

Abstract
The majority of patients with HER2-overexpressing metastatic breast cancer who initially respond to the HER2-targeted antibody trastuzumab demonstrate disease progression within one year. The identification of novel agents that effectively inhibit survival of cancer cells that have progressed on trastuzumab is critical for improving outcome for this patient population. In the current study, we demonstrate that the phenolic compound nordihydroguaiaretic acid (NDGA) promoted cell death of trastuzumab-naïve and trastuzumab-refractory HER2-overexpressing breast cancer cells. NDGA induced DNA fragmentation, cleavage of PARP and caspase 3, and inhibition of colony formation. In addition, NDGA inhibited IGF-I and HER2 signaling in trastuzumab-refractory cells, with reduced downstream PI3K/Akt signaling. Importantly, combination treatment with NDGA and trastuzumab suppressed proliferation and survival of trastuzumab-refractory cells to a greater degree than either agent alone, suggesting that NDGA increases the sensitivity of refractory cells to trastuzumab. Derivatives of NDGA are currently in clinical trial for other solid tumors. Our data strongly support further study of NDGA as a potential therapeutic against breast cancers that have progressed on trastuzumab.
Keywords: Herceptin, drug resistance, erbB2, targeted therapy, apoptosis
INTRODUCTION
The her2 (erbB2/neu) gene is amplified and/or overexpressed in approximately 20% to 30% of invasive breast carcinomas, and is associated with increased metastatic potential and decreased overall survival (1). Trastuzumab (Herceptin™; Genentech, South San Francisco, CA) is a recombinant humanized monoclonal antibody directed against the extracellular domain of the human epidermal growth factor receptor 2 (HER2) tyrosine kinase receptor (2). Clinical studies established that trastuzumab is active against HER2-overexpressing metastatic breast cancer (MBC) (3–5), leading to its approval in 1998 by the U. S. Food and Drug Administration. However, the objective response rates to trastuzumab monotherapy in MBC are low, ranging from 12% to 34% for a median duration of nine months (4,5). Thus, the majority of patients who achieve an initial response to trastuzumab-based therapy demonstrate disease progression within one year of treatment initiation (6,7).
Understanding the mechanisms by which cells evade trastuzumab-mediated growth inhibition has become an increasingly important area of interest in breast cancer research. Most preclinical models have reported that her2 amplification and protein overexpression are maintained in trastuzumab-resistant derivatives of HER2-overexpressing cells (8,9), indicating that the molecular target (HER2) is still intact. Specific mechanisms resulting in trastuzumab resistance are not fully understood, but several proposed models now exist, including but not limited to the inability of trastuzumab to block HER heterodimerization or signaling from other HER receptors (9–12), increased PI3K signaling (13–15), insulin-like growth factor-I receptor (IGF-IR) signaling (16,17), and truncated, kinase-active forms of HER2 (18).
Overexpression of IGF-IR was shown to reduce trastuzumab-mediated growth arrest of HER2-overexpressing breast cancer cells (16). In addition, we demonstrated that IGF-IR induces phosphorylation of HER2 specifically in trastuzumab-refractory breast cancer cells (17). IGF-IR blockade using a specific kinase inhibitor, antibody, or IGF-I binding protein restored trastuzumab response to resistant cells (16,17), supporting the importance of the IGF-I signaling pathway to trastuzumab resistance.
Nordihydroguaiaretic acid (NDGA) is a naturally occurring phenolic compound isolated from the Larrea divaricata creosote bush (19). NDGA has been shown to reduce levels of phosphorylated IGF-IR and HER2 and block proliferation and promote apoptosis in MCF7 breast cancer cells stably transfected with HER2 (20). In addition, NDGA has been shown to function as a global transcription inhibitor, specifically blocking activity of members of the Sp1 transcription factor family, resulting in reduced cdc2 and survivin expression and leading to tumor regression in MCF7 xenograft models (21,22). A methylated analog of NDGA, meso-tetra-O-methyl NDGA (terameprocol), formerly known as EM-1421 and M4N, is in clinical development for use in solid refractory tumors, with phase I and II trials of terameprocol currently being performed in cancers of the prostate, head and neck, and brain.
In the current study, we evaluated NDGA in breast cancer cells that have amplification and overexpression of the her2 gene, and in derivatives of these cells that have become refractory to trastuzumab as a consequence of long-term treatment. We demonstrate here that NDGA induced cell death of parental and trastuzumab-refractory SKBR3 and BT474 HER2-overexpressing breast cancer cells. NDGA inhibited HER2 and IGF-IR signaling, and suppressed IGF-I-mediated proliferation of trastuzumab-refractory cells. Furthermore, co-treatment with NDGA and trastuzumab inhibited proliferation and survival of trastuzumab-refractory cells to a greater degree than either agent alone, suggesting that NDGA restores the growth inhibitory activity of trastuzumab, and that single agent NDGA or the combination of NDGA with trastuzumab may effectively inhibit breast cancers that overexpress HER2 including those that have progressed on trastuzumab.
MATERIALS AND METHODS
Materials
Trastuzumab (Genentech; South San Francisco, CA) was purchased from the Emory Winship Cancer Institute pharmacy and dissolved in sterile water at a stock concentration of 20 mg/ml. NDGA (Sigma-Aldrich, St. Louis, MO) was dissolved at a stock concentration of 165 mM (50 mg/mL). IGF-I (Sigma-Aldrich) was dissolved at 100 μg/mL in PBS and used at 100 ng/mL.
