

Abstract
Objective
Most measures of Aβ are elevated in Alzheimer’s Disease (AD) brain, but correlate inconsistently with disease severity. Since specific forms of Aβ may differentially correlate with clinical features, we segregated Aβ into distinct biochemical pools which may be enriched in biologically-relevant forms of Aβ.
Design
Clinical-pathological correlation
Subjects
27 subjects from a longitudinal study of AD, and 13 age- and gender- matched controls without known history of cognitive impairment or dementia.
Interventions
Temporal and cingulate neocortex were processed using a 4-step extraction, yielding biochemical fractions which are hypothesized to be enriched with proteins from distinct anatomical compartments: Tris (extracellular-soluble), Triton (intracellular), SDS (membrane-associated), Formic Acid (FA) (extracellular-insoluble). Aβ40 and Aβ42 were quantified in each biochemical compartment by ELISA.
Results
Aβ42 from all biochemical compartments was significantly elevated in AD cases vs. controls (p < 0.01). Aβ40 in the Tris and FA fractions were elevated in AD (temporal, p < 0.01; cingulate, p = 0.03), however Triton and SDS Aβ40 were similar in AD and controls. Functional impairment proximal to death correlated with Triton Aβ42 (r = 0.482, p = 0.015) and SDS Aβ42 (r = 0.409, p = 0.042) in temporal cortex. Faster cognitive decline was associated with elevated temporal SDS Aβ42 (p < 0.001) , while slower decline was associated with elevated cingulate FA Aβ42 and SDS Aβ42 (p = 0.02, p = 0.01).
Conclusions
Intracellular and membrane-associated Aβ, especially Aβ42 in temporal neocortex, may be more closely related to AD symptoms than other measured Aβ species.
Introduction
A critical role of the amyloid-forming Aβ peptide in the pathophysiology of Alzheimer’s Disease (AD) has been supported by human, animal, and in vitro studies.1 Most measures of Aβ are markedly elevated in AD brain, yet the extent of total A® accumulation tends to correlate poorly with AD severity.2–4 Since there is now evidence that specific biochemical forms of Aβ (e.g. Aβ42, soluble Aβ, oligomeric Aβ) selectively lead to neuronal dysfunction and neurodegeneration,5–7 and can be more reliable correlates of clinical status,8, 9 identification and reliable measurement of these toxic Aβ species should enhance their utility as biological markers of disease.
Clarifying the dynamics of Aβ production and compartmentalization is also necessary to explain AD pathogenesis. Specific Aβ species may preferentially exert toxic effects as a function of their cellular location. Whereas established histological techniques identify primarily insoluble extracellular and vascular amyloid deposits, novel methods can enhance detection of intraneuronal Aβ, distinguish Aβ pools, and measure changes in Aβ concentration and location over time.10–14
Aβ in brain can be segregated into distinct biochemical compartments defined by sequential extraction procedures. In this study of brain autopsy samples from a well-characterized longitudinal cohort of AD subjects and matched controls, we quantified Aβ40 and Aβ42 in biochemical compartments defined by their solubility in four solutions. Proteins in these biochemical pools are predicted to derive from distinct anatomical compartments within cerebral cortex: extracellular-soluble, intracellular, membrane-associated, and extracellular-insoluble.7 We hypothesized that these measures would differentially correlate with disease diagnosis, progression, and severity.
Methods
Participants
The sample derives from the Predictors Study, consisting of AD subjects recruited at the mild to moderate stage and evaluated every six months at one of three academic centers. The inclusion and exclusion criteria and evaluation procedures have been fully described elsewhere, and were approved by the respective institutional review boards.15 At entry, all patients met NINCDS-ADRDA criteria for probable AD and had a modified Mini-Mental State Examination (mMMSE) score of ≥30 (equivalent to ≥16 on the Folstein MMSE). 27 autopsy cases with confirmed AD pathology were included in the current study. 13 brains of individuals of similar age and gender who were free of neurodegenerative disease by clinical or pathologic criteria were selected as controls.
