

SUMMARY
Heat shock factor 1 (HSF1) is the master regulator of the heat shock response in eukaryotes, a very highly conserved protective mechanism. HSF1 function increases survival under a great many pathophysiological conditions. How it might be involved in malignancy remains largely unexplored. We report that eliminating HSF1 protects mice from tumors induced by mutations of the RAS oncogene or a hot spot mutation in the tumor suppressor p53. In cell culture, HSF1 supports malignant transformation by orchestrating a network of core cellular functions including proliferation, survival, protein synthesis, and glucose metabolism. The striking effects of HSF1 on oncogenic transformation are not limited to mouse systems or tumor initiation; human cancer lines of diverse origins show much greater dependence on HSF1 function to maintain proliferation and survival than their nontransformed counterparts. While it enhances organismal survival and longevity under most circumstances, HSF1 has the opposite effect in supporting the lethal phenomenon of cancer.
INTRODUCTION
The heat shock response is one of the most ancient and evolutionarily conserved protective mechanisms found in nature. Environmental insults provoke a variety of adaptive physiological responses to help organisms cope with specific stressors. The dramatic induction of heat shock proteins (HSPs) is an important unifying component of most of these responses, and this induction has proven to be essential for survival under stressful conditions. Work over the last three decades has revealed that the major HSPs are molecular chaperones that guard against “illicit or promiscuous interactions” between other proteins. Their basal expression facilitates normal protein folding and guards the proteome from the dangers of misfolding and aggregation. In the face of proteotoxic stressors including heat, hypoxia/ischemia, free radicals, ATP depletion, and acidosis, the importance of HSPs in preventing the aggregation and promoting the refolding of other proteins becomes acute. When misfolding exceeds a certain threshold, other HSPs disaggregate proteins and refold them or divert them to the proteasome for destruction (Whitesell and Lindquist, 2005).
Regulation of HSP expression is intricate, with multiple layers of redundancy and feedback control, but a small family of transcription factors called heat shock factors (HSFs) are the primary regulators of stress-inducible expression in eukaryotic cells. The structure and function of HSFs have been conserved for more than a billion years. They bind consensus heat shock elements (HSEs) within the promoter regions of HSP genes (Westerheide and Morimoto, 2005), and this binding is critical to HSP induction. Several HSFs are present in mammalian cells, but HSF1 is clearly the dominant factor controlling cellular responses to stress. Deletion of Hsf1 in mammalian cells allows normal basal expression of HSPs but completely abrogates induction in response to heat shock and a variety of other stresses (Xiao et al., 1999). In mice and Drosophila, HSF1 is dispensable for growth and survival under controlled laboratory conditions but essential for survival following stresses such as high temperature and endotoxin challenge (Jedlicka et al., 1997; Xiao et al., 1999). Hsf1-deficient mouse embryos suffer from defects in placental development and are recovered from crosses in lower numbers than expected by Mendelian segregation. Other than being ∼20% smaller than wild-type mice, however, they display no overt organ system abnormalities and, in the absence of acute stress, live to late adulthood (Xiao et al., 1999; A. Steele and S.L., unpublished data).
Although less well understood, the activities of HSF1 extend far beyond the classical induction of HSPs. In yeast, HSF1 has now been shown to regulate up to 3% of the genome and impact genes ranging in function from energy production to signal transduction, from small molecule transport to carbohydrate metabolism, and from cytoskeletal organization to vesicular transport (Hahn et al., 2004). Immunolocalization and chromatin immunoprecipitation indicate that HSF1 binds to a similarly broad array of non-HSP genes in Drosophila (Westwood et al., 1991; Birch-Machin et al., 2005) and human erythroleukemia cells (Trinklein et al., 2004).
The HSF1-mediated stress response and the activity of specific HSPs have both been implicated in protecting organisms from a broad range of pathophysiological conditions including thermal injury, ischemia/reperfusion, and age-related neurodegeneration (Christians et al., 2002; Westerheide and Morimoto, 2005). Intriguingly, in nematodes, HSF1 promotes longevity under stable laboratory conditions (Hsu et al., 2003; Morley and Morimoto, 2004). Much less is known about the role of HSF1 in cancer. It has long been noted that HSP levels increase in a wide range of tumor types (Jolly and Morimoto, 2000). Many of the signaling pathways and transcription factors that are frequently deranged in cancers display a striking dependence on the chaperone machinery, especially HSP90 (Whitesell and Lindquist, 2005). Moreover, HSF1 expression is elevated in human prostate carcinoma cell lines (Tang et al., 2005). But whether the multifaceted HSF1-mediated stress response plays a causal, supportive, or inhibitory role in mammalian oncogenesis is unknown.
