image stacks were analyzed offline in ImageJ (National Institutes of Health, Bethesda, MD). Single-molecule experiments were videotaped with a super-VHS recorder system. Recorded frames were digitized using a PCI-1407 single-channel monochrome frame-grabber with an 8-bit analog-to-digital conversion and LabView 6.i Software (National Instruments, Austin, TX).
Image analysis
From each image frame, we extracted the microtubule angle, length, and center of mass. This procedure was performed with custom image-analysis software written in MATLAB (The MathWorks, Natick, MA), using raw image stacks.
Data analysis
The average speed in single-molecule assays was calculated from measurements of the distance of the pivoting point (where the motor is located) to the centroid of the microtubule versus time. To be certain that motion was attributable to single molecules, we imposed the following data selection criteria: 1), run length equal to or less than the length of the microtubule; 2), speed close to expected, according to Schief et al. (30); 3), microtubule length between 2–9 μm, to avoid fitting problems (when a microtubule is too short) and bending or moving out of focus (when a microtubule is too long); and 4), visual similarity of angular fluctuations with simulations using a similar torsion elasticity constant. To measure the torsion elasticity constant of the kinesin-1 motor, we calculated a one-sided power spectral density (PSD) of microtubule angular fluctuations. Using an analytical fit to the PSD of the angular microtubule fluctuations, the torsion elasticity constant as well as the drag coefficient of the motor protein can be obtained (31). To avoid artifacts at high frequencies, the fit was made from fmin to fmax/2. The angular variance 〈θ2〉 and the torsion elasticity constant are related by the energy equipartition theorem: kBT/2 = κ 〈θ2〉/2, where kB is the Boltzmann constant, and T is the absolute temperature.
Model peptides derived from the Hinge 1 sequence
The Hinge 1 sequence was cut into five 30-aa-long overlapping pieces (Fig. 1). Each peptide was synthesized, high-performance-liquid-chromatography-purified, and characterized by mass spectroscopy (ThermoElectron, Ulm, Germany). Trifluoroacetic acid was removed, and the peptides were used without further purification. The N-terminal peptide (peptide 1) and C-terminal peptide (peptide 5) contain at least two heptads of the flanking coiled coil regions. One set of five peptides contained a Trp (W) residue at the same relative position (aa 11), serving as an intrinsic fluorophore whose emission monitors changes in its molecular environment in response to thermal unfolding. The Trp at this position in peptide 1 is natural in Dm kinesin-1, whereas the Trp in peptide 2 is naturally found in kinesin-1 from other species (N. crassa, Ustilago maydis, Caenorhabditis elegans, and Syncephalastrum racemosum) replacing the Gln (32) at this position in Dm kinesin. Sequences of the other peptides were chosen such that the Trp also replaced a Gln in each case, motivated by the naturally occurring Gln-to-Trp replacement. In addition, the nonfluorescent native Dm sequences, corresponding to the structurally most different peptides 2 and 3 (shown in this study), and the homologous native (nonfluorescent) sequences of kinesin-1 from Homo sapiens were studied for comparison. The sequences of peptide 2 and 3 derived from Hinge 1 of human kinesin-1 are: RNGETVPIDEQFDKEKANLEAFTVDKDITL (3478 Da) and FTVDKDITLTNDKPATAIGVIGNFTDAERR (3321 Da), respectively.
Fourier transform infrared spectroscopy
Fourier transform infrared (FTIR) spectra were recorded with a Vector 22 (Bruker, Rheinstetten, Germany) equipped with a diamond attenuated-total-reflectance cell (Resultec, Illerkirchberg, Germany). The peptide concentrations used were 6–8 mg/mL in phosphate buffer (100 mM KCl, 50 mM Na2HPO4, pH 8.6). For one spectrum, 256 interferograms were recorded, with a resolution of 2 cm−1 at 20°C, and referenced against pure buffer spectra under identical conditions. Absorption spectra were evaluated by fitting the amide I envelope with five Gaussian/Lorentzian curves. Bandwidths were initially restricted to 30 cm−1. Band frequencies, shapes, and intensities were free to vary in the final fit, using Peakfit software (version 4.12, Seasolve, San Jose, CA), and stayed below 30 cm−1.
Circular dichroism (CD) spectroscopy
A CD Spectroscope Jasco J-810 15OS (Jasco, Gross-Umstadt, Germany) was used at the following settings: nitrogen (N2) flow at 5 L/min, scan speed at 100 nm/min, bandwidth and data pitch at 1 nm, continuous scanning mode (20 scans averaged), and 1-mm cuvette path length. Wavelengths measured from 260 to 190 nm. The CD buffer contained 10 mM KCl and 5 mM Na2HPO4 in ddH2O, pH 8.69. The ellipticity at wavelength λ was given by Greenfield (33), as [θ]λ = 100 θλ/d · m, where θλ is the observed ellipticity (degrees) at wavelength λ, d is the path length (in cm), and m is the molar concentration. The units are deg · cm2 · dmol−1. We used CDSSTR (Dichroweb, Birkbek, UK) to estimate the percentage of secondary structure present for each spectroscopic CD signal. Peptide concentrations ranged from 0.2–0.4 mg/mL. The concentration of tryptophan-containing peptides was calculated by the Beer-Lambert law: A = ɛ · l · m, where A is the measured absorbance, ɛ is the extinction coefficient, l is the path length, and m is the molar concentration. The molar extinction coefficient was calculated in the ExPASy website with ProtParam Tool.