Cell culture
SKBR3 and BT474 parental (American Type Culture Collection, Manassas, VA) and trastuzumab-refractory breast cancer cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum. Trastuzumab-refractory cells derived from SKBR3 and BT474 were developed as previously described (8). Briefly, SKBR3 or BT474 cells were maintained in 4 μg/ml trastuzumab for 3 months, at which point refractory pools were developed by collecting all viable cells on one plate; clones were developed by propagating a single colony of viable cells on a plate. Resistance to trastuzumab was confirmed by trypan blue exclusion assays. All SKBR3 and BT474 trastuzumab-refractory pools and clones are maintained on 4 μg/ml trastuzumab.
Dose-response viability assays
Parental SKBR3 cells (SK-parental), SKBR3 refractory pool 2 (SK-HRp2) and clone 3 (SK-HRc3), parental BT474 (BT-parental), BT474 refractory pool 2 (BT-HRp2) and pool 3 (BT-HRp3) cells were treated with 25, 50, or 100 μM NDGA for 72 hours (h), at which point cell survival was determined by trypan blue exclusion. In addition, SK-parental and SK-HRp2 cells were treated with NDGA +/− IGF-I for 72 h prior to trypan blue exclusion analysis. Experiments were done in triplicate, and repeated at least twice. Cell survival for all experiments is expressed as a percentage of untreated cells, with error bars representing the standard deviation between replicates.
Colony inhibition assays
Cells were plated at 30,000 cells per well in 6-well format in duplicate. After 24 h, cells were untreated or treated with 25 μM or 100 μM NDGA for 48 h. Alternatively, cells were treated with 40 μM NDGA, 20 μg/mL trastuzumab, or a combination of 40 μM NDGA with 20 μg/mL trastuzumab for 72h. Drug-containing media was then removed, cells were washed with PBS, and regular complete media (DMEM plus 10% FCS) was added to cultures. Cells were maintained in complete media for one week, at which point colonies were stained with 1% methylene blue. Assays were done in duplicate each time, and repeated at least twice.
DNA fragmentation ELISA
Cells were plated at 10,000 cells per well in 6-well format and treated with 100 μM NDGA for 48 h. Protein lysates were obtained and analyzed for cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes) representative of apoptosis using the Cell Death Detection ELISA (Roche, Indianapolis, IN) according to manufacturer guidelines. Absorbance was measured at 405 nm. The DNA fragmentation enrichment factor was determined as a ratio of absorbance per treatment group to absorbance of the untreated group per cell line. Experiments were done at least in duplicate.
Immunoblotting
Cells were lysed in RIPA buffer (Cell Signaling; Danvers, MA) supplemented with protease and phosphatase inhibitors (Sigma-Aldrich). Total protein extracts (50 μg) were immunoblotted using the following antibodies at the indicated dilutions: HER2 monoclonal Ab-3 (EMD Chemicals, San Diego, CA) 1:1000; from Cell Signaling, p-Ser 473-Akt (monoclonal 587F11), and polyclonal antibodies against Akt, p-Thr202/Tyr204 p42/p44 MAPK, total p42/p44 MAPK, IGF-IR beta at 1:1000, and caspase 3 and PARP at 1:200, and polyclonal p-Tyr1248 HER2 used at 1:200; β-actin monoclonal AC-15 (Sigma-Aldrich) at 1:10,000; from Biosource, polyclonal p-Tyr1162/1163 IR/IGF-IR used at 1:200. Secondary antibodies were chosen according to species of origin of the primary antibody. Protein bands were detected using the Odyssey Imaging System (Li-Cor Biosciences, Lincoln, NE), and quantitated using NIH imaging software ImageJ. Immunoblots were repeated at least two independent times to ensure reproducible results.
Drug combination proliferation assays
Cells were plated at 5000 per well in 96-well format. After 24 h, cells were treated with 40 μM NDGA, 20 μg/mL trastuzumab, or a combination of 40 μM NDGA with 20 μg/mL trastuzumab for 6 days (d). MTS reagent (Promega; Madison, WI) was then added per manufacturer instructions and absorbance was measured at 490 nm on a 96-well colorimetric plate reader. Cell proliferation for all experiments is expressed as a percentage of untreated cells, with error bars representing standard deviation between replicates. Experiments were done in 6 replicates each time, and repeated at least twice.
RESULTS
NDGA promotes cell death in trastuzumab-refractory breast cancer cells
Trastuzumab-refractory SKBR3 pool 2 (SK-HRp2) and clone 3 (SK-HRc3) cells were developed by long-term treatment of HER2-overexpressing SKBR3 (SK-parental) breast cancer cells with trastuzumab, as previously described (8). SK-parental, SK-HRp2, and SK-HRc3 were treated with 25, 50, or 100 μM NDGA for 72 h. Cell viability was determined by trypan blue exclusion, and is expressed as a percentage of untreated cells per cell line (Fig. 1A). NDGA inhibited survival of trastuzumab-refractory cells in a dose-dependent manner. Inhibition of cell survival in resistant cells was comparable to parental cells, with 50% inhibition achieved at approximately 25 μM NDGA. Colony formation assays in which SK-parental, SK-HRp2, and SK-HRc3 cells were treated with 25 or 100 μM NDGA showed dose-dependent inhibition of colony survival (Fig. 1B). In addition, DNA fragmentation was examined as a hallmark of apoptosis. SK-HRp2 and SK-HRc3 cells were treated with 100 μM NDGA for 48 h. Colorimetric ELISA-based detection of histone-complexed DNA fragments (mono- and oligonucleosomes) demonstrated that NDGA treatment promoted DNA fragmentation in refractory cells (Fig. 1C), confirming induction of apoptosis.