Biochemical compartmentalization
At autopsy, coronal slices from one hemisphere and hemibrainstem were fresh frozen between dry ice-cooled aluminium plates. A 1 cm strip of cortex was dissected from frozen temporal neocortex and cingulate cortex and mechanically homogenized. A four step extraction was utilized.13 The tissue was first extracted in 14 ul / mg wet weight Tris buffer, pH 7.2, (50mM Tris, 200 mM NaCl, 2 mM EDTA, complete protease inhibitors) with 2% protease-free BSA. After centrifugation (15,000 rpm, 21,000 × g, 4°C, 5 min), the supernatant was retained as the Tris-soluble fraction. The pellet was rehomogenized with Tris extraction buffer containing 0.1% Triton X-100, spun (15,000 rpm, 21,000 × g, 4°C, 5 min) and the supernatant retained as the Triton-soluble fraction. The remaining pellet was homogenized in 2% SDS, spun, and the supernatant saved as the SDS-soluble fraction. The remaining pellet was homogenized in 70% formic acid (FA), recentrifuged (22,000 rpm, 44,000 × g, 4°C, 5 min) and the resulting FA-extracted supernatant was neutralized with 1M Tris buffer (pH 11.0), representing the FA-extracted fraction.
These fractions are defined by their biochemical properties, however they are predicted to contain proteins from distinct cellular compartments: extracellular-soluble (Tris), intracellular-soluble (Triton), membrane-associated (SDS), and insoluble (FA) proteins, respectively. Lesne, et al., demonstrated that the Tris fraction was enriched for the extracellular proteins sAPPa and tPA; the Triton fraction was enriched for intracellular proteins c-jun, tau, ERKs, and JNK; the SDS fraction was enriched for full length APP and NMDA receptor subunit NR2 suggesting a membrane protein enriched fraction. The FA fraction contained flotillin-2, suggesting that lipid raft domains as well as insoluble proteins may be enriched in that fraction.7 While the fractions are enriched for proteins from specific cellular compartments, they are unlikely to correspond precisely to these cellular compartments, and Aβ may spill over between biochemical and cellular compartments during the extraction procedure.
Aβ quantification
Aβ40 and Aβ42 in each fraction were determined by sandwich ELISA using capture antibody BNT77 (anti-Aβ11–28) and detector antibodies BA27 (anti-Aβ40) and BC05 (anti-Aβ42) according to published protocols.13, 16 Thus, using two brain regions, four biochemical fractions, and two ELISA assays, sixteen Aβ variables were generated for each subject.
Clinical Measures
Cognition was assessed using the mMMSE.17 Modifications to the Folstein MMSE18 include the addition of digit span forward and backward,19 two calculation items, recall of recent U.S. Presidents, 10 items from the Boston Naming Test,20 one sentence to repeat, one written command and two figures to copy. The mMMSE has a maximum of 57 points with lower scores indicating poorer cognitive function. We used the Blessed Dementia Rating Scale (BDRS) Parts I (IADLs) and II (BADLs) to assess patients’ functional capacity. This is a 17-point scale with higher scores indicating worse functional status.21 Illness duration is the sum of the neurologist’s estimate of duration of symptoms at intake and the time from study entry to death.
Statistical analysis
Cases and controls were compared for group differences in demographics. The means of the 16 Aβ variables were then compared in the two groups using t-tests. Our approach to limiting the liabilities associated with multiple comparisons included applying a p-value of 0.01 to identify Aβ variables which reliably differed between cases and controls. The ten variables which met this criterion are included in subsequent analyses. This approach also addressed the conceptual difficulty of interpreting the significance of an amyloid-related measure which is purported to relate to clinical features of AD, but does not significantly differ from controls.
The Aβ variables were then compared in AD cases with 0, 1, or 2 APOE-ε4 alleles using one-way ANOVA and post-hoc LSD. For cross-sectional analysis, linear regression was used to relate the Aβ measures and clinical features of the AD cases. After log transformation of the data to better approximate normal distributions, the results of the analyses were essentially unchanged; we present the untransformed data.
Rates of cognitive decline were compared in groups of AD cases dichotomized at the median of each of the Aβ measures. Since we were interested in declines in mMMSE which eventuated in high or low Aβ levels at autopsy, the time scale was reversed. Thus, date of death is defined as time zero, and preceding evaluations have positive time values. Analyses of the longitudinal data were carried out by applying generalized estimating equations (GEE).22 GEE takes into account that each subject’s multiple mMMSE measurements are likely to be correlated. In our model, time, Aβ (high/low), and the time×Aβ interaction were included as independent variables. mMMSE was the dependent variable. As a result, all subjects had positive regression coefficients for the time; this corresponds to a decrease in cognition in chronological time, with higher coefficients indicating more rapid decline. A significant time×Aβ interaction term indicates a differential rate of decline in subjects with high or low Aβ measured at autopsy.
