

Abstract
We have studied the initial phase of cell adhesion as a function of the lateral organization of individual integrin molecules with single-cell force microscopy. Nanostructures, consisting of hexagonally ordered gold dots, were prepared with diblock-copolymer micelle lithography and functionalized with arginine- glycine-aspartate peptides, thus defining integrin position with nanometer resolution. Adhesion strength was characterized with an atomic force microscope and both cell detachment forces and work of detachment showed a reinforcement of adhesion if the distance between integrin molecules was <70 nm. This reinforcement had already occurred at cell-substrate contact times <5 min. We believe our results show quantitatively the relevance of the distance between adjacent integrin binding sites rather than their density. Furthermore, we propose a model describing the cooperative stabilization of early integrin clusters as a function of receptor patterning at the nanoscale.
INTRODUCTION
Biological binding is mediated by noncovalent interactions between molecules of high structural complementarity. Due to their relatively low binding energies, single binding events are continually competing with thermal energy, resulting in short lifetimes and high unbinding probabilities. By clustering molecular interactions, however, much stronger mechanical connections can be established (1–3). Molecular clustering occurs in many biological processes; examples include the capture of leukocytes at the blood vessel wall (4), cadherin-mediated cell-cell adhesion (5), and the formation of focal contacts between cells and the extracellular matrix (6–8). The formation of focal contacts in cells is essentially controlled by the activation and clustering of the transmembrane protein integrin. This process is initiated when the integrins bind to the arginine-glycine-aspartate (RGD) sequence in extracellular matrix proteins, such as fibronectin. After the first binding step, the initial clustering of integrin molecules takes place (9) and secondary proteins, such as talin, vinculin, and α-actinin, accumulate in the cluster region and provide a connection between the initial integrin cluster and the cytoskeleton (6,10). Furthermore, these proteins stabilize the adhesion cluster by forming a network of protein interconnections inside the adhesion cluster (11).
Recent studies have shown that the nanoscale clustering of integrin receptors is of utmost importance for cell adhesion and motility (12,13). Due to advances in nanofabrication technology, nanostructured substrates can be produced that not only allow the positioning of molecular binding sites in a certain density, but also in defined patterns (14,15). In this way the spacing between single molecular binding sites can be controlled precisely (16). Recently, such substrates have been used to control the position of single integrin binding sites. With this approach, it has been shown that focal contact and actin stress fiber formation requires an integrin binding site spacing of <73 nm. At larger spacings a decreased cell proliferation rate and an enhanced apoptosis rate have been reported for a large variety of cell types, such as fibroblasts, osteoblasts, and melanocytes (17). Furthermore, cell spreading and focal contact dynamics have proven to be regulated by integrin binding site spacing (18,19). These results imply that structural complementarity of the binding partners is essential not only at the molecular level, but also at larger length scales, i.e., that the intermolecular spatial arrangement of bonds must meet certain requirements to allow cluster formation and the activation of biological functions. A similar phenomenon has been described for T-cells on the micrometer scale, where the spatial patterning of different transmembrane receptors in the synapse alters T-cell activation (20). In contrast to the mature phase of integrin-mediated adhesion, its very early steps and in particular the influence of receptor density and pattern are still poorly understood, although related aspects, such as the role of the glycocalix in early adhesion events, have been studied extensively (21). Only recently, first indications that receptor patterning can cooperatively influence αVβ3 integrin-mediated initial adhesion have been described (22). For the binding of α2β1 integrin to collagen type I, indications for an involvement of cooperativity in integrin binding have been found (23). There, a cooperative binding of the integrin molecules was observed in response to extended adhesion times on homogeneously coated substrates.
In this study, we report on the temporal development of the stability of early integrin-mediated adhesion and how it is cooperatively controlled by spatial receptor patterning. Our approach is based on using biofunctionalized nanostructures that allow controlling the distance between individual integrin molecules with nanometer precision. On such nanostructures, nanometer-sized gold dots are placed in a well-defined hexagonal pattern on a glass substrate with polymer micelle lithography. The region between the gold dots is passivated with a thin protein-resistant layer of poly(L-lysine)-graft-poly(ethylene glycol) (PLL-g-PEG) and the dots themselves are biofunctionalized with the cyclic RGD peptide c(RGDfK)-thiol to achieve a specific and high-affinity binding of integrin molecules to the gold dots. A sketch of the nano-template used in the experiments is shown in Fig. 1. Using these substrates we investigated the adhesion stability of rat embryonic fibroblasts (REF52) as a function of integrin binding site spacing by measuring cell detachment forces with an atomic force microscope (AFM) (Fig. 2). In particular, we focused our experiments on the very early cell-substrate contact, ranging from 5 s to 5 min, to study the role of receptor patterning for the onset of a cooperative stabilization of adhesion. The results of this study show that integrin spacing plays a key role for cell adhesion strength already in the very early stages of adhesion and that this process can be understood with a simple model that describes the dependence of binding stability on the spatial arrangement of integrins at the nanometer scale.
FIGURE 1.

(A) Scanning electron micrograph showing hexagonally ordered gold nanodots on a glass substrate (dot spacing: 55 ± 12 nm, scale bar: 200 nm) (B) Sketch of the adhesion template. The region between the gold dots is coated with a several nanometer thick layer of PLL-g-PEG.
FIGURE 2.