Figure 1. NDGA inhibits survival of SKBR3 parental and trastuzumab-refractory breast cancer cells.

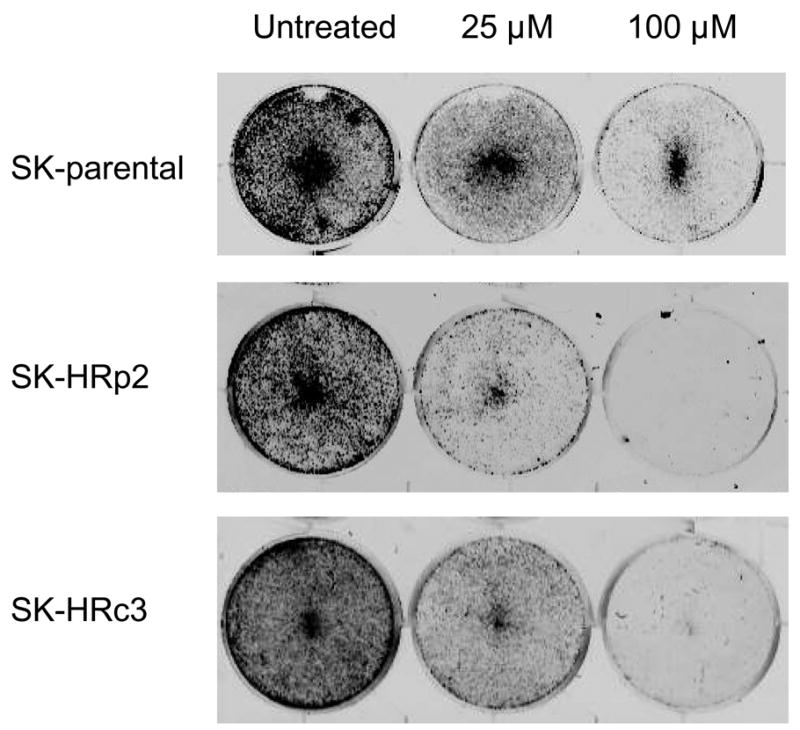

(A) Parental SKBR3 HER2-overexpressing breast cancer cells (SK-parental), SKBR3-derived trastuzumab-refractory pool 2 (SK-HRp2) and SKBR3-derived trastuzumab-refractory clone 3 (SK-HRc3) cells were treated with 0, 25, 50, or 100 μM NDGA for 72 h, at which point trypan blue exclusion analysis was performed. Cell viability is expressed as a percentage of untreated cells per line, with error bars representing standard deviation between replicates. Experiments were done in triplicate, and repeated at least twice. (B) SK-parental, SK-HRp2, and SK-HRc3 were treated with 0, 25, or 100 μM NDGA for 48 h. Drug-containing media was then removed, and cells were maintained in drug-free media for an additional week, at which point colonies were stained with methylene blue. Assays were repeated at least twice, in duplicate cultures each time. NDGA suppressed growth and survival of parental and trastuzumab-refractory HER2-overexpressing SKBR3 breast cancer cells. (C) SK-HRp2 and SK-HRc3 cells were treated with 0 or 100 μM NDGA for 48 h. Cells were lysed for protein and analyzed by ELISA for cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes) representative of apoptosis. Fold change in absorbance which reflects DNA fragmentation is shown relative to untreated cells, with error bars representing standard deviation between replicates. NDGA induced a 2-fold increase in DNA fragmentation, consistent with induction of apoptosis.
In addition to SKBR3-derived cells, we developed trastuzumab-refractory cells from the HER2-overexpressing BT474 cell line by treating BT474 cells with 4 μg/mL trastuzumab for three months, at which point pools were collected and examined for sensitivity to trastuzumab by trypan blue exclusion (Fig. 2A). At the clinically relevant dose of 20 μg/mL trastuzumab, BT-parental cells were growth inhibited by approximately 60% versus untreated cells. In contrast, pool 2 (BT-HRp2) cells continued to grow in the presence of trastuzumab, and pool 3 (BT-HRp3) cells remained 85–90% viable, confirming the relative resistance of these BT474-derived pools to trastuzumab. These cells are now maintained long-term on 4 μg/mL trastuzumab, similar to the SKBR3-derived refractory cells.
Figure 2. NDGA inhibits survival of BT474 parental and trastuzumab-refractory breast cancer cells.


(A) Parental BT474 HER2-overexpressing breast cancer cells (BT-parental), BT474-derived trastuzumab-refractory pool 2 (BT-HRp2) and pool 3 (BT-HRp3) cells were treated with 0 or 20 μg/mL trastuzumab (Tras) for 6 d, and then counted by trypan blue exclusion. Cell viability is expressed as a percentage of untreated cells per line; percentages are shown above each bar with standard deviation between replicates. The results confirm trastuzumab resistance of BT474 pool 2 and pool 3 cells relative to parental cells. (B) BT-parental, BT-HRp2, and BT-HRp3 were treated with 0, 25, 50, or 100 μM NDGA for 72 h, at which point trypan blue exclusion analysis was performed. Experiments were done in triplicate, and repeated at least twice. Cell viability is expressed as a percentage of untreated cells per line, with error bars representing standard deviation between replicates. NDGA suppressed growth and survival of the parental and trastuzumab-refractory HER2-overexpressing BT474 breast cancer cells, with resistant cells showing increased sensitivity to NDGA versus parental cells.