The cross-sectional and longitudinal analyses were repeated with gender and age at death included as covariates. The findings were nearly identical; the unadjusted analyses are presented.
Results
Clinical features of the 27 autopsy-confirmed AD cases are summarized in Table 1. There were 5 male and 8 female controls with a mean age at death of 70.1 (SD = 16.2); neither of these measures differed significantly from the AD cases.
Table 1.
Characteristics of AD cases (n = 27)
| SD | Range | ||
|---|---|---|---|
| Age at death (y) | 78.4 | 9.8 | 57–89 |
| Gender, M:F | 13:14 | ||
| Education (y) | 14.3 | 3.5 | 8–20 |
| Number of ApoE-ε4Alleles
0 (noncarrier) 1 (heterozygous) 2 (homozygous) |
9
14 4 |
||
| Estimated age of symptom onset | 68.3 | 9.8 | 48–83 |
| Illness duration | 10.1 | 4.7 | 3.9–19.6 |
| Last Blessed DRS | 12.3 | 4.0 | 3–17 |
| Time from last evaluation to death(y) | 1.1 | 1.7 | 0.1–6.5 |
| MMMSE at intake | 40.2 | ||
| Number of mMMSE administered | 7.6 | 4.6 | 1–17 |
| Time from last measured mMMSE(y) | 2.3 | 2.4 | 0.1–9.0 |
All are mean values except gender and ε4 status.
Cases vs. Controls
Mean Aβ42 and Aβ40 in biochemical compartments of temporal and cingulate neocortex are given in Table 2. In comparison to controls, AD brains had significantly higher mean concentrations of Aβ42 in the Tris and FA fractions of temporal and cingulate cortex (p < 0.001) and higher mean concentrations of Tris and FA Aβ40 in temporal (p < 0.01) and cingulate (p < 0.03) cortex. Mean Triton and SDS Aβ42 was higher in AD temporal and cingulate cortex (p < 0.01). However, mean Triton and SDS Aβ40 was similar in AD and control brains.
Table 2.
Mean Aβ40 and Aβ42 in biochemical compartments of temporal and cingulate neocortex.
| Temporal | Cingulate | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Aβ40 | Aβ42 | Aβ40 | Aβ42 | ||||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | ||
| Tris | AD | 2.7 | (3.5) | 14.6 | (7.5) | 1.7* | (2.4) | 10.2 | (7.3) |
| Control | 0.8 | (0.4) | 1.6 | (3.2) | 0.6* | (0.5) | 1.1 | (2.5) | |
| Triton | AD | 13.3 | (9.3) | 7.0 | (3.1) | 12.4 | (4.2) | 8.8 | (3.0) |
| Control | 11.9 | (5.2) | 4.0 | (1.6) | 11.6 | (1.5) | 5.0 | (1.4) | |
| SDS | AD | 55.4 | (22.1) | 53.9 | (27.3) | 78.9 | (28.4) | 25.6 | (7.9) |
| Control | 55.8 | (34.9) | 18.6 | (12.7) | 73.6 | (40.3) | 17.1 | (9.1) | |
| FA | AD | 555.6 | (817.2) | 1240.2 | (835.1) | 389.9* | (597.6) | 1088.9 | (520.9) |
| Control | 89.7 | (52.9) | 186.3 | (343.4) | 120.4* | (70.8) | 297.5 | (698.5) | |
All values are means (standard deviation), with units of pmol/gram. Statistically significant case-control differences (p < 0.01) are given in bold. These ten Aβ variables are included in subsequent analyses within the AD group. Marginally significant differences (p < 0.05) are denoted with asterisks.
APOE-ε4 genotype
Of the 27 AD cases, 14 were heterozygous and 4 were homozygous for the APOE-ε4 allele. Subjects with 0, 1, or 2 APOE-ε4 alleles differed in FA Aβ4 0 (p = 0.001) and Tris Aβ40 (p = 0.01) in temporal cortex. Post-hoc analysis revealed that these differences were most pronounced in the four APOE-ε4 homozygotes (Figure 1).