Experimental setup for initial adhesion experiments in the AFM. (A) Bright-field image of a cell immobilized at a cantilever. The cell is located in the center of the dashed circle. (B) Sketch of a cell experiment. The cell is immobilized at a cantilever and brought into contact with the RGD nanopattern.
MATERIALS AND METHODS
Surface preparation
Nanostructured substrates consisting of hexagonally ordered, nanometer-sized gold dots are prepared by the self-assembly of diblock-copolymer micelles, which contain a gold cluster in their core. After the self-assembly of such micelles on a glass substrate, the polymer coat of the micelles is removed by a hydrogen plasma treatment, which also deposits the gold nanoparticles on the surface. The resulting nanoparticles have a diameter of 6–8 nm. The details of this preparation are described elsewhere (15). The distances between single gold dots, d, can be adjusted by the size of the diblock-copolymers used. The precise structure of the pattern is found by scanning electron microscopy. In this study, we worked with distances of 35 ± 6, 55 ± 12, 70 ± 14, and 103 ± 15 nm. A scanning electron micrograph of a nanostructured glass substrate is shown in Fig. 1 A. To prevent nonspecific cell adhesion in the region between the gold dots, this area was covered with a protein-repellent layer of PLL(20)-g[3.5]-PEG(2) (SurfaceSolutions, Dübendorf, Switzerland) by incubating the substrates with a 0.1 mg/ml PLL-g-PEG solution in a HEPES buffer at pH 7.4 for 40 min (24). Previous studies have shown that the height of a hydrated layer of this PLL-g-PEG on the surface is 8.2 nm (25). The gold nanoparticles were functionalized with a cyclic RGD peptide via an alkane linker and a thiol group. For functionalization, a 25-μmol aqueous solution of this peptide was applied to the substrates for 4 h. The full peptide reads c[RGDfK(Ahx-Mpa)] and it was chosen because it serves as a high-affinity ligand for the αVβ3 integrin and is selective against αIIbβ3, the “platelet integrin” (17,26). Fig. 1 B shows a sketch of the surface template. Although several RGD molecules can coat a single gold dot, we assume that not more than one integrin binds per gold dot as the gold dot diameter is 6–8 nm and the integrin head domain has a size of 9 nm (27).
Cell culture
A fibroblast cell line (REF52, YFP Paxillin transfected) was used, which has already been used in a previous study (18). This is one of the standard cell lines used for experiments on the nanostructures. No effect of the transfection on the adhesion behavior of the cells on the nanostructures has been observed. The cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 2% L-glutamine at 37°C and 10% CO2. Because the AFM experiments were carried out in air, CO2- independent medium (buffering system: mono and dibasic sodium phosphate and β-glycerophosphate; Invitrogen (Karlsruhe, Germany) product 18045088) was used to avoid pH changes in the medium during the experiments. To avoid an alteration of the nanostructures by serum accumulation on the surface during the experiments, the medium used in the experiments contained only 2% fetal bovine serum and 2% L-glutamine. Furthermore, 100 μg/mL penicillin-streptomycin was added to reduce the risk for contaminations (all agents: Gibco, Eggenstein, Germany).
Single-cell force microscopy
The force measurements were carried out with an AFM optimized for live cell experiments and installed in conjunction with a standard fluorescence microscope (Nanowizard, CellHesion; JPK Instruments, Berlin, Germany; Axiovert 200 Microscope; Zeiss, Jena, Germany). For the experiments, tipless AFM cantilevers were used (Veeco, Mannheim, Germany; NP-020, MLCT-AU). For practical reasons, if not manufactured without tips, the cantilevers were deprived of their tip as described in Benoit and Gaub (28). The force constants were 0.03–0.3 N/m, as found by individual calibration of the cantilevers with the thermal noise method (29,30). Tipless AFM cantilevers were coated with concanavalin A (conA), which binds to the cellular glycocalix (24). Experiments were carried out in a heated fluid chamber at 36°C (BioCell, JPK Instruments), where a nanostructured substrate was fixed as a base of the chamber. A single cell was immobilized at the free end of the cantilever by picking it up from the surface with a conA-coated cantilever by approaching the cantilever to the cell at a force of a few nN and inducing a close contact for several seconds (Fig. 2 A). Before any experiments, the cell was left to recover for at least 15 min at the cantilever without being subject to any external stress to establish firm binding. In an experiment, the immobilized cell was brought into contact with the substrate (Fig. 2 B) at a load of 300–800 pN and after a defined time period it was detached at a cantilever retraction speed of 5 μm/s. A typical cell detachment curve is shown in Fig. 3 A. After each measurement, the cell was allowed to recover for at least the time span of the adhesive contact. The cell detachment force was calculated from the maximum deflection of the cantilever during cell detachment and the work of detachment was determined by integrating the force-distance curve (Fig. 3 A). All data analysis were carried out with home-written MATLAB (The MathWorks, Natick, MA) programs. To account for cantilever drift, we followed the method described in Franz et al. (31). The total detachment forces from at least five cell detachment curves per substrate type and cell-substrate contact time were evaluated. For small cell-substrate contact times <40 s no significant changes from one single detachment experiment to the other were observed. Thus the adhesion of such cells was probed frequently and the mean detachment forces were calculated from more than 20 cell detachment events. To verify the involvement of the integrin/RGD bond in the binding process, cells were incubated in a 1× PBS solution containing linear RGD (G1269, Sigma-Aldrich, Munich, Germany) for 30 min. A 350-μM RGD solution blocked most of the specific binding events. Furthermore, no specific binding events were observed for a nonfunctionalized PLL-g-PEG substrate (Fig. 3 B). Breakages between cellular polysaccharides and the conA proteins at the cantilever during cell detachment cannot be excluded per se. However, due to the comparably large contact area between cell and cantilever it was never observed that the cell-cantilever contact broke during our experiments, indicating that the majority of the measured force steps is indeed a result of the breakage of integrin/RGD bonds.