BT-parental, BT-HRp2, and BT-HRp3 cells were treated with 25, 50, or 100 μM NDGA for 72 h. Cell viability was determined by trypan blue exclusion, and is expressed as a percentage of untreated cells per cell line (Fig. 2B). Similar to SKBR3 parental and refractory cells, BT474 cells demonstrated dose-dependent cytotoxicity in response to NDGA. However, in contrast to the SKBR3 set of parental and resistant cells, which showed relatively equal responses to NDGA, BT474 trastuzumab-resistant cells demonstrated a slightly increased sensitivity to NDGA versus their parental cells, particularly at the middle dose of 50 μM NDGA. Collectively, the results from the SKBR3 and BT474 sets of cells demonstrate that NDGA is cytotoxic to HER2-overexpressing breast cancer cells, and appears to maintain efficacy in trastuzumab-refractory cells.
To further confirm that NDGA-mediated cytotoxicity was due to induction of apoptosis, cells were treated with 25, 50, or 100 μM of NDGA. Cleavage of poly (ADP-ribose) polymerase (PARP) and caspase 3 was measured by immunoblotting 6 h after drug treatment in SKBR3 (Fig. 3A) and BT474 (Fig. 3B) parental and resistant cells. During apoptosis, caspase 3 is cleaved into 17-kiloDalton (kDa) and 19-kDa proteins, and PARP is cleaved by caspases to produce an 89-kDa fragment from the full-length 116-kDa protein. NDGA treatment induced cleavage of caspase 3 and PARP in trastuzumab-refractory cells consistent with induction of apoptosis.
Figure 3. NDGA induces apoptosis of trastuzumab-refractory cells.


(A) SK-parental, SK-HRp2, and SK-HRc3 cells, and (B) BT-parental and BT-HRp3 cells were treated with 0, 25, 50, or 100 μM NDGA for 6 h. Immunoblotting (50 μg) was performed for PARP and caspase 3 cleavage. Actin served as a loading control. Blots were repeated at least twice to ensure reproducible results, and representative blots are shown. NDGA activated cleavage of PARP from the full-length 116-kDa form to the 89-kDa form, and of caspase 3 into the 19-kDa and 17-kDa forms, indicating induction of apoptosis by NDGA. The cleaved 89-kDa PARP and 19-kDa caspase 3 bands were quantitated using NIH ImageJ software.
NDGA blocks IGF-I and HER2 signaling in trastuzumab-refractory breast cancer cells
IGF-IR signaling has been associated with trastuzumab resistance in HER2-overexpressing breast cancer cells (16,17). To determine if NDGA affected IGF-IR or HER2 signaling in resistant cells, SKBR3 parental and trastuzumab-refractory cells were treated with NDGA (50 μM) for 1 h in the absence or presence of IGF-I (100 ng/mL), which was added for 20 min post-NDGA treatment. In parental and trastuzumab-refractory cells, pre-treatment with NDGA inhibited IGF-I-stimulated phosphorylation of IGF-IR (Fig. 4A). HER2 phosphorylation was also reduced by NDGA treatment in parental and resistant cells (Fig. 4B). Downstream signaling through PI3K was blocked by NDGA as shown by reduced phosphorylation of Akt in SK-HRp2 and SK-HRc3, but, interestingly, was unaffected in parental cells (Fig. 4C). MAPK signaling was not significantly affected by NDGA in parental and SK-HRp2 resistant cells, although SK-HRc3 cells showed reduced phosphorylation of ERK1/2. Thus, NDGA effectively inhibited IGF-I and HER2 signaling, and blocked IGF-I-mediated activation of PI3K/Akt in breast cancer cells that are no longer responsive to trastuzumab.
Figure 4. NDGA inhibits IGF-I and HER2 signaling in trastuzumab-refractory cells.

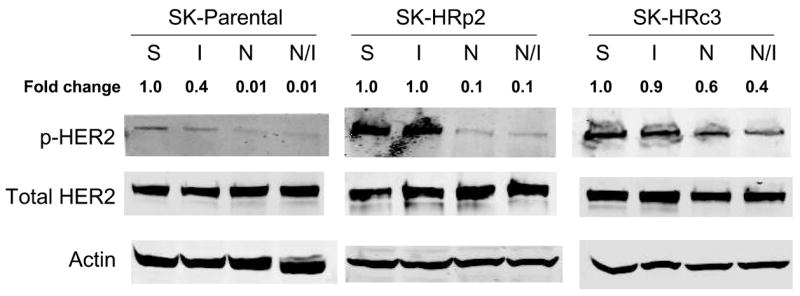


SK-parental, SK-HRp2, and SK-HRc3 cells were serum starved overnight. Cells were treated with NDGA (50 μM) for 1 h, followed by stimulation with IGF-I (100 ng/mL) for 20 min. S, serum-starved control; I, IGF-I stimulation alone; N, NDGA treatment alone; N/I, NDGA treatment followed by IGF-I stimulation. Total protein lysates were immunoblotted (50 μg) for (A) phosphorylated IGF-IR (p-IGF-IR) and total IGF-IR, (B) p-HER2 and total HER2, (C) p-Akt and total Akt, p-ERK1/2 and total ERK1/2. Actin served as a loading control. Blots were repeated at least twice to ensure reproducible results, and representative blots are shown. Bands were quantitated using NIH ImageJ software. NDGA blocked phosphorylation of IGF-IR and HER2 in parental and resistant cells. Akt phosphorylation was inhibited by NDGA in resistant cells, while ERK1/2 phosphorylation was reduced partially in SK-HRc3 cells. (D) SK-parental, SK-HRp2, and SK-HRc3 cells were untreated (0), treated with IGF-I (100 ng/mL) (I), NDGA (50 μM) (N), or combination NDGA (50 μM) plus IGF-I (100 ng/mL) (N+I) for 72 h prior to trypan blue exclusion analysis. Cell viability is expressed as a percentage of untreated cells per line, with error bars representing standard deviation between replicates. Trypan blue experiments were repeated at least twice, in triplicate cultures per group. NDGA suppressed cell survival in the presence of IGF-I in parental and trastuzumab-refractory cells. IGF-I stimulatory effects were more pronounced in resistant cells; IGF-I-mediated proliferation was inhibited by NDGA in resistant cells.