Figure 1. Tris- and FA- extracted Aβ40 in temporal neocortex according to ApoE genotype.
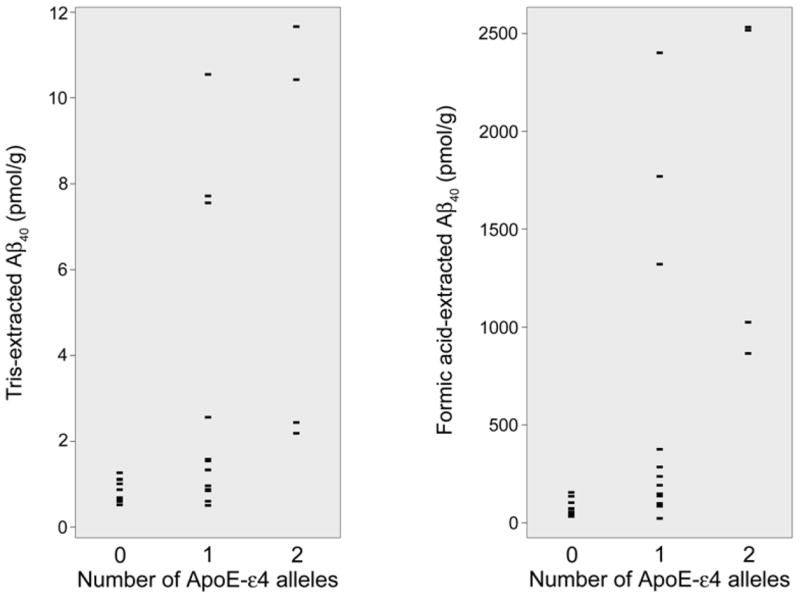
Markedly elevated Aβ40 in the biochemical fractions predicted to contain extracellular proteins is seen in ε4 homozygotes and a subset of heterozygotes.
Homozygotes had greater temporal FA Aβ40 and Tris Aβ40 when compared to either heterozygotes (p = 0.03) or noncarriers (p < 0.01). Mean values were greater in the heterozygotes than noncarriers, but this did not reach statistical significance (p = 0.16). There was no significant difference among ApoE groups in Triton Aβ40, SDS Aβ40, or Aβ42 within any biochemical compartment in either brain region.
Correlations with clinical severity
As shown in Table 3, significant correlations were observed between the last measured Blessed DRS and Triton Aβ42 (r = 0.482, p = 0.015) and SDS Aβ42 (r = 0.409, p = 0.042) in temporal cortex after adjusting for time from last assessment until death. That is, worse terminal functional status was associated with higher Aβ42 in the fractions predicted to contain intracellular and membrane-associated proteins; the unadjusted scatterplots are shown in Figure 2.
Table 3.
Results of cross-sectional and longitudinal analyses
| Temporal | Cingulate | ||||||
|---|---|---|---|---|---|---|---|
| Aβ40 | Aβ42 | Aβ42 | |||||
| r | p | r | P | r | p | ||
| Blessed DRS prior to death | Tris | −0.040 | 0.854 | 0.252 | 0 234 | 0.042 | 0.845 |
| Triton | 0.485 | 0.016 | −0.185 | 0.388 | |||
| SDS | 0.439 | 0.032 | 0.168 | 0.432 | |||
| FA | −0.075 | .728 | −0.065 | 0.763 | −0.189 | 0.376 | |
| Illness Duration | Tris | 0.567 | 0.002 | 0.130 | 0.518 | 0.117 | 0.568 |
| Triton | 0.286 | 0.149 | −0.039 | 0.848 | |||
| SDS | −0.087 | 0.667 | 0.199 | 0.330 | |||
| FA | 0.510 | 0.007 | −0.097 | 0.629 | 0.004 | 0.986 | |
| F | p | F | P | F | p | ||
| ApoE | Tris | 5.154 | 0.014 | 0.226 | 0.799 | 0.508 | 0.608 |
| Triton | 0.874 | 0.430 | 0.603 | 0.556 | |||
| SDS | 1.272 | 0.299 | 0.087 | 0.917 | |||
| FA | 9.343 | 0.001 | 1.695 | 0.205 | 0.278 | 0.760 | |
| β | p | β | P | β | p | ||
| Rate of cognitive decline | Tris | −1.237 | 0.268 | 0.005 | 0.997 | 2.070 | 0.145 |
| Triton | 1.750 | 0.200 | −0.112 | 0.940 | |||
| SDS | 3.506 | <0.001 | −2.747 | 0.011 | |||
| FA | −2.106 | 0.070 | 1.019 | 0.375 | −2.477 | 0.020 | |
All analyses reflect unadjusted models using raw data, except Blessed correlations which include time from last evaluation until death as a covariate. The reported β in the GEE analysis is that of the time × Aβ interaction term; positive values indicate a more rapid cognitive decline for subjects with measured Aβ in the upper median.