FIGURE 3.

Approach (black) and detachment (red) curves of cells from a nanostructured substrate with spacing 55 nm (A) and a pure PLL-g-PEG surface (B) after 5 s of cell-substrate contact. In A, detachment events of single molecules (sm), the total detachment force Ftot and the work of detachment W (gray region) are marked. In B, a reference measurement on a PLL-g-PEG coated, nonbiofunctionalized substrate is shown. There, only nonspecific binding was observed.
RESULTS AND DISCUSSION
Cell detachment from RGD nanopattern
In a single-cell force microscopy experiment, the forces during approach and detachment of cells to and from a substrate are recorded, respectively (Fig. 3). In contrast to a situation where a cantilever is pressed onto a rigid substrate, an elastic response of the cell is observed by the nonlinear force response during its approach to the surface (Fig. 3 A). The detachment curve in Fig. 3 A shows successive rupture events, which are attributed to the rupture of single bonds. A very important parameter for our study is the maximum adhesion force (Ftot, Fig. 3 A), which coincides with the total force necessary for cell detachment in this kind of experiments. As we focus our study on adhesion at the multimolecular level to quantify the total amount of adhesion, we are particularly interested in evaluating these total detachment forces. The detachment curve shown in Fig. 3 A also provides information about the work of detachment (gray area). Furthermore, we found that detachment events of single molecules are only observed on RGD-functionalized nanostructures, and for a PLL-g-PEG sample, purely nonspecific interactions are detected (Fig. 3 B).
Cell detachment forces and work of detachment are reinforced for integrin spacings smaller than 70 nm
To study the role of receptor patterning for the onset of a cooperative stabilization of adhesion, we characterized the temporal changes of initial cell adhesion by measuring cell detachment forces as a function of integrin binding site spacing (Fig. 4 A). Although the detachment force increases nonlinearly with time for all data sets, a significant difference is observed between nanostructures providing an integrin spacing smaller (solid circles and squares) or larger (open triangles and “×”) than 60 nm. At 35 and 55 nm spacings an increase in detachment force to ∼1 nN is observed within 40 s, whereas for larger spacings the cell detachment forces neither exceed 500 pN nor increase significantly with time. The differences in adhesion strength on the particular substrates increase with cell-substrate contact time, as shown in Fig. 4 B. There, cell detachment force is plotted as a function of integrin binding site spacing for cell-substrate contact times of 40 s and more. From this representation it is evident that adhesion is only significantly reinforced for d ≤ 55 nm and increases markedly with time. To test the effect of receptor patterning on a more comprehensive measure, we calculated the work of detachment as a function of cell-substrate contact time and binding site spacing (Fig. 4 C). This graph shows that for the work of detachment a similar reinforcement occurs for d ≤ 55 nm as for the cell detachment forces.
FIGURE 4.

(A) Cell detachment force versus cell-substrate contact time for different integrin binding site spacings. At 35 and 55 nm spacings the detachment force increases with time to 1 nN and more after ∼40 s. Detachment forces increase only slightly with time for 70 and 103 nm integrin spacings. No significant difference is observed between the forces at 70 and 103 nm spacings. Error bars: SE. Cell detachment force (B) and work of detachment (C) as a function of integrin binding site spacing and cell-substrate contact time. Weak binding is observed for spacings above a transition region (55 nm ≤ d ≤ 70 nm; gray zone), binding is reinforced for smaller spacings.
Recently, long-term experiments have shown that adhesion sites are only connected to actin stress fibers for d ≤ 55 nm (17,18). A difference in the cytoskeletal coupling of the integrin molecules, i.e., a more viscous coupling for d > 55 nm than for d ≤ 55 nm, implies that the amount of energy dissipated during cell detachment is larger for d > 55 nm than for d ≤ 55 nm. Such an additional amount of energy could obscure differences in the work of detachment for different spacings. Fantner et al. (32) showed that the adhesion energies measured for a system of two plates connected by a layer of proteins can exceed the sum of the individual binding energies by orders of magnitude. This is because an unbinding event might be followed by a relaxation that requires a viscous restretching. Such an effect can, in principle, also play a role in our cell experiments. Thus, the measured work of adhesion is a complex function of cell mechanics, binding energy, contact area, and possible active movements of the cell. However, a significant viscoelastic relaxation, and hence a change of the stiffness of the whole cell during detachment, can not be expected in our experiments due to the rather high detachment speed of 5 μm/s, i.e., the cell is detached within 1–2 s. Furthermore, we found that the coupling of integrins to cytoskeletal elements is weak compared to the c[RGDfK(Ahx-Mpa)]-integrin bond or even absent and no large-scale cytoskeletal rearrangements are taking place at the timescale of our experiments because membrane tethers are frequently pulled before unbinding events on all probed substrates. The phenomenon of tether-pulling has been described frequently in experiments on live cells (33).