We then examined whether NDGA was able to reduce growth of trastuzumab-refractory cells in the presence of exogenous IGF-I. SK-parental, pool 2 and clone 3 cells were treated with NDGA (50 μM) and/or IGF-I (100 ng/mL) for 72 hours prior to trypan blue exclusion analysis. NDGA suppressed cell survival even in the presence of IGF-I (Fig. 4D), suggesting that NDGA prevents IGF-I-mediated proliferative and mitogenic effects in HER2-overexpressing breast cancer cells. Thus, NDGA can overcome the molecular and biological effects of IGF-I signaling in trastuzumab-resistant cells.
NDGA increases trastuzumab efficacy
Novel agents that are developed for clinical use against trastuzumab-refractory HER2-overexpressing breast cancers are most likely to be combined with trastuzumab, rather than replace the HER2-targeted antibody. We thus examined the combination of NDGA with trastuzumab in SK-parental (Fig. 5A), SK-HRp2 (Fig. 5B), BT-parental (Fig. 5C), and BT-HRp3 (Fig. 5D) cells. Cells were treated with 40 μM NDGA, 20 μg/mL trastuzumab, or a combination of both drugs for 6 d, at which point cell proliferation was measured by MTS colorimetric assay. All cells showed increased inhibition of proliferation with the combination of NDGA plus trastuzumab versus either agent alone, with trastuzumab-refractory cells showing a stronger difference in response to the combination versus single agents when compared to parental cells. Similarly, colony formation assays were performed, in which SK-parental and SK-HRp2 (Fig. 6A) and BT-parental and BT-HRp3 (Fig. 6B) cells were treated with 40 μM NDGA, 20 μg/mL trastuzumab, or a combination of both drugs for 72h. Colony inhibition was most pronounced in the combination treatment groups versus single agent groups. These results suggest that trastuzumab response is restored, at least partially, by NDGA treatment. Hence, the combination of NDGA and trastuzumab is a potentially novel therapeutic approach for treating trastuzumab-refractory HER2-overexpressing breast cancer.
Figure 5. Combination NDGA plus trastuzumab inhibits proliferation of trastuzumab-refractory breast cancer cells.


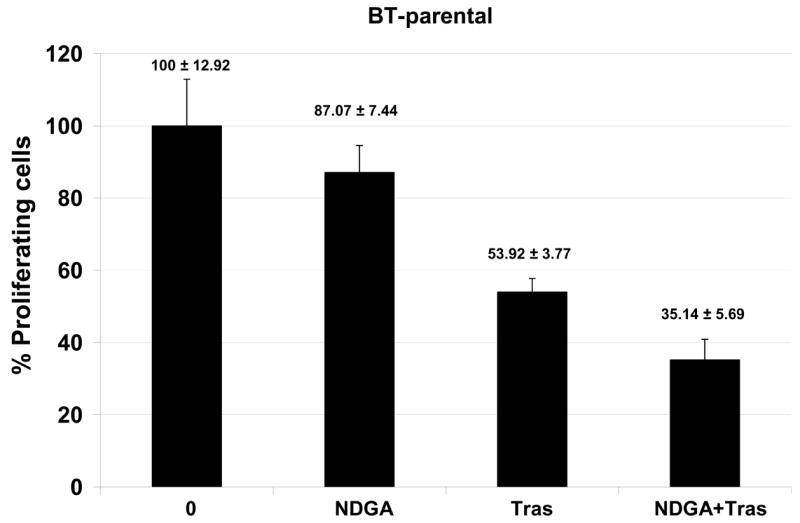

(A) SK-parental, (B) SK-HRp2, (C) BT-parental, and (D) BT-HRp3 cells were treated with NDGA (40 μM), trastuzumab (20 μg/mL), or a combination of both drugs for 6 d, at which point cell proliferation was measured by standard colorimetric MTS assay. Experiments were done at least twice, in 6 replicates per group each time. Cell proliferation is expressed as a percentage of untreated cells per line, with error bars representing standard deviation between replicates; values are shown above each bar. The combination of NDGA plus trastuzumab inhibited cell proliferation to a greater degree than either agent alone, suggesting that NDGA partially restores trastuzumab efficacy to trastuzumab-refractory breast cancer cells.
Figure 6. Combination NDGA plus trastuzumab blocks survival of trastuzumab-refractory breast cancer cells.