Fluorescence spectroscopy
We used the intrinsic fluorescence of Trp to monitor temperature-induced structural transitions in the model peptides. The emission of peptides was excited at 280 nm, and was measured from 295–400 nm at a 2-nm slit width, using a Perkin Elmer LS 55 instrument (Perkin Elmer, Vaudreuil, Quebec, Canada), equipped with a temperature-controlled cuvette connected to a water-bath thermostat (Hake, Frankfurt, Germany). The temperature dependence of Trp emission, however, is an intrinsic property of this residue, and is not per se an indication of a structure-dependent change in the Trp environment within a polypeptide chain. Therefore, we analyzed the structure-sensitive contribution by normalizing the emission of peptides relative to the corresponding intensity recorded under identical conditions from free Trp in aqueous solution. The emission of the single Trp present in the model peptides was excited at 280 nm, and was measured from 295–400 nm in buffer (100 mM KCl, 50 mM phosphate, pH 8.6) at temperatures between 23–70°C. The fluorescence of free Trp was measured at identical temperatures in the same buffer. The ratio of the emission at 345 nm from the peptide (Epep345) relative to that of free Trp in aqueous solution EW345 was determined as:
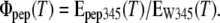 |
(1) |
In the same way, the emission of free Trp in ethanol/water mixtures was scaled to that in water, yielding the ratio:
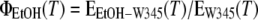 |
(2) |
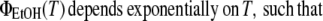 |
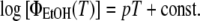 |
(3) |
This is shown in Fig. 6A (inset), which also provides evidence that the slope p increases with an increasing admixture of ethanol, i.e., with increasing hydrophobicity of the Trp environment. We used this dependency to determine ΔH and ΔS for the unfolding of peptides in a two-state model, assuming different hydrophobicity in the initial native state (concentration N) and thermally unfolded state (concentration U), whose fractional concentration is defined as:
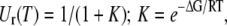 |
(4) |
where K is the equilibrium constant of the unfolded and native state U/N (reversibility was found for all peptides in the assessed range of 23–70°C). The measured temperature dependence of logΦpep(T) was reconstructed by assigning temperature-independent slopes pN and pU to the native and unfolded peptide states, respectively, in the plot of log Φpep(T) versus T:
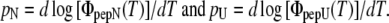 |
(5) |
Thus, the temperature dependence in the measured trace was fitted by
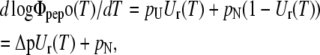 |
(6) |
and the pair of ΔH and ΔS values was determined that gave the least quadratic deviation from the data (with Δp = pU − pN, where pN was the initial slope of the plot at room temperature, and pU was a fit parameter equal to or greater than the slope at 70°C).
FIGURE 6.

Thermal denaturation and oligomerization of peptides. (A) Tryptophan fluorescence intensity plotted against temperature for five Dm peptides. Solid lines are fits to a model in which equilibration occurs between a folded and unfolded state (see Materials and Methods). Plots obtained with the most stable peptides 3 and 4 (black lines) exhibit strongest curvature (i.e., largest change in Ur = U/(N+U), according to Eq. 6) at higher temperatures than the curves of less stable peptides (gray lines). Corresponding thermodynamic parameters used for the fits are shown in Table 1. (Inset) Normalized fluorescence intensity of free Trp in 0%, 10%, 20%, and 50% ethanol in water mixtures shows an increase of temperature sensitivity with increasing solvent hydrophobicity. Temperature-dependence of Trp emission from the peptides corresponds to an increasingly hydrophobic environment, as mimicked by high ethanol content. (B) Elution profiles of five peptides (1 mg/mL) from a gel-filtration column. Peptides 3 and 4 elute in monomeric and dimeric fractions. We interpret the elution profiles of peptides 1, 2, and 5 as unfolded proteins (see text). Upper arrows show locations of standards ribonuclease A (A), dimeric insulin A chain (B), aprotinin (C), and dimeric insulin B chain (D).
Gel filtration
A Superdex Peptide 10/300 GL column (Amersham Biosciences, GE HealthCare, Uppsala, Sweden) was used. The running buffer (100 mM KCl and 100 mM Na2HPO4, pH 8.6) and all liquids were filtered with a 0.22-μm filter and degassed in bottles rinsed three times with filtered water. As standards, we used: 1), ribonuclease (mol wt 13700); 2), aprotinin (MW 6512); 3), insulin chain A (mol wt 2531.64); and 4), insulin chain B (mol wt 3495.89). Before samples were run, the column was cleaned (at 0.25 mL/min) with 4 column volumes (CVs) of water, 2 CVs of EtOH, and 2 CVs of water, finally equilibrated with 2 CVs of running buffer. Each sample was filtered with a 0.22-μm filter before loading. One hundred microliters of sample (1 mg/mL) were loaded for each peptide, and run at 0.25 mL/min.
RESULTS
Hinge 1 is necessary for maximal microtubule speed at high motor densities
Measurements of microtubule gliding speeds at different surface densities of the motors were performed at saturating ATP concentrations (1 mM). At high densities (>1000 μm−2), where it was estimated that up to 100 kinesin molecules are interacting with each microtubule (1), full-length kinesin-1 moved microtubules at a speed of 702 ± 7 nm/s (mean ± SE, n = 55, Fig. 2, open circles). The speed did not change significantly as motor density was reduced (p < 0.01, Fig. 2, dotted line). At the lowest densities (<10 μm−2), the moving microtubules exhibited angular fluctuations around a single point on the surface, and rarely moved more than their own length before dissociating from the surface. By these criteria, the low-density movement was driven by single kinesin molecules (1), and the independence of speed from length confirms the earlier finding that the speed of movement of kinesin-1 is independent of the number of motors driving the motion.
FIGURE 2.

Microtubule gliding speeds versus kinesin density. As kinesin density increases, gliding speed remains constant for full-length (open circles; dotted line shows average speed) and ΔT kinesin (solid squares; solid black line shows average speed), but decreases for construct ΔTΔH (solid triangles and solid gray line indicate Hill-type curve 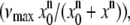 where x0 = 460 and n = 0.8). Error bars are standard deviations.
where x0 = 460 and n = 0.8). Error bars are standard deviations.
The truncated construct ΔT behaved similarly to full-length kinesin. At high kinesin density, the gliding speed was 700 ± 32 nm/s (mean ± SD, n = 60), whereas at low density, the gliding speed was 820 ± 80 nm/s (mean ± SD, n = 40). Thus speed was almost independent of density (Fig. 2, solid black squares, solid black line), though the small decrease was statistically significant (p < 0.01). Evidently, as in the full-length construct, these truncated motors, which lack Hinge 2 and the C-terminal domains, can cooperate at high densities to move microtubules at high speeds. In contrast, the deletion construct that lacked Hinge 1, ΔTΔH, moved significantly more slowly at high densities (>1000 μm−2), at 151 ± 32 nm/s (n = 50), than at low densities (<10 μm−2), at 657 ± 23 nm/s (n = 45) (Fig. 2, solid gray triangles, solid gray line). The high speed at low density indicates that ΔTΔH moves normally under single-molecule conditions. Indeed, the distances that microtubules moved before dissociating from surfaces coated with ΔTΔH at low density were similar to those of full-length and ΔT kinesins: 2.4 ± 0.4 μm (n = 8) for full-length, 1.9 ± 0.3 μm (n = 20) for ΔT, and 1.8 ± 0.2 μm (n = 18) for ΔTΔH, showing that deleting Hinge 1 has no effect on processivity. On the other hand, the low speed of ΔTΔH at high density indicates that the Hinge 1 domain is important for preventing motors from interfering with each other at high densities. We estimate that 10–100 motors reduce the speed of ΔTΔH to 50% of is maximum value (assuming that an average microtubule interacts with ΔTΔH motors over an area of 5 μm × 30 nm, and that all the ΔTΔH motors are active).