Cell detachment forces and their relation to cell spreading
It has been reported recently that cell spreading depends significantly on the nanostructured substrates used (18,19). However, these studies investigated cell spreading on a very different timescale (i.e., several hours) compared to the timescale studied here. Such long-term experiments suffer from less experimental problems compared to initial adhesion studies, because studies on the hour scale can easily detect the cell spreading area with phase contrast microscopy. In contrast, in our study the cells remained in a spherical shape throughout the experiment and no spreading activity at all was observed. It is noteworthy that cell spreading is influenced by the extent of initial binding; if a lot of stable bonds form in the beginning of cell-substrate contact, adhesion is stabilized, the cell membrane can flatten, and additional bonds can be formed. This can, in turn, enhance the amount of cell spreading. In contrast, if the initial binding is too weak and unstable, the cell will not be able to spread out. Thus, initial binding strength and cell spreading show a complex interplay, which will only be possible to evaluate if surface-sensitive microscopy techniques, such as total internal reflection microscopy, are combined with force microscopy. However, the observation that initial integrin binding strongly affects adhesion strength can be a starting point for understanding the role of integrin positioning in cell spreading.
Cooperativity in initial integrin adhesion
If the integrin binding site density was the decisive factor for the increase in detachment force and not the distance of individual integrin molecules, the detachment force would be a linear function of binding site density. Instead, adhesion is nonlinearly reinforced for d ≤ 55 nm, as shown in a plot of cell detachment forces as a function of binding site density (see Fig. 6). We interpret this transition to result from a failure of the cooperative clustering of integrin molecules at d ≥ 70 nm. Such a cooperative interaction is necessary for the hierarchical assembly of the focal contact, where the integrin molecules are connected with each other. If the integrin molecules are positioned far apart, this linkage might fail. A prominent candidate for an integrin cross-linker is talin, an ∼60 nm long focal adhesion protein that provides two high-affinity binding sites for integrin in its antiparallel, homodimeric configuration (Fig. 5 A) (34). Furthermore, talin has proven to be involved in early adhesion events (35). An alternative candidate is α-actinin, which interconnects actin fibers and also carries an integrin binding unit (1). It has a length of 24 nm in its heterodimeric configuration (36) so that two α-actinin molecules and an actin fiber could interconnect two integrin proteins. We introduce a simple model that explains our experimental data by the failure of integrin cross-linking for certain integrin binding site spacings.
FIGURE 6.

Mean cell detachment force as a function of the density (bottom scale) and spacing (top scale) of integrin binding sites, including a fit curve provided by our model. The gray zone (55 nm ≤ d ≤ 70 nm) marks the cooperative transition in adhesion stability. The values used for the fit parameters are Fsm = 0.032 nN, A = 2 μm2, λ = 0.01147 1/s and F0 = 0.09 nN, which are typical for cell experiments. The scaling factors Sn(d > d0) = 0.02, Sn(d ≤ d0) = 0.1, and Sc(d ≤ d0) = 1 are heuristically assumed. The number of noncross-linked bonds and cross-linked bonds Nn,max(d) + Nc,max(d) is equal to the density of total binding sites in the cell-substrate contact area ρ(d)A. α changes with cell-substrate contact time and the fitted values for α are: α(5 s) = 1 ± 0.09, α(40 s) = 0.74 ± 0.02, α (160 s) = 0.68 ± 0.01, α (310 s) = 0.65 ± 0.01.
FIGURE 5.

(A) Sketch showing the integrin binding sites at the antiparallel talin homodimer (according to Critchley (34)). The high-affinity integrin binding site is located at the N-terminal talin headgroup. A third integrin binding site is found in the talin rod domain (48). However, in our model we only take into account the high-affinity integrin binding sites. (B) Bond stabilization by talin through integrin cross-linking at appropriate distances of integrin binding sites. The black lines correspond to stabilization through the integrin binding sites at both ends of the talin dimer. The red curve gives the bond stabilization probability used for fitting the experimental data.
To describe the kinetics of the cell-substrate contact we divide the total population of bonds into two categories: the intracellularly cross-linked bonds (c) and the noncross-linked bonds (n). Bonds can only be cross-linked, if the distance between them fits the cross-linker length, d0. Even for binding site spacings d ≤ d0 only molecules with a spacing of d0 can be cross-linked, the remaining bonds stay noncross-linked and their number depends on the available binding site density. The maximum number of cross-linked bonds, Nc,max(d), and the maximum number of noncross-linked bonds, Nn,max(d), are related to the binding site density, ρ(d), and the cell-substrate contact area, A, through Nn,max(d) + Nc,max(d) = ρ(d)A. Cross-linked bonds are more stable than noncross-linked bonds, thus we introduce the scaling factors Sc(d) and Sn(d) that describe the stability of individual cross-linked and noncross-linked bonds as a function of d, respectively. Then, the maximum number of stable bonds becomes N*c,max(d) = Sc(d)Nc,max(d) in the cross-linked case and N*n,max(d) = Sn(d)Nn,max(d) in the noncross-linked case. Cluster formation reduces membrane fluctuations and increases the rebinding rate of bonds, hence noncross-linked bonds are expected to be more stable at d ≤ d0 than at d > d0, so we take Sn(d ≤ d0) > Sn(d > d0). Due to the weakness of a single biological bond, we assume that only a fairly small percentage of the noncross-linked bonds is closed and that the closed bonds are highly dynamic. This should be permitted, as recently, an enormous binding dynamics and turnover has been shown for the molecules in a focal contact (37,38). Finally, we scale our data with the scaling factors Sn(d > d0) = 0.02 and Sn(d ≤ d0) = 0.1. Sn(d) is defined as a step function because we expect the stability of noncross-linked bonds to be highly influenced by cluster formation, which can only take place for d ≤ d0.