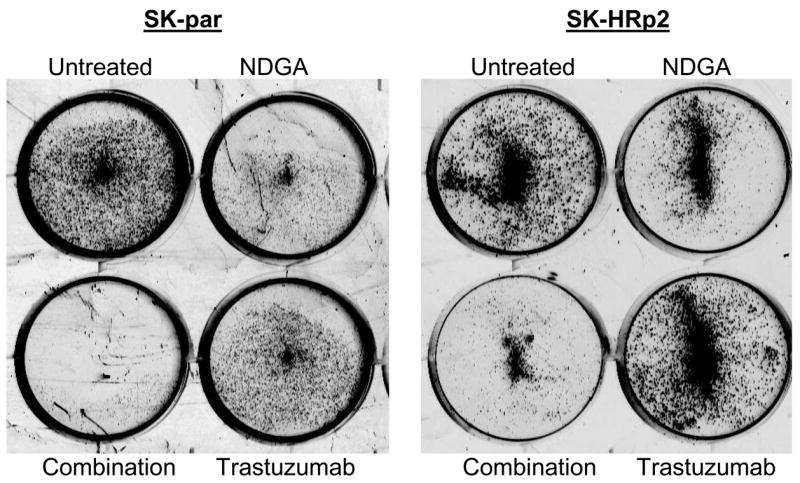
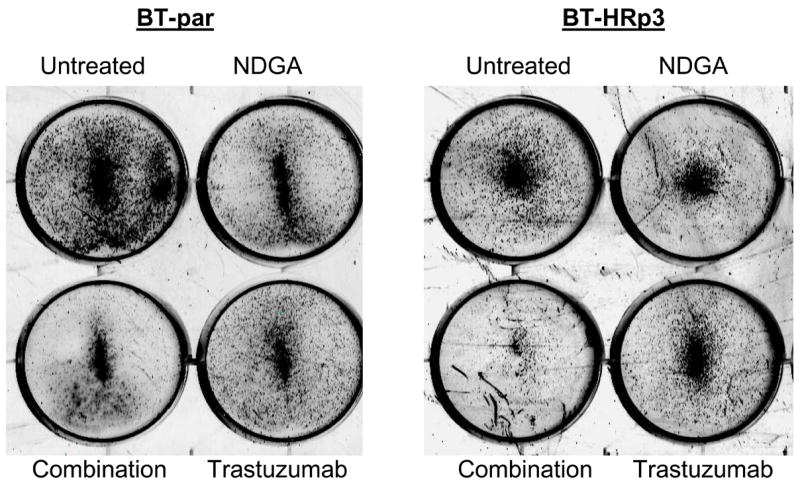
(A) SK-parental and SK-HRp2, (B) BT-parental and BT-HRp3 cells were treated with NDGA (40 μM), trastuzumab (20 μg/mL), or a combination of both drugs for 72 h, at which point drug-containing media was removed and cells were maintained in drug-free media for an additional week. Colonies were then stained with methylene blue. Experiments were repeated at least twice, in duplicate each time. Representative colony assays are shown. The combination of NDGA plus trastuzumab inhibited cell survival to a greater degree than either agent alone, suggesting that NDGA partially restores trastuzumab efficacy to trastuzumab-refractory breast cancer cells.
DISCUSSION
Trastuzumab resistance is a clinically significant concern, as at least one-third of HER2-overexpressing MBC patients demonstrate de novo resistance, and a majority of MBC patients who initially respond demonstrate disease progression within one year (4–7). In the adjuvant setting, the use of trastuzumab, either in combination with or following chemotherapy, improves disease-free and overall survival rates for patients with early-stage HER2-overexpressing breast cancer (23–25). However, approximately 15% of patients receiving trastuzumab-based adjuvant chemotherapy progress to develop metastatic disease. Therefore, resistance to trastuzumab, both primary (de novo) resistance and acquired (treatment-induced) resistance, is a major clinical concern facing breast oncologists today.
Preclinical studies of novel experimental agents in trastuzumab-refractory cancer are critical to improving patient outcome. We made the following observations regarding NDGA in our trastuzumab-resistant cell culture models. 1) NDGA promoted cell death of HER2-overexpressing breast cancer cells that are naïve (parental cells) or refractory (resistant) to trastuzumab. 2) IGF-IR and HER2 signaling were inhibited by NDGA in HER2-overexpressing parental and trastuzumab-resistant breast cancer cells, with downstream PI3K signaling diminished in resistant cells. 3) Co-treatment with NDGA increased trastuzumab-mediated anti-proliferative and growth inhibitory effects. These results suggest that NDGA is a potentially promising therapy for trastuzumab-refractory breast cancer. Further, NDGA-mediated cytotoxicity in resistant cells may be due in part to inhibition of the IGF-IR and HER2 signaling pathways.
Our results indicate that NDGA-mediated co-targeting of IGF-IR and HER2 may be beneficial in trastuzumab-refractory HER2-overexpressing breast cancers. Preclinical studies support the concept of co-targeting IGF-IR and HER2, as trastuzumab and a dominant negative IGF-IR construct produced synergistic growth inhibition in MCF7 cells stably transfected with HER2 (MCF7/HER18 cells), which also have endogenous overexpression of IGF-IR (26). Treatment with recombinant human IGF-binding protein 3, which inhibits IGF-I signaling, restored or potentiated the response to trastuzumab in vitro and in xenograft mouse models using either MCF7/HER18 cells or SKBR3 HER2-overexpressing cells stably transfected with IGF-IR (27). Chakraborty et al. (28) recently demonstrated that dual inhibition of HER2 and IGF-IR using trastuzumab plus either a commercially available antibody or kinase inhibitor of IGF-IR resulted in synergistic inhibition of anchorage-independent growth, apoptosis, and diminished PI3K and MAPK signaling not only in HER2-overexpressing cells, but also in breast cancer cells that express normal levels of the HER2 protein. These results suggest that this co-targeting strategy may be effective in multiple subtypes of breast cancer. Increased PI3K signaling has also been associated with trastuzumab resistance (13–15). Akt inhibition has previously been shown to induce apoptosis and promote tumor regression in trastuzumab-resistant breast cancer models (29). Hence, the finding that NDGA inhibits phosphorylation of Akt specifically in resistant cells, not in parental cells, is of potential therapeutic significance.