Figure 2. Relation between Triton- and SDS-extracted A β42 in temporal neocortex and functional disability prior to death.

Raw data are shown, unadjusted for time from last assessment to death. The Triton and SDS compartments may be enriched with intracellular and membrane-associated proteins.
Illness duration
Significant correlations were observed between illness duration and FA Aβ40 (r = 0.510; p = 0.007) and Tris Aβ40 (r = 0.567, p = 0.002) in temporal cortex. No significant correlation was observed between illness duration and any Aβ42 measurement. There was no significant correlation between illness duration and age at onset, age at death, or functional impairment at last evaluation (data not shown).
Rate of cognitive decline
GEE was used to compare rates of cognitive decline in groups split at the median of measured Aβ at death; results are given in Table 3 and Figure 3. The mean number of cognitive assessments was 7.6 ± 4.6 per subject. Elevated SDS Aβ42 in temporal neocortex was associated with more rapid decline (p < 0.001). In cingulate cortex, however, higher FA Aβ42 and SDS Aβ42 were related to slower decline (p = 0.02, p = 0.01, respectively).
Figure 3. GEE-derived models of estimated course of cognitive decline.
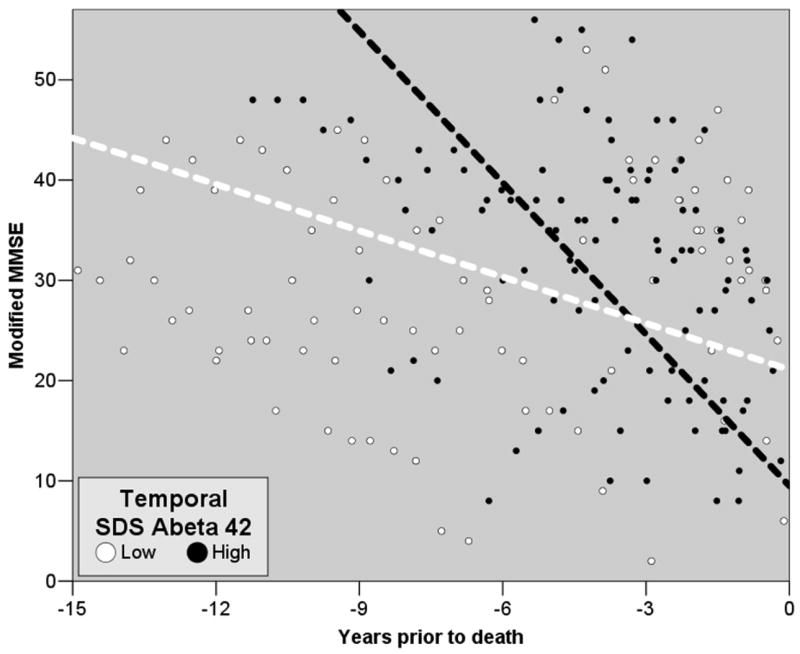
Lines depict the differential rate of decline in mMMSE scores prior to death as predicted by the GEE model for subjects whose SDS-extracted Aβ42 in temporal cortex was above (black) or below (white) the median. Circles represent subjects’ mMMSE scores at all times prior to death in the two groups. Note that the raw data are consonant with the GEE models.