To describe the time evolution of adhesion, we assume that the number of stable bonds (b) formed per time, is in direct proportion to the number of free binding sites (f), i.e., dNc,b(t,d)/dt = λNc,f(t,d) for cross-linked and dNn,b(t,d)/dt = λNn,f(t,d) for noncross-linked bonds. Lambda describes the binding rate of an individual bond and is hence not a function of d. The total number of free and occupied binding sites is constant for each bond type, i.e., N*c,max(d) = Nc,f(t,d) + Nc,b(t,d) and N*n,max(d) = Nn,f(t,d) + Nn,b(t,d), thus the total number of stable bonds as a function of time and binding site spacing becomes
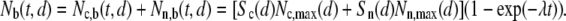 |
This description assumes that the number of integrin molecules is not a limiting factor, a condition that should be fulfilled as diffusion leads to a continuous integrin transport into the contact zone (37) and receptor densities in the cell membrane of up to 1000/μm2 have been reported (39).
With the talin as relevant cross-linking protein, substrates providing 60, 34, 30, and 17 nm integrin spacing, for example, can reinforce adhesion, because then the spacing between direct or higher-order integrin neighbors equals the length of the talin cross-linker. Thus, we let Sc(d) = 1 for these spacings, representing a maximum stability of the bond. Furthermore, we account for fluctuations of the integrin ligands on the substrate. In our experiments, the integrin ligands (the RGD molecules) are bound to the gold dots via a linker of ∼1.1 nm length. A surface of ∼2 nm radius is available for integrin attachment at a gold dot. In consequence, integrins can even be cross-linked if they are bound to gold dots that are off the ideal position for cross-linking. In the model, this is accounted for by describing Sc(d) with normal distributions around the ideal binding site positions with a standard deviation of 4 nm. According to this model, adhesion should be reinforced at several dot distances below 60 nm (Fig. 5 B), a hypothesis that has not yet been investigated experimentally. For simplicity, we set this issue aside for this study and let Sc(d ≤ d0) = 1.
The model described thus far requires that integrin molecules are available at any binding site with equal probability. This model does not fit the data very well, presumably because the random walk that the integrins carry out while diffusing in the membrane is blocked by an increasing number of obstacles (i.e., bonds, clusters) in the contact zone for d ≤ d0. Without any obstacles, the integrins could carry out this random walk with their mean-square displacement being proportional to the time diffused, according to the normal description of a random walk: 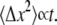 However, in the general diffusion situation the mean-square displacement is described by
However, in the general diffusion situation the mean-square displacement is described by 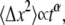 where α = 1 for normal diffusion, α > 1 for superdiffusion, and α < 1 for subdiffusion (40,41). In our situation, where integrin diffusion can be disturbed by obstacles, subdiffusion is expected to occur (42). Consequently, a receptor needs more time to find free binding sites compared to normal diffusion. In our model we account for this effect by introducing the effective time, tα, which is a measure for the area the receptor could scan for a new binding site in time t. α decreases with cell-substrate contact time as more and more obstacles arise in the cell-substrate contact region, thus interfering with receptor diffusion. The existence of such subdiffusive effects in the context of molecular crowding has been reported recently (43) and is theoretically well-described (44).
where α = 1 for normal diffusion, α > 1 for superdiffusion, and α < 1 for subdiffusion (40,41). In our situation, where integrin diffusion can be disturbed by obstacles, subdiffusion is expected to occur (42). Consequently, a receptor needs more time to find free binding sites compared to normal diffusion. In our model we account for this effect by introducing the effective time, tα, which is a measure for the area the receptor could scan for a new binding site in time t. α decreases with cell-substrate contact time as more and more obstacles arise in the cell-substrate contact region, thus interfering with receptor diffusion. The existence of such subdiffusive effects in the context of molecular crowding has been reported recently (43) and is theoretically well-described (44).
Finally, the force can be calculated from the number of closed bonds by multiplying it with the rupture force of a single bond, Fsm, and adding a constant force, F0, that accounts for nonspecific cell-substrate interactions. F0 is a fit parameter and its value corresponds to the values measured for the nonspecific interaction between a cell and a layer of PLL-g-PEG. Corrections introduced by cluster theories (45) and the relative stiffness between the bonds, the matrix and the adhesion plaque (46) are neglected here because both cluster and adhesion plaque size are small in early adhesion contacts compared to mature focal contacts. The final fit function for the cell detachment forces reads F(t, d, α) = Nb(tα, d) Fsm + F0, which describes the experimental data very well (Fig. 6). The fit also gives a temporal decrease of the exponent α, which could be expected due to the considerations presented above. For a typical contact zone of A = 2 μm2 on a substrate with 55 nm gold dot spacing, 750 binding sites are available and the measured force for t = 310 s corresponds to ∼60 bonds. This relates to an obstacle density of 8%, where a decrease of the diffusion coefficient is indeed predicted by simulations (44).