Torsional flexibility of Hinge 1
Because Hinge 1 corresponds to a clear break between two coiled coil domains, we hypothesized that it might be an unstructured domain, and therefore have high flexibility. Such flexibility could allow many kinesins to operate simultaneously without interfering with each other. Loss of this domain in ΔTΔH could therefore provide a structural basis for its low gliding speed at high density.
To test this hypothesis, we measured the torsional flexibility of individual kinesin molecules by analyzing the pivoting motions of microtubules gliding over surfaces coated with kinesin at low densities (Fig. 3 A). To identify an unambiguously functional motor, ATP was present at 5 μM, i.e., at a concentration high enough to observe motor operation, but low enough to allow a sufficiently long observation time so that the rotational fluctuations of moving microtubules could be measured (see Materials and Methods for additional single-molecule criteria). Based on our hypothesis, we expected that microtubules driven by single full-length and ΔT kinesins would undergo large angular fluctuations because they contained Hinge 1. Smaller angular fluctuations were expected for ΔTΔH, where Hinge 1 had been removed.
FIGURE 3.

Thermally driven rotational Brownian motion and gliding of a microtubule bound to a single kinesin molecule in presence of 5 μM ATP. (A) Left, single frame of rhodamine-labeled microtubule exhibiting random rotations and motion along its major axis. Right, scheme of analysis. The fit gives an angle (θ), measured from an arbitrary x axis to the major axis, the center (asterisk), and the length of the ellipse. Cross, pivoting point. Scale bar, 2 μm. (B) Six original angular traces of freely rotating and gliding microtubules tethered by full-length kinesin-1 molecules. (C) Six original angular traces of microtubules, using ΔT kinesin construct. (D) Six original angular traces, using ΔTΔH kinesin.
Our results were in general agreement with the expectation that ΔTΔH would be less flexible than full-length and ΔT kinesins. The microtubules driven by single full-length kinesins underwent large rotational motions (Fig. 3 B). However, the motions were not consistent with a rotation attributable to thermal (i.e., Brownian) fluctuations of a single torsional spring. Instead, the microtubules spent much of their time undergoing comparatively small rotational fluctuations (e.g., Fig. 3 B, traces i, iv, v, and vi), with peak-to-peak amplitudes of less than 20°. Occasionally, however, the mean angle jumped through angles of 20° or more (Fig. 3 B, traces ii and iii). These transitions were infrequent, 0.2 ± 0.03 s−1, and slow, taking ∼1 s to reach a new mean angle for full-length kinesin-1. The ΔT kinesin constructs behaved similarly, exhibiting jumps between mean levels (Fig. 3 C, traces i, ii, v, and vi) about which there were comparatively small fluctuations (Fig. 3 C, traces iii and iv). In contrast, microtubules driven by a single molecule of the ΔTΔH construct lacked large angular motions (Fig. 3 D). All measured traces of ΔTΔH corresponded to the long-lived stiff state. Thus, deletion of Hinge 1 specifically abolishes slow and infrequent large-angle angular transitions. Furthermore, the absence of slow transitions in only the deletion mutant demonstrates that these transitions are not attributable to the unbinding and rebinding of kinesin to the anti-His antibody used for immobilization, or to the unbinding of one head from the microtubule and rebinding with a twist. If either of these phenomena had occurred, large fluctuations would have also been observed in ΔTΔH, which binds to antibody and microtubule in the same way as ΔT.
Torsion elasticity of kinesin constructs
To determine whether the small-amplitude angular fluctuations of microtubules could be accounted for by thermal fluctuations of a linear torsionally elastic element within the kinesin molecule, we performed Fourier analysis on recordings made of the long-lived stiff state. An example of the PSD obtained from a ΔT construct is shown in Fig. 4. At low frequencies, the PSD has a large amplitude, and decreases with a slope of approximately −2 in the log-log plot. The curve is well-described by a Lorentzian function, which is characteristic of thermal fluctuations in a linear elastic element, i.e., a spring, subject to damping. For this example, the torsional spring constant was 0.020 ± 0.006 kBT/deg, and the drag coefficient was 0.50 ± 0.02 × 10−21 N · m · s/deg (fit value ± 95% confidence interval) (Fig. 4, solid curve). The drag coefficient was similar to that measured by Hunt and Howard (16). This torsional spring constant corresponds to a standard deviation of the torsional angle of 7°. The standard deviations were 6 ± 2° (n = 8) for full-length kinesin, 7 ± 3° (n = 14) for ΔT, and 8 ± 2° (n = 8) for ΔTΔH. Thus, the rotational fluctuations of the long-lived “small-amplitude states” were similar.
FIGURE 4.

Measurement of torsion-elasticity constant of a single kinesin-1 molecule. Power spectral density of angular fluctuations is from Fig. 3 C, trace i; solid line is a Levenberg-Marquadt least-squares weighted fit to the data with the Lorentzian: 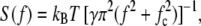 where S(f) is the power at frequency f, fc is the corner frequency, kB is the Boltzmann constant, T is the absolute temperature, and γ is the drag coefficient. The torsion elasticity constant is calculated as κ = 2π fcγ. (Inset) Fit residuals. Solid lines represent expected standard deviation (1/n1/2, where n is number of points, and each point is an average of 20 data/fit points).
where S(f) is the power at frequency f, fc is the corner frequency, kB is the Boltzmann constant, T is the absolute temperature, and γ is the drag coefficient. The torsion elasticity constant is calculated as κ = 2π fcγ. (Inset) Fit residuals. Solid lines represent expected standard deviation (1/n1/2, where n is number of points, and each point is an average of 20 data/fit points).