In addition to its abilities to inhibit IGF-I and HER2 signaling, NDGA has been shown to function as a global transcription inhibitor, specifically blocking activity of members of the Sp1 transcription factor family, and resulting in reduced cdc2 and survivin expression (21,22). We did not observe any changes in cdc2 expression levels at the doses and durations of exposure tested in these cell lines (not shown). Expression of the anti-apoptotic protein survivin was slightly down-regulated in response to NDGA in parental and resistant cells (not shown), which may be related to induction of cell death rather than inhibition of Sp1-dependent transcription, since the Sp1 target gene cdc2 was unaffected at the same doses of NDGA at which survivin expression was affected. Hence, the major mechanisms involved in NDGA-mediated cytotoxicity in our cell system may not involve global inhibition of transcription, but are likely to involve IGF-I and HER2 signaling blockade, according to our results.
An NDGA analog is being tested in early phase clinical trials and preliminary results are promising. The methylated analog of NDGA, terameprocol, was injected directly into refractory head and neck tumors in three patients (30). Two patients showed complete tumor necrosis at the sites of injection, although progressive disease occurred in the surrounding tumor areas. Terameprocol was well-tolerated in 29 patients with solid malignancies including gynecologic, lung, and colorectal, and all of whom were refractory to cytotoxic chemotherapy and radiation (31). One partial response and 6 patients with stable disease were reported in this study. In 11 patients with non-metastatic androgen-dependent prostate cancer, NDGA suppressed rising PSA levels, with one patient showing a 50% decrease in the PSA value (32). Based on these initial clinical studies, NDGA analog terameprocol is continuing to be studied as a potential novel anti-neoplastic agent. Our studies indicate that a trastuzumab-refractory population should be included in these studies.
In summary, trastuzumab is the first targeted agent approved for use in patients with HER2-overexpressing breast cancer. Disease progression despite being treated with trastuzumab necessitates the identification of novel agents. Our studies strongly suggest that NDGA, a dual IGF-IR/HER2 inhibitor in trastuzumab-resistant cells, should be explored as a novel therapy specifically in HER2-overexpressing breast cancers that have progressed on trastuzumab.
Acknowledgments
Grant Support: National Cancer Institute K01CA118174 (R Nahta); Georgia Cancer Coalition Distinguished Cancer Scholar Award (R Nahta)
ABBREVIATIONS
- NDGA
nordihydroguaiaretic acid
- HER2
human epidermal growth factor receptor 2
- DNA
deoxyribonucleic acid
- PARP
poly (ADP-ribose) polymerase
- IGF-I
insulin-like growth factor-I
- IGF-IR
IGF-I receptor
- PI3K
phosphoinositide 3 kinase
- MBC
metastatic breast cancer
- SK-parental
SKBR3 breast cancer cells
- SK-HRp2
SKBR3 Herceptin-resistant pool 2 cells
- SK-HRc3
SKBR3 Herceptin-resistant clone 3 cells
- BT-parental
BT474 breast cancer cells
- BT-HRp2
BT474 Herceptin-resistant pool 2 cells
- BT-HRp3
BT474 Herceptin-resistant pool 3 cells h, hours
- ELISA
enzyme-linked immunosorbent assay
- d
days
- MAPK
mitogen-activated protein kinase
- kDa
kilodaton
References
- 1.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 2.Carter P, Presta L, Gorman CM, et al. Humanization of an Anti-p185HER2 Antibody for Human Cancer Treatment. Proc Natl Acad Sci USA. 1992;89:4285–9. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baselga J, Tripathy D, Mendelsohn J, et al. Phase II study of weekly intravenous recombinant humanized anti-p185HER2 monoclonal antibody in patients with HER2/neu-overexpressing metastatic breast cancer. J Clin Oncol. 1996;14:737–44. doi: 10.1200/JCO.1996.14.3.737. [DOI] [PubMed] [Google Scholar]
- 4.Cobleigh MA, Vogel CL, Tripathy D, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER-2 overexpressing metastatic breast cancer that has progresssed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17:2639–48. doi: 10.1200/JCO.1999.17.9.2639. [DOI] [PubMed] [Google Scholar]
- 5.Vogel CL, Cobleigh MA, Tripathy D, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:719–26. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 6.Albanell J, Baselga J. Unraveling resistance to trastuzumab (Trastuzumab): Insulin-like growth factor-I receptor, a new suspect. J Natl Cancer Inst. 2001;93:1830–2. doi: 10.1093/jnci/93.24.1830. [DOI] [PubMed] [Google Scholar]
- 7.Esteva FJ, Valero V, Booser D, et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20:1800–8. doi: 10.1200/JCO.2002.07.058. [DOI] [PubMed] [Google Scholar]
- 8.Nahta R, Takahashi T, Ueno NT, Hung MC, Esteva FJ. P27(kip1) down-regulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res. 2004;64:3981–6. doi: 10.1158/0008-5472.CAN-03-3900. [DOI] [PubMed] [Google Scholar]
- 9.Ritter CA, Perez-Torres M, Rinehart C, et al. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007;13:4909–19. doi: 10.1158/1078-0432.CCR-07-0701. [DOI] [PubMed] [Google Scholar]