CONCLUSIONS
The aim of this study was to investigate the effect of nanoscale receptor patterning on the initial phase of cell adhesion. Focal contact formation and cell spreading are influenced strongly by the spacing of integrin binding sites (17,18), but an important question left open by previous studies is at what timescale variations in integrin binding site spacing start to affect cell adhesion. To answer this question, a characterization of interaction forces between cells and nanopatterns can provide important information. Thus, we used single-cell force microscopy to resolve cooperative integrin interactions during the first seconds and minutes of adhesion. This is long before conventional optical techniques are capable of detecting adhesion clusters. With our approach, we can show that nanoscale receptor patterning controls the reinforcement of adhesion already in the initial phase of cell adhesion. At this stage, few bonds have formed, compared to mature focal contacts, where thousands of molecules are involved and where even a connection between the integrins and the cytoskeleton is present. We believe our results reflect the amazingly fast nature of adhesion processes and adhesion cluster formation and prove that the spatial arrangement of single bonds is already important in the very early stages of adhesion. This finding is in agreement with earlier experiments, where first indications for the role of receptor patterning in initial adhesion were reported (22). Previously, the role of clusters for adhesion reinforcement has only been known for much longer adhesion times of 8 h (13) and for homogeneously coated substrates (23). In all these studies, the precise geometry of integrin binding was not controlled. Consequently, the situation described in our investigation must be clearly distinguished from these previous investigations.
Early fibroblast adhesion on homogeneous fibronectin substrates has been studied by Garcia and Boettiger for the first 15 min of adhesion (47). They report the absence of cooperative effects during early integrin binding. Instead, a linear increase of bound integrins with fibronectin density was observed. This is no surprise. In contrast to our measurements on nanostructures, the usage of homogeneously coated fibronectin surfaces only allows a definition of the global integrin binding site density. The distance of single integrin binding sites cannot be tuned. This is an essential aspect, because integrin clustering is assumed to be switched on and off by integrin binding site spacing, and not necessarily by the global integrin binding site density (17). For studying integrin clustering, nanopatterns are a unique tool because they allow local binding site densities and spacings to be imprinted precisely onto the surface. Although our results are well-explained by the cross-linking of individual integrin molecules with an intracellular linker protein, a detailed molecular model remains necessary for a deeper understanding of the dynamics and stability of cell adhesion in the context of receptor patterning. We believe our experimental setup, and in particular the usage of nanostructured substrates, makes it possible for the first time to quantify the cooperative strengthening of adhesion from the nonclustered to the clustered integrin configuration during the first 5 min of adhesion. We feel that this result is very far-reaching because no other experiment has thus far studied the effect of cooperative integrin clustering due to the intermolecular spatial arrangement in early cell adhesion events.
Acknowledgments
We thank E. Sackmann, B. Geiger, J. Vörös, U. S. Schwarz, A.-S. Smith and I. Dunlop for stimulating discussions. M. Kapp, M. Sycha and I. Grigoridis at the Max Planck Institute for Metals Research are acknowledged for the preparation of nanostructures.
The project was financially supported by the Landesstiftung Baden-Württemberg (Spitzenprogramm Baden-Württemberg), EU-IP NanoEar, NIH Roadmap for Medical Research (PN2 EY016586), the Alfried-Krupp von Bohlen und Halbach-Stiftung, the German Excellence Initiative (IGSSE and CIPSM) and the Max-Planck-Society. C. S.-U. was supported by the Boehringer Ingelheim Fonds, M. L.-G. holds a fellowship from the Alexander von Humboldt Foundation.
Christine Selhuber-Unkel's present address is The Niels Bohr Institute, University of Copenhagen, Denmark.
Editor: Michael Edidin.