Secondary structural features of the Hinge 1
To assess the secondary structure-forming propensity of the Hinge 1 sequence, we designed five overlapping 30mer peptides that spanned the hinge region (60 aa). The first peptide started at the penultimate heptad repeat of the neck coiled coil, and the fifth peptide ended after the second heptad repeat of Coil 1. The peptides overlapped by at least 10 aa, and contained an introduced tryptophan at position 11, as shown in Fig. 5 A. These peptides were analyzed by CD, fluorescence, and FTIR spectroscopy. Qualitatively, only peptides 3 and 4 exhibited CD spectra that were indicative of partial α-helix formation at room temperature, resulting in the characteristic α-helical signature at 222 nm (34), in addition to the contribution from unordered conformations showing negative CD below 205 nm (Fig. 5 B). The helicity of peptides 3 was further enhanced by cooling to 2°C, and was lost upon heating to 60°C (Fig. 5 B, inset). Peptide 4 behaved almost identically (not shown). This is in contrast to peptides 1, 2, and 5, which exhibited only marginal ellipticity at 222 nm at room temperature.
FIGURE 5.

Ultraviolet (UV) circular dichroism and infrared spectra of model peptides. (A) Amino-acid sequences of synthesized 30-aa-peptides. Each peptide (1–5, left to right, with molecular weights of 3602, 3375, 3071, 3070, and 3526, respectively) contains a tryptophan residue (W) in position 11. (B) Far UV circular dichroism spectra of peptides 1–5 at 20°C in mean residue molar ellipticity. Circles correspond to fits from CDSSTR used for ranking helical propensity, as shown in Table 1. (Inset) Change of CD spectral signature during denaturation of peptide 3 with temperature (bottom to top, 2°C, 10°C, 20°C, 30°C, 40°C, 50°C, and 60°C). (C) Infrared absorption spectra of peptide 3 (top), and peptides 2 and 4 (bottom). Major peaks in spectra indicate that peptides 3 and 4 can be fitted with identical spectral components. Higher absorption at 1643 cm−1 relative to 1656 cm−1 may indicate helix-helix interactions. See text for details of peptides 3 and 4. For comparison, broad amide absorptions of unstructured peptide 2 are superimposed (bottom). (D) Overlaid CD spectra of peptide 2 (lower traces) and peptide 3 (upper traces) with either native sequences (black) or tryptophan substitution at position 11 (gray; see Fig. 1 A). (Inset) Thermal unfolding and refolding of peptide 3 carrying native kinesin-1 sequence. After being heated to 90°C, CD spectrum at 20°C (gray) reproduces initial trace measured at 20°C (black). (E) Comparison of induction of secondary structure by TFE at 2°C for peptides 2 and 3 derived from Hinge 1 of Dm (upper traces) and Hs (lower traces). Thin lines, without TFE; thick lines, with 32% TFE. Circles indicate fit by CDSSTR resulting in 70% helicity for Dm peptide 2.
Helix-forming peptides 3 and 4 were further analyzed by FTIR spectroscopy (Fig. 5 C). The structure-sensitive amide I absorption band (i.e., the peptide carbonyl stretching vibration) was decomposed into five different spectral features at 1622 cm−1, 1643 cm−1, 1655 cm−1, 1670 cm−1, and 1684 cm−1 for both peptides. The peaks around 1655 cm−1 are indicative of the peptide carbonyl absorption of α-helices. Absorptions at 1684 and 1622 cm−1 are typically associated with β-sheet structure (35,36), whereas the 1670-cm−1 absorption can be associated with turns. For comparison, and in agreement with the CD results, the broadening of the amide I and II absorptions of peptide 2 (Fig. 5 C, bottom) and its lower amide I frequency confirm the virtual absence of secondary structure, as opposed to peptides 3 and 4. However, the corrected (37) relative integral intensity in the 1652–1656-cm−1 range (15–20% at 23°C) for peptides 3 and 4 is only ∼60% of that predicted from CD (30–35%), and the main infrared absorption lies in the 1640–1645-cm−1 range, usually assigned to random structure. Similar relations between CD and FTIR spectra were observed with other peptides and proteins, and are considered indicative of coiled coil formation (38). Almost identical results were obtained for peptide 4 (Fig. 5 C, bottom). Although the distinct helix-forming potential of peptides 3 and 4 is obvious from CD, both peptides were only partially folded at any of the conditions tested. Therefore, the FTIR data may indicate helix-helix interactions in peptides 3 and 4 under the high-concentration conditions of the FTIR experiment (6–8 mg/mL), but should not be regarded as proof of tertiary structure, and particularly of coiled coil formation. In the lower-concentration regime used in CD and fluorescence (see below), helix interactions are also not suggested by the data. The more general distinction between secondary structure-forming potential in peptides 3 and 4 versus the largely unstructured peptide 2 is consistent with the FTIR results.
To assess possible structural consequences of the Trp substitution (used for the fluorescence monitoring of unfolding; see below), CD spectra of the structurally most different peptides 2 and 3 were compared with spectra of the native Dm sequences. The pairwise superposition of the CD spectra of both peptides (Fig. 5 D) demonstrates that the Gln-to-Trp replacement causes a slight reduction in the helical content of peptide 3 (reduction of the 222-nm band), rather than acting as a helix-inducing residue. The preceding unsubstituted peptide 2 is again disordered (virtual absence of ellipticity at 222 nm). Thus, the distinct gradient of structure-forming potential along the region sampled by peptides 2 and 3 is a robust intrinsic property of Hinge 1. The helix-forming propensity of peptide 3 is further underscored by its high refolding potential: Fig. 5 D, inset, shows the full recovery of the CD signature at 20°C of the unsubstituted sequence after heating to 90°C. Furthermore, the native sequence can adopt up to 70% helicity, whereas even in the presence of 32% trifluoroethanol and at 2°C, the helicity of peptide 2 does not exceed 30% (Fig. 5 E, upper traces). Given the overlap between the peptides, their distinct differences in helical propensity reveal an abrupt transition in the structure-forming potential between the central and flanking region of Hinge 1. This suggests that the functionally required flexibility of Hinge 1 does not originate in an intrinsically disordered domain.