- 10.Motoyama AB, Hynes NE, Lane HA. The efficacy of ErbB receptor-targeted anticancer therapeutics is influenced by the availability of epidermal growth factor-related peptides. Cancer Res. 2002;62:3151–8. [PubMed] [Google Scholar]
- 11.Moulder SL, Yakes FM, Muthuswamy SK, Bianco R, Simpson JF, Arteaga CL. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and in vivo. Cancer Res. 2001;61:8887–95. [PubMed] [Google Scholar]
- 12.Anastasi S, Sala G, Huiping C, et al. Loss of RALT/MIG-6 expression in ERBB2-amplified breast carcinomas enhances ErbB-2 oncogenic potency and favors resistance to Herceptin. Oncogene. 2005;24:4540–8. doi: 10.1038/sj.onc.1208658. [DOI] [PubMed] [Google Scholar]
- 13.Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002;62:4132–41. [PubMed] [Google Scholar]
- 14.Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 15.Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 16.Lu YH, Zi XL, Zhao YH, Mascarenhas D, Pollak M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Trastuzumab) J Natl Cancer Inst. 2001;93:1852–7. doi: 10.1093/jnci/93.24.1852. [DOI] [PubMed] [Google Scholar]
- 17.Nahta R, Yuan LXH, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65:11118–28. doi: 10.1158/0008-5472.CAN-04-3841. [DOI] [PubMed] [Google Scholar]
- 18.Anido J, Scaltriti M, Bech Serra JJ, et al. Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. EMBO J. 2006;25:3234–44. doi: 10.1038/sj.emboj.7601191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer GE, Chesler L, Liu D, et al. Nordihydroguaiaretic acid inhibits insulin-like growth factor signaling, growth, and survival in human neuroblastoma cells. J Cell Biochem. 2007 doi: 10.1002/jcb.21373. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zavodovskaya M, Campbell MJ, Maddux BA, et al. Nordihydroguaiaretic acid (NDGA), an inhibitor of the HER2 and IGF-1 receptor tyrosine kinases, blocks the growth of HER2-overexpressing human breast cancer cells. J Cell Biochem. 2007 doi: 10.1002/jcb.21435. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 21.Chang CC, Heller JD, Kuo J, Huang RC. Tetra-O-methyl nordihydroguaiaretic acid induces growth arrest and cellular apoptosis by inhibiting Cdc2 and survivin expression. Proc Natl Acad Sci U S A. 2004;101:13239–44. doi: 10.1073/pnas.0405407101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park R, Chang CC, Liang YC, et al. Systemic treatment with tetra-O-methyl nordihydroguaiaretic acid suppresses the growth of human xenograft tumors. Clin Cancer Res. 2005;11:4601–9. doi: 10.1158/1078-0432.CCR-04-2188. [DOI] [PubMed] [Google Scholar]
- 23.Buzdar AU, Ibrahim NK, Francis D, et al. Significantly higher pathologic complete remission rate after neoadjuvant therapy with trastuzumab, paclitaxel, and epirubicin chemotherapy: results of a randomized trial in human epidermal growth factor receptor 2-positive operable breast cancer. J Clin Oncol. 2005;23:3676–85. doi: 10.1200/JCO.2005.07.032. [DOI] [PubMed] [Google Scholar]
- 24.Piccart-Gebhart MJ, Procter M, Leyland-Jones B, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353:1659–72. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- 25.Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353:1673–84. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- 26.Camirand A, Lu Y, Pollak M. Co-targeting HER2/ErbB2 and insulin-like growth factor-1 receptors causes synergistic inhibition of growth in HER2-overexpressing breast cancer cells. Med Sci Monit. 2002;8:BR521–6. [PubMed] [Google Scholar]
- 27.Jerome L, Alami N, Belanger S, et al. Recombinant human insulin-like growth factor binding protein 3 inhibits growth of human epidermal growth factor receptor-2-overexpressing breast tumors and potentiates herceptin activity in vivo. Cancer Res. 2006;66:7245–52. doi: 10.1158/0008-5472.CAN-05-3555. [DOI] [PubMed] [Google Scholar]
- 28.Chakraborty AK, Liang K, DiGiovanna MP. Co-targeting insulin-like growth factor I receptor and HER2: dramatic effects of HER2 inhibitors on nonoverexpressing breast cancer. Cancer Res. 2008;68:1538–45. doi: 10.1158/0008-5472.CAN-07-5935. [DOI] [PubMed] [Google Scholar]
- 29.Lu CH, Wyszomierski SL, Tseng LM, et al. Preclinical testing of clinically applicable strategies for overcoming trastuzumab resistance caused by PTEN deficiency. Clin Cancer Res. 2007;13:5883–8. doi: 10.1158/1078-0432.CCR-06-2837. [DOI] [PubMed] [Google Scholar]
- 30.Dunphy FR, Dukelow KK, Provenzal J, Crawford J. Phase I clinical results of intralesional injection of tetra-O-methyl nordihydroguairetic acid (M4N) in refractory head and neck cancer. J Clin Oncol. 2004;22:5614. [Google Scholar]
- 31.Goel S, Burris H, Mendelsohn D, et al. A phase I study of intravenous tetra-O-methyl nordihydroguaiaretic acid in patients with refractory malignancy. J Clin Oncol. 2007;25:3584. [Google Scholar]
- 32.Harzstark AL, Ryan C, Diamond M, et al. A phase I trial of nordihydroguareacetic acid (NDGA) in patients with non-metastatic prostate cancer and rising PSA. J Clin Oncol. 2007;25:15500. [Google Scholar]