On the one hand, given its prominent role in helping cells cope with stressful insults, HSF1 might promote oncogenesis by facilitating cellular adaptation to the malignant lifestyle. On the other hand, given its general role in enhancing longevity, HSF1 might assist organisms in combating malignancy. To investigate these possibilities, we used both whole-animal and cell-culture models in which HSF1 expression could be disrupted by genetic techniques. We find that HSF1 is a remarkably potent modifier of tumor-free survival in whole animals. Further, it modulates oncogenesis by coordinating a diverse array of core cellular functions and supports the aberrant proliferation and survival of human tumor cell lines carrying a wide range of molecular genetic defects. As a very ancient adaptive mechanism, the HSF1-dependent stress response has evolved to enhance survival in the face of environmental challenges from without and disease processes within such as ischemic injury and neurodegeneration. These broadly recognized beneficial effects, however, contrast sharply with its lethal role in the phenomenon of cancer that we now report.
RESULTS
HSF1 Deficiency Suppresses Chemical Skin Carcinogenesis in Mice
To begin investigating the role of Hsf1 as a modifier of tumorigenesis, we used a classical multistep chemical skin carcinogenesis protocol. In this mouse model, somatic mutations are induced in epidermal cells by a single topical application of the mutagen dimethylbenzanthracene (DMBA). Tumor promotion is then achieved by repeated applications of the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA). Early on, the overwhelming majority of the resulting skin tumors are benign papillomas. A small portion of these tumors spontaneously progress to become malignant squamous cell carcinomas, which are invasive and sometimes metastatic (Yuspa, 1994). When Hsf1 wild-type mice (Hsf1+/+) and their Hsf1 null littermates (Hsf1-/-) were treated with DMBA and TPA, no obvious skin damage or irritation was noticed in either genotype after topical application of the chemicals. There was, however, a striking difference in carcinogen-induced tumorigenesis.
Hsf1-/- mice were far more resistant to tumor formation than Hsf1+/+ mice (Figure 1A), and this difference was manifested in several ways. First, the latency period before the development of any tumors was 5 weeks longer in Hsf1-/- mice than in Hsf1+/+ mice (Figure 1B). Second, Hsf1-/- mice exhibited a marked reduction in tumor incidence (Hsf1+/+ 93.1% versus Hsf1-/- 60.9% at week 24, p = 0.0047, chi-square test) (Figure 1B). Third, they had a much lower overall tumor burden. This applied both to the number of tumors that arose (Figure 1C) and to the size the tumors achieved (Figure 1D). Fourth, and most importantly, Hsf1-/- mice survived much longer than their wild-type counterparts (Figure 1E).
Figure 1. HSF1 Deficiency Suppresses Chemical Skin Carcinogenesis.

(A) Representative images of mouse skin tumors 25 weeks after topical DMBA application.
(B) Lower skin tumor incidence and longer incubation time in Hsf1-/- mice (p < 0.0001, two-way ANOVA).
(C) Lower tumor burden in Hsf1-/- mice. The data are presented as the number of skin tumors per mouse (mean ± SE, p < 0.0001, two-way ANOVA).
(D) Smaller tumor volumes in Hsf1-/- mice (the lines indicate geometric means; p = 0.0003, Mann Whitney test).
(E) The survival curves of Hsf1+/+ and Hsf1-/- mice following skin carcinogenesis (median survival: Hsf1+/+ 41 weeks; Hsf1-/- undefined; p = 0.0073, Logrank test).
To further investigate the extraordinary resistance of Hsf1-/- mice to carcinogen-induced tumorigenesis, an independent experiment was performed in which Hsf1-/- mice and their wild-type littermates were treated with a second mutagen, NMMG (N-methyl-N’-nitro-N-nitrosoguanidine), at week 25 to promote tumor progression. Once again, Hsf1-/- mice developed many fewer tumors. They also had a very strong survival advantage over Hsf1+/+ mice (see Figure S1A in the Supplemental Data available with this article online). Thus, in sharp contrast to the many circumstances under which Hsf1-deficient organisms are at a survival disadvantage relative to wild-type organisms, in survival after chemically induced skin carcinogenesis they have a profound advantage.