We asked whether a sharp transition in the structure-forming potential exists in the Hinge 1 domain of kinesin from other species. Fig. 5 E (lower traces) shows CD spectra of the homologous peptides 2 and 3 derived from Hinge 1 of kinesin-1 from Homo sapiens (Hs, without Trp substitution). Whereas human peptide 3 does not exceed 8% helicity even in the presence of 32% TFE at 2°C, the preceding human peptide 2 sequence adopts 33% helicity under these conditions. Although less pronounced than in Dm, a distinct difference of helical propensity within a 15-amino-acid window also resides in human Hinge 1. The presence of a proline in the center of human peptide 3 is probably the reason its helicity is low, whereas a proline is situated at the more marginal position 7 in the more helical human peptide 2.
Thermal stability of Hinge 1 model peptides
In the context of the proposed function of Hinge 1 as a flexibility-providing segment, we addressed the energetics of structural transitions in the model peptides, using thermal unfolding experiments in which the fluorescence of the introduced Trp-residues at position 11 was monitored (see Materials and Methods). The structure of both the most helical and the least helical peptide was very little affected by the introduced Trp. This allows for the description of unfolding by a two-state transition where the fluorescence-based assessment of thermal stability is more appropriate than in CD, because unfolding has to proceed at least up to position 11 to become observable. A more distinct temperature dependence and a more stringent definition of the unfolded state is thus achieved as partially (C-terminally and N-terminally) unfolded states are not monitored which otherwise broaden the temperature profiles in CD, leading to ambiguous results for peptides that never adopt a 100% helical structure.
All Trp-containing peptides exhibited a temperature-induced decrease of Trp emission measured at 340 nm. However, the normalized emission Φ(T) relative to that of free Trp in aqueous solution increased strongly with temperature (T) for all peptides (Fig. 6 A), such that a plot of logΦp(T) versus T (see Materials and Methods) deviated from linearity by a distinct upward curvature. In contrast, at a given degree of hydrophobicity of the Trp environment adjusted by water/ethanol mixtures, logΦEtOH(T) increased linearly with T (Fig. 6 A, inset). The steepness of the linear relation depended on solvent polarity, and became larger at higher hydrophobicity. Thus, we assigned the upward curvature in the logΦp(T) plot of the peptides to the transition of Trp from a more aqueous to a more hydrophobic environment formed upon the denaturation/aggregation of peptides. We modeled the T-dependence of this fluorescence change by a corresponding free energy of unfolding that caused the transition between the two Trp environments. The ΔH and ΔS values were determined such that the observed curvature of the logΦ(T) plot was reproduced (see Materials and Methods). Peptide 3 showed the highest enthalpy for unfolding and the largest gain in entropy (Fig. 6 A and Table 1). Peptide 1 showed the smallest values for both ΔH and ΔS, whereas peptides 2, 4, and 5 fell within an intermediate range. Thus, the ranking of peptides by thermal stability parallels their degree of helix-forming potential, as summarized in Table 1. Based on these data, we propose that Hinge 1 exhibits a central region with a large helical propensity (mainly sampled by peptide 3), flanked by less structured and less stable regions (mainly sampled by peptides 1, 2, and 5). The ΔH and ΔS values for the unfolding of the most stable and most helical stretch in Hinge 1 correspond to an equilibrium concentration of the unfolded state of ∼5% at room temperature. Interestingly, a probability in the low percentage range also describes the occurrence of the large-amplitude fluctuations in ΔT and full-length kinesin-1, suggesting a possible correlation between the thermal unfolding of Hinge 1 and the appearance of high-compliance, non-Brownian fluctuations.
TABLE 1.
Thermodynamic and structural parameters of Hinge 1 model peptides
| Peptide 1 | Peptide 2 | Peptide 3 | Peptide 4 | Peptide 5 | |
|---|---|---|---|---|---|
| ΔH (kJ) | 52.6 | 78.6 | 115.5 | 75.0 | 80.5 |
| ΔS (JK−1) | 141 | 244 | 367 | 221 | 254 |
| Helical propensity | m | l (m) | h (l) | h | l |
Unfolding enthalpies and entropies of Dm peptides were derived from the temperature sensitivity of florescence emission of tryptophan (see Eq. 4 and Eq. 6 in Materials and Methods, and Fig. 6) Helicity was determined by evaluation of CD spectra (Fig. 5) using CDSSTR (Dichroweb). The ranking of helical propensity is expressed as follows. High (h), helicity >35% without or >65% with TFE (32%) at 20°C; medium (m), helicity >15% at 20°C (no TFE) or >30% with TFE (32%) at 2°C; and low (l), helicity <15% under all conditions. Helical propensity of the Hs peptides is given in parentheses.
Homo-oligomerization of the model peptides
The structure and stability profile in Hinge 1 may support specific tertiary contacts in native kinesin where the corresponding sequences are aligned next to each other. We addressed this question by gel-filtration assays to determine the oligomerization state of the model peptides. Peptide 3 and 4 exhibit similar elution peaks (Fig. 6 B). The right-hand peak (∼12 mL) is the first elution, and corresponds to the monomeric state. The next peak (∼11 mL) elutes close to aprotinin and the dimeric insulin chains, suggesting the dimeric state of the peptide. Dimerization for peptides 3 and 4 corresponds roughly to 47% and 57%, respectively. Peptides 1, 2, and 5 elute earlier, as expected for unfolded peptides, which typically elute with a volume larger than expected from the nominal peptide mass. The results from gel filtration are consistent with the CD spectroscopic evidence that peptides 3 and 4 are α-helices, whereas peptides 1, 2, and 5 lack a defined structure.
DISCUSSION
We studied the functional role and structural properties of the Hinge 1 region adjacent to the kinesin neck. The deletion of Hinge 1 has no effect on the speed of microtubules driven by single kinesin molecules, indicating that it is not essential for hand-over-hand motility, at least under the low force conditions of our gliding assays. However, the deletion of Hinge 1 impairs microtubule motility at high kinesin densities. This suggests that Hinge 1 is required to provide sufficient flexibility to compensate for the strain generated when many motors are attached to, and moving, the same microtubule. Surprisingly, the compliance measurements of single kinesin molecules do not indicate a simple fit with this picture. Instead, all motor constructs spent most of their time in a low-flexibility state whose torsional rigidity was almost unaffected by the deletion of Hinge 1. The torsional stiffness of this state, 8 kBT, based on an angular variance of 7°, is similar to that expected of a coiled coil (assuming a persistence length of 100 nm, and a coiled coil length of 10 nm (25)). Rather than exhibiting continuous high compliance, the presence of Hinge 1 is associated with rare, non-Brownian angular fluctuations of large amplitude occurring on a time scale of seconds. The CD spectra show that the central part of the Hinge 1 sequence exhibits a high α-helix-forming potential, flanked by less ordered segments. The ensemble of these data demonstrates that Hinge 1 is required for motor cooperativity, but neither its structural nor mechanical properties are consistent with a disordered domain.