Discussion
In this clinical-pathological correlation study, we observed all Tris- and FA- extracted Aβ isoforms to be elevated in AD compared to controls; these fractions are predicted to contain extracellular soluble Aβ (Tris) and insoluble Aβ associated with parenchymal and vascular amyloid deposition (FA). In contrast, in the biochemical compartments predicted to contain intracellular (Triton) and membrane-associated (SDS) protein pools, Aβ42, but not Aβ40, was elevated in AD. These findings are consistent with previous work which established that a range of extracellular Aβ measurements are elevated in AD brain. Our data suggest that intracellular and membrane-associated Aβ42 may be more closely related to the expression of symptoms in AD than other measured Aβ species.
Among AD cases, we found that only Triton- and SDS- extracted Aβ42 in temporal neocortex correlated with dementia severity at last evaluation prior to death. These measures are predicted to be enriched with intracellular and membrane-associated Aβ42. This lends support to the contention that accumulation of intracellular Aβ is not simply a marker of disease state, but progresses with clinical severity.
In our longitudinal analyses, elevated SDS Aβ42 in temporal cortex at autopsy was associated with a more rapid cognitive decline observed during the mild-moderate stages of dementia. This was the finding of greatest magnitude and statistical significance, and supports the contention that Aβ42 accumulation in the membrane-associated intracellular compartments is closely tied to disease symptoms, such as cognitive changes early in the clinical course.
ApoE-ε4 is a well-recognized genetic risk factor for AD which is associated with lower age of symptom onset. In our study, there was an ε4 allele dose-related increase in Aβ40 in the Tris and FA fractions. This is in agreement with previous findings23, 24; the pronounced increase among ε4 homozygotes was previously reported using methodology similar to the current study.25 While the molecular mechanism of the ε4 effect is uncertain, as a genetic factor, it likely exerts its influence for years prior to symptom emergence. Furthermore, we also found Tris- and FA-extracted Aβ40 to be the strongest correlates of illness duration in our study. Thus, constitutive extracellular Aβ40 accumulation may be trait of individuals destined to develop AD. Although their levels appear to increase as a function of genetic risk and illness duration in AD, we could not relate these Aβ40 species to the clinical state of our subjects. It has been shown that mice that produce only Aβ40 do not produce cerebral amyloid deposits.26 Thus, additional factors, including Aβ42 production, appear necessary to generate toxic amyloid and clinical manifestation of disease.
Recent work has led to increased recognition of the presence and importance of intraneuronal Aβ. These studies were enabled, by the development of antibodies which could differentiate Aβ40 and Aβ42 from the trans-membrane APP from which they derive.27 In human and mouse brain studies, intraneuronal Aβ has been detected prior to the emergence of extracellular plaques.28–30 In a recent animal study, appearance of intraneuronal Aβ coincided with emergence of cognitive impairment which was reversible with immunotherapy.31 Oligomerization of Aβ associated with increased neurotoxicity has been identified within neurons.32, 33
While the sources of intraneuronal Aβ are not well-defined, accumulation is known to occur at subcellular compartments of the endosomal pathway30, 34 and is associated with impairment of intracellular protein trafficking following endocytosis.35 Recently, genetic studies have also implicated alterations of intraneuronal protein recycling and sorting mechanisms in the pathogenesis of AD.36, 37 Elsewhere, it has been hypothesized that endocytosed Aβ42 is not degraded as efficiently as Aβ40.38
The results of the present study support a selective accrual of intraneuronal Aβ42 with progression of AD. Additional work is necessary to elaborate the mechanisms of extracellular Aβ40 accumulation and their relation to the protein misprocessing leading to intracellular Aβ42 accumulation. A potential link can be sought at the retromer complex, which shuttles proteins from the endosomal system to the secretory pathway. Selective retention of Aβ42 in the endosomal organelles and/or facilitated transfer of Aβ40 to the secretory system could account for such findings.
In the longitudinal analysis, results from the cingulate cortex appear discrepant with those of the temporal cortex. We have focused on the temporal cortex data in the figures and discussion for several reasons. Temporal association cortex is more likely to be involved in our cases who were recruited at early stages. Beyond this, the factors that contribute to differential regional vulnerability and alternate patterns of disease progression in AD are poorly understood. Thus, different results by region can be expected and informative. As such, we caution against modeling the whole brain as a homogenous biochemical compartment. In fact, future studies using serial extraction procedures on multiple brain regions may be suitable for analyzing regional covariance in toxic Aβ.