Nature of Carcinogen-Induced Tumors
Although there was a large difference in the number of tumors formed in the Hsf1+/+ and Hsf1-/- mice, the percentage of benign versus malignant tumors (papilloma versus squamous cell carcinoma) was comparable (Figures S1C and S1D). Next we asked if the tumors harbored mutations in the H-Ras proto-oncogene. This gene is almost always activated during chemical skin carcinogenesis. Furthermore, activating mutations of RAS occur in approximately 30% of all human cancers including skin cancers (Balmain et al., 1984; Sebti and Adjei, 2004). Fourteen skin lesions were randomly sampled in both genotypes. All harbored activating mutations in H-Ras. Strikingly, these occurred at positions that are known hot spots in human malignancies (Table S1).
HSF1 Deficiency Suppresses Tumorigenesis Driven by Mutant p53
To test the generality of the detrimental effects of HSF1 on tumor-free survival, we examined its impact on the development of tumors in mice carrying a germline mutation in the tumor suppressor p53, the most frequently mutated gene in human cancers. We crossed mice heterozygous for a clinically relevant hot spot mutation (p53R172H; Olive et al., 2004) with Hsf1+/- mice. Three genotypes, all heterozygous for p53 R172H, were examined: (1) Hsf1+/+, (2) Hsf1+/-, and (3) Hsf1-/-. Mice were allowed to age with no intervention and were monitored for tumor formation and overall survival. Moribund mice were sacrificed and subjected to full necropsy to detect potential tumor formation at the gross and microscopic levels in all major tissues and organs.
Tumor-free survival was dramatically prolonged in Hsf1-/- mice carrying a mutant p53 allele (Figure 2A). Strikingly, even Hsf1+/- mice survived longer than the Hsf1+/+ mice, indicating a dosage-dependent effect of HSF1. Median survivals were 427 days for Hsf1+/+ mice, 470 days for Hsf1+/- , and >622 days for Hsf1-/- mice. Thus, as with the skin carcinogenesis model, Hsf1-deficient mice had a surprising and profound survival advantage compared to wild-type animals.
Figure 2. HSF1 Deficiency Suppresses Tumorigenesis Driven by Mutant p53.
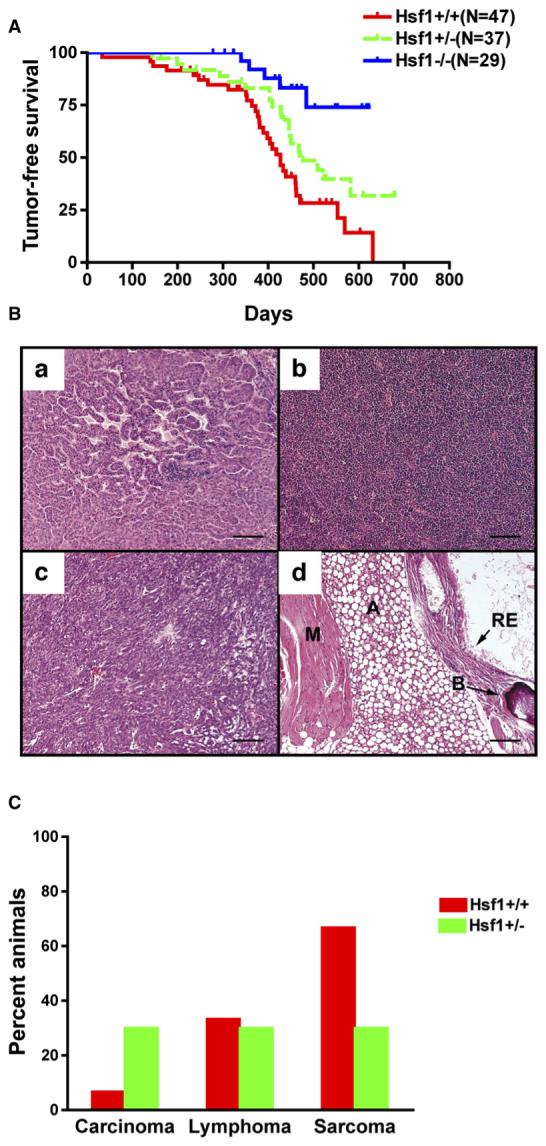
(A) Tumor-free survival curves of p53R172H knockin mice (Hsf1+/+ versus Hsf1-/-, p = 0.0001; Hsf1+/- versus Hsf1-/-, p = 0.0185;Hsf1+/+ versus Hsf1+/- , p = 0.0387; Logrank test).
(B) Representative micrographs of tumors (scale bars, 160 mm). (Ba) Carcinoma, (Bb) lymphoma, (Bc) soft tissue sarcoma, (Bd) teratoma (A, adipose; B, bone; M, muscle; RE, respiratory epithelium).