How can this be related to a structure-based understanding of the measured torsion elasticity? The presence of Hinge 1 allows transitions between two different torsional states. The predominant long-lived, low-amplitude Brownian fluctuations in the Hinge 1-containing proteins indicate that the hinge domains have low compliance. Such low compliance is consistent with the formation of secondary structure, which reduces flexibility within Hinge 1. Our spectroscopic data show that a strong helical propensity resides in the central region of Hinge 1, giving rise to low compliance because of stabilizing intrastrand interactions. We interpret the occurrence of an additional state with high compliance as evidence for rare unfolding events, leading to the formation of a more flexible, disordered domain that mechanically separates the coiled coil domain of the neck from that of the stalk. This non-Brownian response is specific to Hinge 1 because it is abolished in the Hinge 1 deletion construct ΔTΔH. Hence, the flexibility of Hinge 1, required for maximal speed at high motor density, is not directly reflected in its torsion elasticity. Instead, “rare” unfolding events appear to generate a state of high compliance that is observed in single-molecule experiments as an infrequent deviation from the overall coiled coil-like behavior. Our spectroscopic results provide a reasonable structural basis to reconcile the unexpected existence of secondary structure in Hinge 1 with the requirement of flexibility, for which a disordered domain intuitively appears to be the better candidate. The data provide evidence for a distinct profile of secondary structure-forming propensity in the Hinge 1 sequence, exhibiting a central part with a high tendency for helix formation and a large enthalpy of unfolding (peptides 3 and 4), approaching the reported mean H-bond enthalpies of ∼4 kJ per residue (39,40). The N-terminal and C-terminal extensions from the central region of Hinge 1 are essentially structureless, which is again paralleled by the reduced unfolding entropies. Thus, the mechanical, structural, and thermodynamic data are in agreement. The CD and fluorescence data clearly show the formation of secondary structure and the presence of intrastrand stabilizing enthalpic interactions, respectively, but do not indicate coiled coil formation. In full-length kinesin and ΔT, however, the two kinesin heavy chains are in close proximity, and favorably preoriented by the dimeric heavy chains. This may promote secondary structure formation and dimerization of the central ∼30-aa-long α-helical stretch within Hinge 1. Although interstrand interactions in the central hinge region cannot be proven or disproven based on the present peptide studies alone, such interactions would agree with the observed high-rigidity state of full-length kinesin-1 and ΔT. This hypothetical coiled coil-like arrangement is depicted in Fig. 1 E. If such a dimeric segment exists within Hinge 1, it is probably less stable than “canonical” coiled coil regions, and importantly, it lacks additional stabilization from the unstructured flanking domains. Therefore, we ascribe the less frequent high-compliance state of full-length kinesin and the ΔT construct to the unfolded state of Hinge 1, even when the rather low dimerizing potential seen with the isolated peptides becomes enhanced upon dimerization of the full-length kinesin-1 heavy chains.
Previous work on full-length bovine-brain kinesin, carried out in the absence of ATP, measured a higher compliance than in this study (16). A possible explanation for this discrepancy is that the earlier measurements were made over much longer times (tens of minutes rather than the tens of seconds here), such that the unfolded state(s) may have been sampled more frequently. On the other hand, Hua and Gelles also used nonzero ATP (5 and 400 nM) concentrations, and measured a root mean-square (rms) angle of 20–30° (26), some threefold larger than the rms we observed for the low-compliance state. The larger rms angle may also be due to greater sampling of the high-compliance state in their measurements, made at lower ATP concentrations over longer times.
Based on our functional and structural studies, we suggest that at high motor densities, the strain exerted by the force-generating motor domain in Hinge 1 induces a transition of the structured regions from helical to disordered stretches. We propose that with the increasing torque and strain exerted during multiple-motor motion, α-helical segments adjacent to the most stable central sequence will increasingly unfold. Finally, the entire Hinge 1 may adopt an entropic spring-like behavior, after the secondary structure has fully unfolded at larger torques, as is the case at high motor density. In this model, the Hinge 1 region can be thought of as a “flexibility reservoir” that is accessed by the gradual growth of flanking disordered domains in a strain-dependent unfolding process. Hence, reorientation of misaligned motor heads into a more favorable position for microtubule attachment can be achieved during motor cooperativity in a strain-dependent manner, as depicted in Fig. 7. The function-dependent increase in flexibility causes an overall reduction of drag during motor cooperation as the infrequently occurring high-compliance state seen in the single-molecule experiment becomes populated, thereby preventing motor interference. In contrast, in the absence of strain exerted by other motors, there is no functional requirement of either flexibility or secondary structure in the hinge for single-molecule microtubule attachment and processivity.
FIGURE 7.

Model of functional role of strain-dependent unfolding of Hinge 1 region during motor cooperation. (A) Under single-molecule conditions, kinesin can bind and perform its mechanochemical cycle without mechanical hindrance. (B) In a multimotor situation, a kinesin molecule bound to a surface or cargo has a given orientation that does not allow following the microtubule protofilament axis (α-tubulin, light gray; β-tubulin, dark gray). (C) If wild-type or ΔT binds with one head to the microtubule, the force exerted by other motors through the cargo unfolds Hinge 1, reducing drag. This will allow forward stepping because the neck and both head domains work independently of any other motor bound to the same surface or cargo. In contrast, if ΔTΔH binds to the microtubule, an attached but misaligned motor cannot step forward, and it will exert a force (F) against the motor ensemble mediated by the common cargo. Therefore, a reduction of speed (v), i.e., an increase in drag, will be observed. φ, nucleotide-free; D, ADP; DP, ADP·Pi.