References
- 1.Geiger, B., A. Bershadsky, R. Pankov, and K. M. Yamada. 2001. Transmembrane extracellular matrix-cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2:793–805. [DOI] [PubMed] [Google Scholar]
- 2.Mammen, M., S.-K. Choi, and G. M. Whitesides. 1998. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 37:2754–2794. [DOI] [PubMed] [Google Scholar]
- 3.Kiessling, L. L., J. E. Gestwicki, and L. E. Strong. 2006. Synthetic multivalent ligands as probes of signal transduction. Angew. Chem. Int. Ed. 45:2348–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dwir, O., A. Solomon, S. Mangan, G. S. Kansas, U. S. Schwarz, and R. Alon. 2003. Avidity enhancement of L-selectin bonds by flow: shear-promoted rotation of leukocytes turn labile bonds into functional tethers. J. Cell Biol. 163:649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adams, C., W. Nelson, and S. Smith. 1996. Quantitative analysis of cadherin-catenin-actin reorganization during development of cell-cell adhesion. J. Cell Biol. 135:1899–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geiger, B., and A. Bershadsky. 2001. Assembly and mechanosensory function of focal contacts. Curr. Opin. Cell Biol. 13:584–592. [DOI] [PubMed] [Google Scholar]
- 7.Zaidel-Bar, R., M. Cohen, L. Addadi, and B. Geiger. 2004. Hierarchical assembly of cell-matrix adhesion complexes. Biochem. Soc. Trans. 32:416–420. [DOI] [PubMed] [Google Scholar]
- 8.Zamir, E., M. Katz, Y. Posen, N. Erez, K. M. Yamada, B. Z. Katz, S. Lin, A. Bershadsky, Z. Kam, and B. Geiger. 2000. Dynamics and segregation of cell-matrix adhesions in cultured fibroblasts. Nat. Cell Biol. 2:191–196. [DOI] [PubMed] [Google Scholar]
- 9.Gottschalk, K.-E., and H. Kessler. 2004. A computational model of transmembrane integrin clustering. Structure. 12:1109–1116. [DOI] [PubMed] [Google Scholar]
- 10.Vogel, V., and M. Sheetz. 2006. Local force and geometry sensing regulate cell functions. Nat. Rev. Mol. Cell Biol. 7:265–275. [DOI] [PubMed] [Google Scholar]
- 11.Cluzel, C., F. Saltel, J. Lussi, F. Paulhe, B. A. Imhof, and B. Wehrle-Haller. 2005. The mechanisms and dynamics of (α)v(β)3 integrin clustering in living cells.. J. Cell Biol. 171:383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maheshwari, G., G. Brown, D. Lauffenburger, A. Wells, and L. Griffith. 2000. Cell adhesion and motility depend on nanoscale RGD clustering. J. Cell Sci. 113:1677–1686. [DOI] [PubMed] [Google Scholar]
- 13.Koo, L., D. Irvine, A. M. Mayes, D. Lauffenburger, and L. Griffith. 2002. Co-regulation of cell adhesion by nanoscale RGD organization and mechanical stimulus. J. Cell Sci. 115:1423–1433. [DOI] [PubMed] [Google Scholar]
- 14.Spatz, J. P., S. Mößmer, C. Hartmann, M. Möller, T. Herzog, M. Krieger, H. Boyen, P. Ziemann, and B. Kabius. 2000. Ordered deposition of inorganic clusters from micellar block copolymer films. Langmuir. 16:407–415. [Google Scholar]
- 15.Glass, R., M. Möller, and J. P. Spatz. 2003. Block copolymer micelle nanolithography. Nanotechnology. 14:1153–1160. [Google Scholar]
- 16.Wolfram, T., F. Belz, T. Schoen, and J. P. Spatz. 2007. Site-specific presentation of single recombinant proteins in defined nanoarrays. Biointerphases. 2:44–48. [DOI] [PubMed] [Google Scholar]
- 17.Arnold, M., E. A. Cavalcanti-Adam, R. Glass, J. Blümmel, W. Eck, M. Kantlehner, H. Kessler, and J. P. Spatz. 2004. Activation of integrin function by nanopatterned adhesive interfaces. ChemPhysChem. 5:383–388. [DOI] [PubMed] [Google Scholar]
- 18.Cavalcanti-Adam, E. A., R. Volberg, A. Micoulet, H. Kessler, B. Geiger, and J. P. Spatz. 2007. Cell spreading and focal adhesion dynamics are regulated by spacing of integrin ligands. Biophys. J. 92:2964–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cavalcanti-Adam, E. A., A. Micoulet, J. Blümmel, J. Auernheimer, H. Kessler, and J. P. Spatz. 2006. Lateral spacing of integrin ligands influences cell spreading and focal adhesion assembly. Eur. J. Cell Biol. 85:219–224. [DOI] [PubMed] [Google Scholar]
- 20.Mossman, K., G. Campi, J. Groves, and M. Dustin. 2005. Altered TCR signaling from geometrically repatterned immunological synapses. Science. 310:1191–1193. [DOI] [PubMed] [Google Scholar]
- 21.Cohen, M., D. Joester, B. Geiger, and L. Addadi. 2004. Spatial and temporal sequence of events in cell adhesion: from molecular recognition to focal adhesion assembly. ChemBioChem. 5:1393–1399. [DOI] [PubMed] [Google Scholar]
- 22.Walter, N., C. Selhuber, J. Blümmel, H. Kessler, and J. P. Spatz. 2006. Cellular unbinding forces of initial adhesion processes on nanopatterned surfaces probed with magnetic tweezers. Nano Lett. 6:398–402. [DOI] [PubMed] [Google Scholar]
- 23.Taubenberger, A., D. A. Cisneros, J. Friedrichs, P.-H. Puech, D. J. Muller, and C. M. Franz. 2007. Revealing early steps of alpha2beta1 integrin-mediated adhesion to collagen type I by using single-cell force spectroscopy. Mol. Biol. Cell. 18:1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lussi, J. W., D. Falconnet, J. A. Hubbell, M. Textor, and G. Csucsd. 2006. Pattern stability under cell culture conditions—a comparative study of patterning methods based on PLL-g-PEG background passivation. Biomaterials. 27:2534–2541. [DOI] [PubMed] [Google Scholar]