(C) Tumor spectra of Hsf1+/+ and Hsf1+/- mice(Hsf1+/+, n = 15; Hsf1+/- , n = 10).
Nature of p53 Mutant Tumors
Histopathological review revealed that both Hsf1+/+ and Hsf1+/- mice produced a broad spectrum of tumor types when carrying the p53R172H hot spot mutation. These included sarcomas, lymphomas, and carcinomas (Figure 2B). Intriguingly, in this model, compromise of Hsf1 function appeared to alter the distribution of tumor types. Hsf1+/- mice had an increase in carcinoma frequency and a decrease in sarcoma frequency compared to Hsf1+/+ mice (Figure 2C). Note, however, that changes in the tumor spectrum of Hsf1-/- mice could not be determined because so very few mice of this genotype developed tumors.
Hsf1 Status Does Not Alter Intrinsic Cell Growth Rates
To pursue observations made in mice at a cellular and molecular level, we examined the effect of Hsf1 status on several classical parameters of neoplastic transformation in cell culture. First, we examined freshly isolated mouse embryonic fibroblasts (MEFs). Hsf1+/+ and Hsf1-/- cells had comparable growth rates in vitro (data not shown). Moreover, staining cells for DNA content followed by flow cytometry revealed similar cell-cycle profiles in both genotypes (Figure S1B). Thus, the dramatic resistance of Hsf1-/- mice to tumor formation was not due to an intrinsic defect in cell proliferation or cell-cycle progression. Next, we used MEFs that had been previously immortalized, but not transformed, by stable expression of the E6 and E7 proteins of human papilloma virus (HPV)(McMillan et al., 1998) and transformed them with a variety of oncogenes.
HSF1 Enables Cellular Transformation Initiated by Oncogenic RAS
To directly investigate susceptibility to transformation by RAS, MEFs were transduced with retroviruses encoding green fluorescent protein (GFP), mouse Hsf1, or oncogenic H-RASV12D, a RAS mutation commonly found in human cancers. After several weeks in culture, cells were fixed and stained to visualize the transformed foci that had arisen as a consequence of the classic RAS-mediated loss of contact inhibition of growth.
Transduction with H-RASV12D induced high rates of focus formation in wild-type cells, but such foci were rare in cells derived from mice carrying the germline deletion (Hsf1-/- cells; Figure 3A and Figure S2A). Cells from both the abundant Hsf1+/+ foci and the rare Hsf1-/- foci were fully transformed as measured by soft agar cloning and tumor formation following subcutaneous injection into nude mice (data not shown). As expected, transduction with virus encoding GFP did not induce focus formation. Importantly, focus formation was also not observed with Hsf1 overexpression (Figure 3A and Figure S2A). Thus, by this criterion, Hsf1 acts as a powerful modifier of tumorigenesis rather than as an oncogene per se.
Figure 3. HSF1 Enables Cellular Transformation Initiated by Oncogenic RAS and PDGF-B.

(A) Hsf1-/- MEFs are relatively resistant to focus formation driven by oncogenic H-RASV12D. Immortalized MEFs were plated and transduced with retroviruses encoding the genes indicated. Foci were fixed and visualized by dye staining. The number of foci per well was quantified as shown in Figure S2. All experiments were repeated once with similar results.
(B) Hsf1-/- MEFs are relatively resistant to focus formation driven by the proto-oncogene PDGF-B.
(C) Hsf1-/- MEFs are refractory to proliferation driven by oncogenic RAS and PDGF-B. Equal numbers of immortalized Hsf1+/+ and Hsf1-/- MEFs were transduced with retroviruses encoding GFP, H-RASV12D,or PDGF-B. The cells were fixed on the indicated days and the number of cells per well determined by fluorescent DNA staining. Relative cell number was calculated by normalizing the values against the GFP-transduced group at each time point (mean ± SD, n = 5, ***p < 0.001, two-way ANOVA).
(D) Expression of c-MYC and LTA does not drive marked proliferation in immortalized Hsf1+/+ and Hsf1-/- MEFs.
(E) Hsf1-/- MEFs show no enhanced survival in response to RAS and PDGF/B expression but reduced survival in response to c-MYC and LTA expression. Viability of immortalized Hsf1+/+ and Hsf1-/- MEFs was determined by flow cytometry 36 hr after transduction. The data are presented as percent nonviable cells (mean ± SD, n = 5, *p < 0.05; ***p < 0.001, Student’s t test).