Because of the divergence of the Hinge 1 sequence, it is difficult to predict whether 1), helical stretches are interspersed in the corresponding regions of other kinesins; and 2), whether similar flanking sequences exist that would support the unfolding of the encompassed helical structures during function. Remarkably, the homologous sequence of Hinge 1 from human kinesin-1 sampled by peptides 2 and 3 also exhibits a clear secondary structure-forming potential adjacent to an essentially unordered region (Table 1). The presence of a proline in the center of the human peptide 3 is probably the reason why its helicity is low, whereas a proline is situated in the more marginal position 7 in the more helical human peptide 2. In both organisms, the functionally required flexibility of Hinge 1 appears to originate in local alterations of helical propensity in a 15–30-amino-acid window, rather than being the consequence of a contiguous, intrinsically disordered domain.
Our hypothesis that the force generated by the motor domain could lead to partial unfolding of a protein sequence may apply in other situations. For example, studies of peptides from the dimerization domains of kinesin-1 provided evidence that the dimerization domain (also called the neck) might unfold during motility (28). Such unfolding would make it easier for the two heads in a kinesin dimer to span the 8 nm between adjacent tubulin dimers in the microtubule. However, later work showed that single kinesin-1 molecules could still move processively when dimerization was strengthened by using more stable coiled coils (41,42) or by cysteine-mediated cross-linking (43). This suggested that unfolding is not necessary for motility. However, if motor forces are capable of unwinding protein domains, it is possible that partial unfolding of the neck sustains normal processivity under load (42). Similarly, the model proposed here explains why the lack of the Hinge 1 in ΔTΔH does not affect the processivity of an individual motor, but causes a reduction in speed when many motors attach to the same microtubule but can no longer generate compliance in response to an increasing torque.
Acknowledgments
We thank Dr. Jörg Fitter from the Forschungszentrum Jülich for access to and support of the CD measurements. We also thank the members of the laboratory of J. Howard for their helpful comments on earlier versions of the manuscript.
A.H.C., D.N.C., M.W., and J.H. were supported by the Max Planck Society and the National Institutes of Health. S.M. and K.F. were supported by the Deutsche Forschungsgemeinschaft (grant Fa248).
Alvaro H. Crevenna's present address is the Max Planck Institute for Biochemistry, Am Klopferspitz 18, 81252 Martinsried, Germany.
Daniel N. Cohen's present address is the Medical Scientist Training Program, Vanderbilt University Medical School, Light Hall Box 111, Nashville, TN 37232.
Editor: David D. Hackney.
References
- 1.Howard, J., A. J. Hudspeth, and R. D. Vale. 1989. Movement of microtubules by single kinesin molecules. Nature. 342:154–158. [DOI] [PubMed] [Google Scholar]
- 2.Zhang, Y., and W. O. Hancock. 2004. The two motor domains of KIF3A/B coordinate for processive motility and move at different speeds. Biophys. J. 87:1795–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King, S. J., and T. A. Schroer. 2000. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat. Cell Biol. 2:20–24. [DOI] [PubMed] [Google Scholar]
- 4.Sakamoto, T., I. Amitani, E. Yokota, and T. Ando. 2000. Direct observation of processive movement by individual myosin V molecules. Biochem. Biophys. Res. Commun. 272:586–590. [DOI] [PubMed] [Google Scholar]
- 5.Meyhofer, E., and J. Howard. 1995. The force generated by a single kinesin molecule against an elastic load. Proc. Natl. Acad. Sci. USA. 92:574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hancock, W. O., and J. Howard. 1998. Processivity of the motor protein kinesin requires two heads. J. Cell Biol. 140:1395–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaseda, K., H. Higuchi, and K. Hirose. 2003. Alternate fast and slow stepping of a heterodimeric kinesin molecule. Nat. Cell Biol. 5:1079–1082. [DOI] [PubMed] [Google Scholar]
- 8.Asbury, C. L., A. N. Fehr, and S. M. Block. 2003. Kinesin moves by an asymmetric hand-over-hand mechanism. Science. 302:2130–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yildiz, A., J. N. Forkey, S. A. McKinney, T. Ha, Y. E. Goldman, and P. R. Selvin. 2003. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 300:2061–2065. [DOI] [PubMed] [Google Scholar]
- 10.Hancock, W. O., and J. Howard. 1999. Kinesin's processivity results from mechanical and chemical coordination between the ATP hydrolysis cycles of the two motor domains. Proc. Natl. Acad. Sci. USA. 96:13147–13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leopold, P. L., A. W. McDowall, K. K. Pfister, G. S. Bloom, and S. T. Brady. 1992. Association of kinesin with characterized membrane-bounded organelles. Cell Motil. Cytoskeleton. 23:19–33. [DOI] [PubMed] [Google Scholar]
- 12.Zhou, H. M., I. Brust-Mascher, and J. M. Scholey. 2001. Direct visualization of the movement of the monomeric axonal transport motor UNC-104 along neuronal processes in living Caenorhabditis elegans. J. Neurosci. 21:3749–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cole, D. G., D. R. Diener, A. L. Himelblau, P. L. Beech, J. C. Fuster, and J. L. Rosenbaum. 1998. Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J. Cell Biol. 141:993–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan, X., G. Ou, G. Civelekoglu-Scholey, O. E. Blacque, N. F. Endres, L. Tao, A. Mogilner, M. R. Leroux, R. D. Vale, and J. M. Scholey. 2006. Mechanism of transport of IFT particles in C. elegans cilia by the concerted action of kinesin-II and OSM-3 motors. J. Cell Biol. 174:1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Block, S. M., L. S. Goldstein, and B. J. Schnapp. 1990. Bead movement by single kinesin molecules studied with optical tweezers. Nature. 348:348–352. [DOI] [PubMed] [Google Scholar]
- 16.Hunt, A. J., and J. Howard. 1993. Kinesin swivels to permit microtubule movement in any direction. Proc. Natl. Acad. Sci. USA. 90:11653–11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheetz, M. P., and S. C. Kuo. 1993. Tracking nanometer movements of single motor molecules. Methods Cell Biol. 39:129–136. [DOI] [PubMed] [Google Scholar]
- 18.Leduc, C., F. Ruhnow, J. Howard, and S. Diez. 2007. From the cover: detection of fractional steps in cargo movement by the collective operation of kinesin-1 motors. Proc. Natl. Acad. Sci. USA. 104:10847–10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mehta, A. D., R. S. Rock, M. Rief, J. A. Spudich, M. S. Mooseker, and R. E. Cheney. 1999. Myosin-V is a processive actin-based motor. Nature. 400:590–593. [DOI] [PubMed] [Google Scholar]
- 20.Nishizaka, T., R. Seo, H. Tadakuma, K. Kinosita, Jr., and S. Ishiwata. 2000. Characterization of single actomyosin rigor bonds: load dependence of lifetime and mechanical properties. Biophys. J. 79:962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozielski, F., S. Sack, A. Marx, M. Thormahlen, E. Schonbrunn, V. Biou, A. Thompson, E. M. Mandelkow, and E. Mandelkow. 1997. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 91:985–994. [DOI] [PubMed] [Google Scholar]
- 22.de Cuevas, M., T. Tao, and L. S. Goldstein. 1992. Evidence that the stalk of Drosophila kinesin heavy chain is an alpha-helical coiled coil. J. Cell Biol. 116:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lupas, A., M. Van Dyke, and J. Stock. 1991. Predicting coiled coils from protein sequences. Science. 252:1162–1164. [DOI] [PubMed] [Google Scholar]
- 24.Wolf, E., P. S. Kim, and B. Berger. 1997. MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci. 6:1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howard, J. 2001. Mechanics of Motor Proteins and the Cytoskeleton. Sinauer Associates, Sunderland, MA.