- 25.Pasche, S., M. Textor, L. Meagher, N. D. Spencer, and H. J. Griesser. 2005. Relationship between interfacial forces measured by colloid–probe atomic force microscopy and protein resistance of poly(ethylene glycol)-grafted poly(L-lysine) adlayers on niobia surfaces. Langmuir. 21:6508–6520. [DOI] [PubMed] [Google Scholar]
- 26.Pfaff, M., K. Tangemann, B. Müller, M. Gurrath, G. Müller, H. Kessler, R. Timpl, and J. Engel. 1994. Selective recognition of cyclic RGD peptides of NMR defined conformation by alphaIIbbeta3, alphaVbeta3, and alpha5beta1 integrins. J. Biol. Chem. 269:20233–20238. [PubMed] [Google Scholar]
- 27.Xiong, J.-P., T. Stehle, B. Diefenbach, R. Zhang, R. Dunker, D. L. Scott, A. Joachimiak, S. L. Goodman, and M. A. Arnaout. 2001. Crystal structure of the extracellular segment of integrin αVβ3. Science. 294:339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benoit, M., and H. E. Gaub. 2002. Measuring cell adhesion forces with the atomic force microscope at the molecular level. Cells Tissues Organs. 172:174–189. [DOI] [PubMed] [Google Scholar]
- 29.Hutter, J. L., and J. Bechhofer. 1994. Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 64:1868–1873. [Google Scholar]
- 30.Cleveland, J., S. Manne, D. Bocek, and P. Hansma. 1993. A nondestructive method for determining the spring constant of cantilevers for scanning force microscopy. Rev. Sci. Instrum. 64:403–405. [Google Scholar]
- 31.Franz, C. M., A. Taubenberger, P.-H. Puech, and D. J. Muller. 2007. Studying integrin-mediated cell adhesion at the single-molecule level using AFM force spectroscopy. Sci. STKE. 2007:pl5. [DOI] [PubMed] [Google Scholar]
- 32.Fantner, G. E., T. Hassenkam, J. H. Kindt, J. C. Weaver, H. Birkedal, J. A. Cutroni, G. A. G. Cidade, G. D. Stucky, D. E. Morse, and P. K. Hansma. 2005. Sacrificial bonds and hidden length dissipate energy as mineralized fibrils separate during bone fracture. Nat. Mater. 4:612–616. [DOI] [PubMed] [Google Scholar]
- 33.Schmitz, J., M. Benoit, and K.-E. Gottschalk. 2008. The viscoelasticity of membrane tethers and its importance for cell adhesion. Biophys. J. 95:1448–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Critchley, D. 2000. Focal adhesions—the cytoskeletal connection. Curr. Opin. Cell Biol. 12:133–138. [DOI] [PubMed] [Google Scholar]
- 35.Jiang, G., G. Giannone, D. R. Critchley, E. Fukumoto, and M. P. Sheetz. 2003. Two-picoheadgroupton slip bond between fibronectin and the cytoskeleton depends on talin. Nature. 424:334–337. [DOI] [PubMed] [Google Scholar]
- 36.Sjöblom, B., A. Salmazo, and K. Djinoviæ-Carugo. 2008. α-Actinin structure and regulation. Cell. Mol. Life Sci. 65:2688–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ballestrem, C., B. Hinz, B. A. Imhof, and B. Wehrle-Haller. 2001. Marching at the front and dragging behind: differential alphaVbeta3-integrin turnover regulates focal adhesion behavior. J. Cell Biol. 155:1319–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Digman, M. A., C. M. Brown, A. R. Horwitz, W. W. Mantulin, and E. Gratton. 2008. Paxillin dynamics measured during adhesion assembly and disassembly by correlation spectroscopy. Biophys. J. 94:2819–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akiyama, S. K., and K. M. Yamada. 1985. The interaction of plasma suspension fibronectin with fibroblastic cells in suspension. J. Biol. Chem. 260:4492–4500. [PubMed] [Google Scholar]
- 40.Saxton, M. J., and K. Jacobson. 1997. Single-particle tracking: applications to membrane dynamics. Annu. Rev. Biophys. Biomol. Struct. 26:373–399. [DOI] [PubMed] [Google Scholar]
- 41.Metzler, R., and J. Klafter. 2000. The random walk's guide to anomalous diffusion: a fractional dynamics approach. Phys. Rep. 339:1–77. [Google Scholar]
- 42.Saxton, M. 1993. Anomalous diffusion due to obstacles: a Monte Carlo study. Biophys. J. 66:394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weiss, M., M. Elsner, F. Kartberg, and T. Nilsson. 2004. Anomalous subdiffusion is a measure for cytoplasmic crowding in living cells. Biophys. J. 87:3518–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Höfling, F., T. Franosch, and E. Frey. 2006. Localization transition of the three-dimensional Lorentz model and continuum percolation. Phys. Rev. Lett. 96:165901. [DOI] [PubMed] [Google Scholar]
- 45.Erdmann, T., and U. S. Schwarz. 2004. Adhesion clusters under shared linear loading: a stochastic analysis. Europhys. Lett. 66:603–609. [Google Scholar]
- 46.Qian, J., J. Wang, and H. Gao. 2008. Lifetime and strength of adhesive molecular bond clusters between elastic media. Langmuir. 24:1262–1270. [DOI] [PubMed] [Google Scholar]
- 47.Garcia, A. J., and D. Boettiger. 1999. Integrin-fibronectin interactions at the cell-material interface: initial integrin binding and signaling. Biomaterials. 20:2427–2433. [DOI] [PubMed] [Google Scholar]
- 48.Calderwood, D. A. 2004. Talin controls integrin activation. Biochem. Soc. Trans. 32:434–437. [DOI] [PubMed] [Google Scholar]