Reduced transformation of Hsf1-/- cells was not due to an intrinsic growth defect, nor was it due to reduced viral transduction efficiency. Both immortalized cell types displayed comparable saturation densities (Figure 3A, mean ± SD, Hsf1+/+ 2832 ± 267 nuclei per field versus Hsf1-/- 3411 ± 275, n = 5, p = 0.0096) and proliferation characteristics (Figure S4). If anything, Hsf1-/- MEFs displayed slightly greater gene transfer efficiencies as measured by flow cytometry after transduction with GFP-encoding retrovirus (mean ± SD, Hsf1+/+, 9.0% ± 0.6% positive versus Hsf1-/-, 12.7% ± 1.4% positive, n = 4, p = 0.003).
To control for the unlikely possibility that reduced transformation in Hsf1-/- MEFs was due to a potent but unknown polymorphism that happened to be closely linked to the HSF1 gene, we took advantage of short hairpin RNA interference (shRNAi) technology. Independent stable Hsf1 knockdown cell lines were generated from Hsf1+/+ MEFs using five different lentiviral vectors, each encoding a distinct Hsf1-targeted sequence. Two knockdown lines with differential gene silencing, C2 and C3, were chosen for further experiments. C3 cells, in which HSF1 levels were only partially reduced, behaved like wild-type cells in the RAS transformation assay. Isogenic C2 cells, in which HSF1 levels were dramatically reduced, behaved like Hsf1-/- MEFs from the germline knockout (Figures S3A-S3C).
HSF1 Enables Cellular Transformation by the Proto-Oncogene PDGF-B
To determine whether transformation-permissive effects of HSF1 at the cellular level extend beyond activating mutations of H-RAS, we tested another potent protooncogene: platelet-derived growth factor B (PDGF-B). As with RAS, Hsf1-/- MEFs displayed dramatic resistance to focus formation induced by PDGF-B compared to Hsf1+/+ cells (Figure 3B and Figure S2B). These findings, too, were confirmed with shRNAi experiments (Figures S3D-S3F). Thus, HSF1 also exerts a marked effect on cellular transformation initiated by overexpression of PDGF-B, an oncoprotein that activates multiple signaling cascades in addition to the RAS/MAPK pathway.
HSF1 Enhances Proliferation and Survival in Response to Diverse Oncogenic Stimuli
Neoplastic stimuli can increase the rates of cell proliferation, cell survival, or both. To determine which of these processes is influenced by HSF1, immortalized Hsf1+/+ and Hsf1-/- MEFs were transduced with retroviruses encoding GFP or several mechanistically distinct oncogenes. Retroviral transduction of H-RAS and PDGF-B drove a marked increase in cell number in Hsf1+/+ cells, but not in Hsf1-/- cells (Figure 3C). This was due to increased proliferation of Hsf1+/+ cells rather than increased death in Hsf1-/- cells (Figure 3E).
Unlike RAS and PDGF-B, which act as mitogenic signal transducers, c-MYC and LTA act primarily as regulators of cell-cycle progression and might not be expected to dramatically increase proliferation in these already-immortalized cell lines. Indeed, c-MYC and SV40 Large T Antigen (LTA) did not significantly increase cell accumulation (Figure 3D) or induce focus formation (data not shown). LTA and c-MYC can, however, predispose cells to apoptosis (Evan et al., 1992; Yin et al., 1997). Indeed, in contrast with RAS and PDGF,c-MYC and LTA sharply increased cell death in Hsf1-/- cultures, but not in Hsf1+/+ cultures (Figure 3E). Thus, depending upon the nature of the oncogenic stimuli involved, HSF1 enables oncogenic transformation in at least two ways, by permitting increased cell proliferation and/or by decreasing cell death.
HSF1 Modulates Signal Transduction
The ability to sustain dysregulated signaling is crucial to human cancers. In light of our observation that Hsf1-/- MEFs are resistant to RAS-driven transformation, we sampled downstream effectors in the RAS/MAPK signaling pathways. HSF1 deficiency caused a marked reduction in the levels of kinase suppressor of RAS 1 (KSR1) protein, both in Hsf1-/- MEFs and in shRNAi knockdown lines (Figure 4A). Furthermore, activation of the downstream effector, ERK, was blunted in Hsf1-/- MEFs following serum stimulation (Figure 4B).
Figure 4. HSF1 Modulates Signal Transduction.

(A) Hsf1-/- and C2-transduced MEFs show remarkably decreased expression of KSR1 protein by immunoblotting. Ponceau red staining was used to confirm equal protein loading.