- 26.Hua, W., J. Chung, and J. Gelles. 2002. Distinguishing inchworm and hand-over-hand processive kinesin movement by neck rotation measurements. Science. 295:844–848. [DOI] [PubMed] [Google Scholar]
- 27.Grummt, M., G. Woehlke, U. Henningsen, S. Fuchs, M. Schleicher, and M. Schliwa. 1998. Importance of a flexible hinge near the motor domain in kinesin-driven motility. EMBO J. 17:5536–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tripet, B., R. D. Vale, and R. S. Hodges. 1997. Demonstration of coiled-coil interactions within the kinesin neck region using synthetic peptides. Implications for motor activity. J. Biol. Chem. 272:8946–8956. [DOI] [PubMed] [Google Scholar]
- 29.Coy, D. L., W. O. Hancock, M. Wagenbach, and J. Howard. 1999. Kinesin's tail domain is an inhibitory regulator of the motor domain. Nat. Cell Biol. 1:288–292. [DOI] [PubMed] [Google Scholar]
- 30.Schief, W. R., R. H. Clark, A. H. Crevenna, and J. Howard. 2004. Inhibition of kinesin motility by ADP and phosphate supports a hand-over-hand mechanism. Proc. Natl. Acad. Sci. USA. 101:1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirstine, B.-S., and F. Henrik. 2004. Power spectrum analysis for optical tweezers. Rev. Sci. Instrum. 75:594–612. [Google Scholar]
- 32.Uyeda, T. Q., S. J. Kron, and J. A. Spudich. 1990. Myosin step size. Estimation from slow sliding movement of actin over low densities of heavy meromyosin. J. Mol. Biol. 214:699–710. [DOI] [PubMed] [Google Scholar]
- 33.Greenfield, N. J. 2004. Analysis of circular dichroism data. Methods Enzymol. 383:282–317. [DOI] [PubMed] [Google Scholar]
- 34.Kelly, S. M., and N. C. Price. 2000. The use of circular dichroism in the investigation of protein structure and function. Curr. Protein Pept. Sci. 1:349–384. [DOI] [PubMed] [Google Scholar]
- 35.Krimm, S., and J. Bandekar. 1986. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 38:181–364. [DOI] [PubMed] [Google Scholar]
- 36.Barth, A., and C. Zscherp. 2002. What vibrations tell us about proteins. Q. Rev. Biophys. 35:369–430. [DOI] [PubMed] [Google Scholar]
- 37.de Jongh, H. H., E. Goormaghtigh, and J. M. Ruysschaert. 1996. The different molar absorptivities of the secondary structure types in the amide I region: an attenuated total reflection infrared study on globular proteins. Anal. Biochem. 242:95–103. [DOI] [PubMed] [Google Scholar]
- 38.Reisdorf, W. C., Jr., and S. Krimm. 1996. Infrared amide I′ band of the coiled coil. Biochemistry. 35:1383–1386. [DOI] [PubMed] [Google Scholar]
- 39.Scholtz, J. M., S. Marqusee, R. L. Baldwin, E. J. York, J. M. Stewart, M. Santoro, and D. W. Bolen. 1991. Calorimetric determination of the enthalpy change for the alpha-helix to coil transition of an alanine peptide in water. Proc. Natl. Acad. Sci. USA. 88:2854–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheu, S. Y., D. Y. Yang, H. L. Selzle, and E. W. Schlag. 2003. Energetics of hydrogen bonds in peptides. Proc. Natl. Acad. Sci. USA. 100:12683–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romberg, L., D. W. Pierce, and R. D. Vale. 1998. Role of the kinesin neck region in processive microtubule-based motility. J. Cell Biol. 140:1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaud, J., F. Bathe, M. Schliwa, M. Rief, and G. Woehlke. 2006. Flexibility of the neck domain enhances kinesin-1 motility under load. Biophys. J. 91:1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomishige, M., and R. D. Vale. 2000. Controlling kinesin by reversible disulfide cross-linking. Identifying the motility-producing conformational change. J. Cell Biol. 151:1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tripet, B., and R. S. Hodges. 2002. Helix capping interactions stabilize the N-terminus of the kinesin neck coiled-coil. J. Struct. Biol. 137:220–235. [DOI] [PubMed] [Google Scholar]
- 45.Seeberger, C., E. Mandelkow, and B. Meyer. 2000. Conformational preferences of a synthetic 30mer peptide from the interface between the neck and stalk regions of kinesin. Biochemistry. 39:12558–12567. [DOI] [PubMed] [Google Scholar]
- 46.Berger, B., D. B. Wilson, E. Wolf, T. Tonchev, M. Milla, and P. S. Kim. 1995. Predicting coiled coils by use of pairwise residue correlations. Proc. Natl. Acad. Sci. USA. 92:8259–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones, D. T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292:195–202. [DOI] [PubMed] [Google Scholar]
- 48.Rost, B., and C. Sander. 1993. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 232:584–599. [DOI] [PubMed] [Google Scholar]