A major contribution of the present analyses lies in the careful diagnosis and clinical follow-up that patients received. Clinical diagnosis took place via consensus conference in university hospitals with specific expertise in dementia. The patients were observed prospectively, which eliminates the potential biases of retrospective chart reviews. Evaluations were performed semiannually, and included assessments closely proximate to death. Finally, the novelty of the Aβ measures is a significant strength. We are not aware of other studies of human AD brain which include biochemical pools predicted to contain intracellular and membrane-associated proteins.
Relative weaknesses include the limited number of subjects studied, which resulted in reduced statistical power; however, multiple data points per subject increased the power of our longitudinal analysis. We do not have detailed clinical information on the control subjects, who were not part of the Predictors cohort. However, the control data was used only for between-group comparison and was not included in our cross-sectional or longitudinal analyses of AD subjects. We recognize the exploratory nature of the investigation, and the problems associated with multiple comparisons. We have attempted to mitigate these by only including in subsequent analyses Aβ variables which, in the initial case-control comparison, satisfied a moderately-conservative correction for multiple comparisons. Nevertheless, the reported findings should be considered hypothesis-generating and require replication and refinement in future studies. Additionally, it is likely that our Aβ ELISA assay is insensitive to certain biologically relevant species of cerebral amyloid.39 For example, it is expected to quantify monomers only and does not distinguish multimeric forms or N-terminal modifications of the Aβ peptide.
We expect that detailed biochemical fractionation of Aβ pools will significantly enhance future clinical-pathological investigations of AD. Our study confirms the relevance of Aβ42 in the intracellular and membrane-associated compartments to disease manifestations. Constitutive accumulation of extracellular Aβ40 appears to be an AD trait which correlates with illness duration and is accentuated among ApoE-ε4 positive subjects. Further study of the covariance of Aβ measures across biochemical compartments and brain regions, and more detailed study of Aβ length and conformation within the intracellular and membrane-associated pools may contribute to updated models of amyloid dynamics.
Acknowledgments
This work was supported by NIH R01AG07370, P50AG05134, T32MH020004, and the Charles L. and Ann L. S. Brown Fellowship. The sponsors played no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript. Dr. Stern had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Contributor Information
Joshua R. Steinerman, Sergievsky Center, Department of Neurology, Columbia University Medical Center, New York, NY.
Michael Irizarry, Massachusetts General Hospital, Charlestown, MA.
Nikolaos Scarmeas, Sergievsky Center, Department of Neurology, Columbia University Medical Center, New York, NY.
Susan Raju, Massachusetts General Hospital, Charlestown, MA.
Jason Brandt, Johns Hopkins School of Medicine, Baltimore, MD.
Marilyn Albert, Johns Hopkins School of Medicine, Baltimore, MD.
Deborah Blacker, Massachusetts General Hospital, Charlestown, MA.
Bradley Hyman, Massachusetts General Hospital, Charlestown, MA.
Yaakov Stern, Sergievsky Center, Departments of Neurology, Psychiatry, and Psychology, Columbia University Medical Center, New York, NY.