(B) Hsf1-/- MEFs display blunted ERK activation in response to serum stimulation. Cells were stimulated with 20% FBS for the periods of time indicated, then fixed and stained with phospho-ERK1/2 antibody (green) and the DNA stain TO-PRO-3 iodide (red), which was used to normalize for relative cell number. The plate was scanned, and data are presented as relative phospho-ERK1/2 level by setting values at 0 min as 100% (mean ± SD, n = 5, **p < 0.01; ***p < 0.001; two-way ANOVA).
(C) Hsf1-/- and Hsf1 knockdown cells show markedly reduced phosphorylation of endogenous PKA substrates. Equal amounts of total protein were loaded and probed with antibodies recognizing phospho-(Ser/Thr) PKA substrate, HSF1, and GAPDH, respectively.
(D) Hsf1-/- MEFs possess lower PKA activity. The PKA activity in lysates prepared from Hsf1+/+ and Hsf1-/- MEFs was measured using the classical substrate Kemptide. Reaction mixtures were fractionated by agarose gel electrophoresis to visualize the extent of substrate phosphorylation. PKA, recombinant PKA as positive control. NC, peptide substrate only without lysate or recombinant PKA. The relative amount of phosphorylated peptide in each lane was quantitated by fluorometry.
We also asked whether Hsf1 affects the G protein-coupled receptor (GPCR) pathway, which increases cAMP levels, drives PKA activation, and is implicated in many human cancers (Bossis et al., 2004). A marked reduction in the phosphorylation of endogenous PKA substrates was observed in cells with germline Hsf1 deletion (Figure 4C). shRNAi-mediated Hsf1 knockdown in wild-type cells produced a similar effect. (In this case, dosage sensitivity was apparent.) These differences in PKA activity were verified by directly measuring the phosphorylation of a standard peptide substrate. Lysates from Hsf1-/- cells demonstrated less than half the PKA activity of lysates from Hsf1+/+ cells (mean ± SD, Hsf1+/+ 2452 ± 451 versus Hsf1-/- 917 ± 100, n = 2, p < 0.05) (Figure 4D). Having found that HSF1 modulates at least two classical oncogenic signaling pathways, we asked if it affects other crucial, but more recently recognized, cancer-related processes: ribosomal biogenesis and translation control.
HSF1 Modulates the Translation Machinery
The dysregulated growth of cancer cells requires growth factor independence in the control of ribosome biogenesis and protein translation. To reveal a potential role for HSF1 in this process, we cultured MEFs under growth factor-depleted conditions—that is, serum starvation. HSF1 status caused no difference in the levels of eIF4E, an mRNA capbinding protein, β-actin, or glyceraldehyde-3-phosphate dehydrogenase (GAPDH). However, HSF1-deficient MEFs consistently had reduced levels of the three ribo-somal subunits tested, L26RP, L28RP, and S6RP, whether they were deficient from germline knockout or from an shRNAi (Figures 5A and 5B). Furthermore, much lower levels of phosphorylated ribosomal protein S6 kinase (p70 S6K), a potent regulator of translational activity, were observed in Hsf1-deficient cells compared to wild-type cells (Figures 5A and 5B). Importantly, in both germline-deleted and shRNAi knockdown cells, comparable levels of these ribosomal proteins were restored when the cells were returned to culture in serum-replete medium (Figures 5A and 5B). Thus, HSF1 deficiency enforces tighter growth factor dependency on the translational machinery.
Figure 5. HSF1 Modulates Translation Machinery.
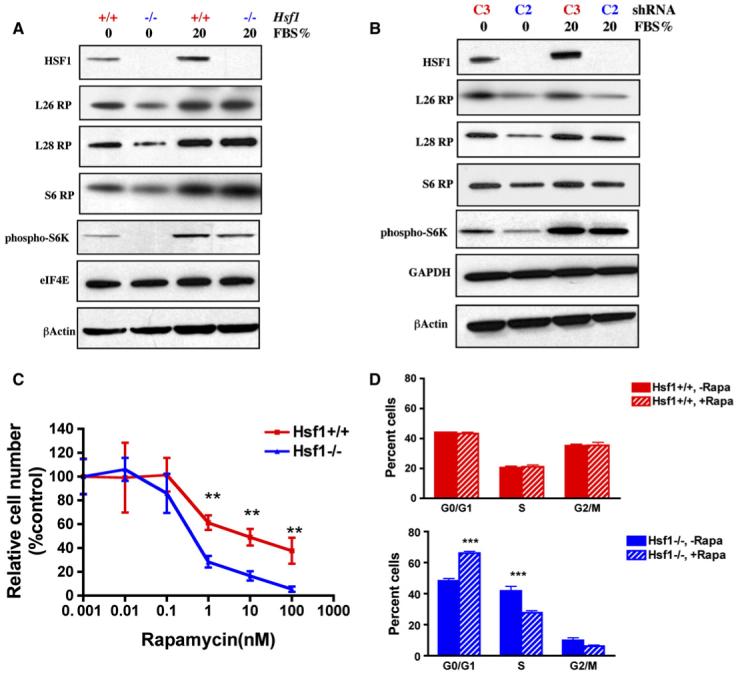
(A and B) HSF1 maintains p70 S6K phosphorylation and ribosomal protein expression during serum starvation. Cells were first serum starved for 2 days, then either stimulated with 20% FBS overnight or maintained under serum-free conditions.