References
- 1.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Giannakopoulos P, Herrmann FR, Bussiere T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 3.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 4.Berg L, McKeel DW, Jr, Miller JP, et al. Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol. 1998;55:326–335. doi: 10.1001/archneur.55.3.326. [DOI] [PubMed] [Google Scholar]
- 5.Kuo YM, Emmerling MR, Vigo-Pelfrey C, et al. Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271:4077–4081. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- 6.Gong Y, Chang L, Viola KL, et al. Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lesne S, Koh MT, Kotilinek L, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 8.Naslund J, Haroutunian V, Mohs R, et al. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. Jama. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 9.McLean CA, Cherny RA, Fraser FW, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 10.Wertkin AM, Turner RS, Pleasure SJ, et al. Human neurons derived from a teratocarcinoma cell line express solely the 695-amino acid amyloid precursor protein and produce intracellular beta-amyloid or A4 peptides. Proc Natl Acad Sci U S A. 1993;90:9513–9517. doi: 10.1073/pnas.90.20.9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skovronsky DM, Doms RW, Lee VM. Detection of a novel intraneuronal pool of insoluble amyloid beta protein that accumulates with time in culture. J Cell Biol. 1998;141:1031–1039. doi: 10.1083/jcb.141.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bateman RJ, Munsell LY, Morris JC, et al. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 2006;12:856–861. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawarabayashi T, Younkin LH, Saido TC, et al. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bateman RJ, Wen G, Morris JC, Holtzman DM. Fluctuations of CSF amyloid-beta levels: implications for a diagnostic and therapeutic biomarker. Neurology. 2007;68:666–669. doi: 10.1212/01.wnl.0000256043.50901.e3. [DOI] [PubMed] [Google Scholar]
- 15.Richards M, Folstein M, Albert M, et al. Multicenter study of predictors of disease course in Alzheimer disease (the “predictors study”). II. Neurological, psychiatric, and demographic influences on baseline measures of disease severity. Alzheimer Dis Assoc Disord. 1993;7:22–32. doi: 10.1097/00002093-199307010-00003. [DOI] [PubMed] [Google Scholar]
- 16.Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- 17.Stern YSM, Paulson J, Mayeux R. Modified Mini-Mental State Examination: validity and reliability [abstract] Neurology. 1987;37(suppl 1):179. [Google Scholar]
- 18.Folstein MF, Folstein SE, McHugh PR. Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 19.Wechsler D. Wechsler Adult Intelligence Scale Revised. New York, NY: Psychological Corp; 1981. [Google Scholar]
- 20.Kaplan EGH, Weintraub S. Boston Naming Test. Philadelphia, PA: Lea & Febiger; 1983. [Google Scholar]
- 21.Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry. 1968;114:797–811. doi: 10.1192/bjp.114.512.797. [DOI] [PubMed] [Google Scholar]
- 22.Zeger SL, Liang KY. Longitudinal data analysis for discrete and continuous outcomes. Biometrics. 1986;42:121–130. [PubMed] [Google Scholar]
- 23.Gearing M, Mori H, Mirra SS. Abeta-peptide length and apolipoprotein E genotype in Alzheimer’s disease. Ann Neurol. 1996;39:395–399. doi: 10.1002/ana.410390320. [DOI] [PubMed] [Google Scholar]
- 24.Ishii K, Tamaoka A, Mizusawa H, et al. Abeta1–40 but not Abeta1–42 levels in cortex correlate with apolipoprotein E epsilon4 allele dosage in sporadic Alzheimer’s disease. Brain Res. 1997;748:250–252. doi: 10.1016/s0006-8993(96)01363-7. [DOI] [PubMed] [Google Scholar]
- 25.Ingelsson M, Fukumoto H, Newell KL, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 26.McGowan E, Pickford F, Kim J, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol Aging. 2005;26:1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 28.Gyure KA, Durham R, Stewart WF, et al. Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med. 2001;125:489–492. doi: 10.5858/2001-125-0489-IAAPDO. [DOI] [PubMed] [Google Scholar]
- 29.Oakley H, Cole SL, Logan S, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cataldo AM, Petanceska S, Terio NB, et al. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging. 2004;25:1263–1272. doi: 10.1016/j.neurobiolaging.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 31.Billings LM, Oddo S, Green KN, et al. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 32.Walsh DM, Tseng BP, Rydel RE, et al. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39:10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi RH, Almeida CG, Kearney PF, et al. Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004;24:3592–3599. doi: 10.1523/JNEUROSCI.5167-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takahashi RH, Milner TA, Li F, et al. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006;26:4277–4288. doi: 10.1523/JNEUROSCI.5078-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rogaeva E, Meng Y, Lee JH, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Small SA, Gandy S. Sorting through the cell biology of Alzheimer’s disease: intracellular pathways to pathogenesis. Neuron. 2006;52:15–31. doi: 10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zerbinatti CV, Wahrle SE, Kim H, et al. Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J Biol Chem. 2006;281:36180–36186. doi: 10.1074/jbc.M604436200. [DOI] [PubMed] [Google Scholar]
- 39.Stenh C, Englund H, Lord A, et al. Amyloid-beta oligomers are inefficiently measured by enzyme-linked immunosorbent assay. Ann Neurol. 2005;58:147–150. doi: 10.1002/ana.20524. [DOI] [PubMed] [Google Scholar]