(C) Hsf1-/- MEFs are more sensitive to the proliferation-inhibitory effect of rapamycin. Cells were exposed to a series of rapamycin concentrations as indicated for 5 days. After fixation, the relative number of cells in each well was determined by DNA staining. Data are presented as percent control by normalizing the values of rapamycin-treated wells against those of solvent-treated wells (mean ± SD, n = 5, **p < 0.01, two-way ANOVA).
(D) Hsf1-/- MEFs are more susceptible to cell-cycle arrest induced by rapamycin. Cells were treated with either methanol vehicle or rapamycin (100 nM) for 24 hr. Cell-cycle distribution was determined by flow cytometry. Data are presented as percent cells in each phase of the cell cycle (mean ± SD, n = 3, ***p < 0.001, two-way ANOVA).
Inhibition of mTOR function by rapamycin impairs protein translation and reduces cell size. Eliminating its downstream effector, p70 S6K, recapitulates this phenotype (Fingar et al., 2002). The lower levels of p70 S6K phosphorylation in Hsf1-deficient cells, together with the smaller size of Hsf1-/- mice (Xiao et al., 1999; and data not shown) led us to ask if individual cells derived from Hsf1-/- mice are smaller than Hsf1+/+ cells. Indeed, the mean cell volume (pL) of Hsf1-/- MEFs was 20% less than that of Hsf1+/+ MEFs (1925 ± 49 versus 2401 ± 184, mean ± SD, n = 3, p < 0.05). This suggested that the mTOR pathway might be affected by Hsf1 status. Indeed, Hsf1-/- cells were significantly more sensitive than Hsf1+/+ cells to rapamycin-induced growth inhibition (Figure 5C and Figure S5A). The hypersensitivity of Hsf1-/- cells was not due to increased cell death (data not shown). Instead, rapamycin caused the same type of cell-cycle arrest in G1 typical of mTOR inhibition, but it was more profound in Hsf1-/- than in Hsf1+/+ cells (Figure 5D). Notably, rapamycin neither induced a heat shock response nor impaired it (Figure S5B). Thus, independently of a classic proteotoxic stress response, HSF1 maintains the activity of the translation machinery and permits continued cell-cycle progression in immortalized but nontransformed cells under growth factor-reduced conditions in a manner that likely involves the role of mTOR in regulating protein translation.
HSF1 Modulates Glucose Metabolism
Unlike normal cells, virtually all cancer cells preferentially catabolize glucose by glycolysis, even under normoxic conditions, and thereby produce high levels of lactic acid (Bissell et al., 1976; Gatenby and Gillies, 2004). Recent evidence indicates that increased glycolysis is a consequence of oncogenic transformation and is advantageous to tumor growth and survival (Fantin et al., 2006; Matoba et al., 2006). To determine whether Hsf1 status alters glucose metabolism, we first examined glucose uptake, which is almost universally increased in cancers.
Hsf1+/+ and Hsf1-/- MEFs were cultured overnight in the presence of a fluorescent, noncleavable glucose analog, 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxyglucose (2-NBDG). Hsf1-/- MEFs accumulated much less 2-NBDG than did Hsf1+/+ cells (Figure 6A). Hsf1 knockdown MEFs exhibited the same trend, although greater variability was observed, as expected in the heterogeneous population of knockdown cells (Figure 6B).
Figure 6. HSF1 Modulates Glucose Metabolism.

(A and B) HSF1 regulates glucose uptake. Cells were incubated in medium containing either PBS (dotted lines) or 100 μM 2-NBDG (solid lines) overnight. The extent of glucose uptake was measured by flow cytometry. Data are presented as frequency histograms of relative tracer uptake. (A) depicts Hsf1+/+ (red line) and Hsf1-/- MEFs (blue line). (B) depicts C2 (blue line)- and C3 (red line)-transduced MEFs. The mean fluorescence intensity of each cell population is indicated (mean ± SD, n = 5, Hsf1+/+ versus Hsf1-/- 2-NBDG p < 0.0001; C3 versus C2 2-NBDG p < 0.0001, Student’s t test).
(C and D) Hsf1-/- MEFs are relatively resistant to glucose deprivation. Cells were incubated with either glucose-replete or -reduced medium for 4 days. The number and viability of cells in each well were determined by flow cytometry. Data are presented as relative cell numbers and percent nonviable cells (mean ± SD, n = 5, ***p < 0.001, two-way ANOVA).
Tumor cells, which utilize glucose at a much higher rate than normal cells, are generally more sensitive to glucose deprivation. We asked if Hsf1-/- cells are less addicted to glucose and less sensitive to its deprivation. Culturing Hsf1+/+ MEFs for 4 days in glucose-reduced medium led to a drastic decrease in total cell number and cell viability relative to cells cultured in standard high-glucose medium (Figures 6C and 6D). Hsf1-/- MEFs tolerated low glucose conditions much better. Thus, HSF1 deficiency leads to reduced dependence on glucose to support cellular energy needs and/or more efficient use of the sugar. Two other findings support this hypothesis. First, the nonmetabolizable glucose analog 2-deoxy-glucose (2-DG), which competes with glucose for the key glycolytic enzyme phosphohexose isomerase, killed Hsf1+/+ cells more efficiently than Hsf1-/- cells (Figure S6A). Second, under glucose-replete conditions, Hsf1-/- and shRNAi C2 cells generated significantly less lactate and possessed lower lactate dehydrogenase (LDH) activity than Hsf1+/+ cells (Figures S6B and S6C). Thus, the resistance of Hsf1-deficient cells to malignant transformation that we have demonstrated is associated with a pattern of basal glucose metabolism that could make it more difficult for them to undergo the glycolytic shift characteristic of cancers.
HSF1 Maintains the Transformed Phenotype
Having established in whole mice and cultured mouse cells that HSF1 is required in multifaceted ways for the initiation of transformation, we asked if it was also required for the maintenance of transformed phenotypes in established oncogenic cell lines and, at the same time, whether it was relevant to human cancers. We chose human lines of varying malignant potential, lines created in the laboratory with known oncogenes and lines derived from naturally occurring tumors with a diversity of histopathological origins and a broad spectrum of molecular genetic abnormalities. To modulate HSF1 expression, we evaluated three independent human HSF1-targeted shRNAi constructs, hA8, hA9, and hA6, and two control constructs (a GFP-targeted shRNAi and an shRNAi with no known homology within the human genome), all of the same infectious titers (Figure S8). Only the three HSF1-targeted vectors lowered HSF1 levels (Figure 7A); the most efficacious, hA6 and hA9, were selected for further analysis.
Figure 7. HSF1 Is Required for Maintenance of the Transformed Phenotype.

(A) ShRNA constructs exhibit differential HSF1-targeting efficacy. 293T cells were transfected with equal amounts of the indicated shRNA plasmid DNAs, and MCF-7 cells were transduced with the same shRNA constructs but packaged as lentiviral supernatants. Cells were harvested for immunoblotting after 3 days of puromycin selection.
(B) The effect of HSF1 compromise on cell growth and survival correlates with malignant state. Cells were plated in 96-well format and transduced with viral supernatants as indicated. Viable cell number in each well was measured 4 days after viral transduction. Data are presented as relative viable cell number by normalizing the values of each transduction group against those of GFP shRNA-transduced wells (mean ± SD, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, two-way ANOVA).
(C) Compromise of HSF1 impairs the growth and survival of established human breast cancer cells (mean ± SD, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, two-way ANOVA).
(D) Compromise of HSF1 impairs the growth and survival of human cancer cells derived from diverse histological origins (mean ± SD, n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, two-way ANOVA). All experiments were repeated at least once with similar results.
We first examined three breast cell lines representing progressively more oncogenic states (Figure 7B): (1) primary human mammary epithelial (PHME) cells; (2) human mammary epithelial (HME) cells made immortal, but nontumorigenic, by expression of hTERT (telomerase); and (3) HME cells rendered fully transformed and tumorigenic by introduction of LTA and H-RAS in addition to hTERT (HMLER) (Elenbaas et al., 2001). PHME cells were little affected by transduction with HSF1-targeted constructs. Tumorigenic HMLER cells were strongly affected. Immortalized, but nontumorigenic, cells (HME) were intermediate in sensitivity, suggesting a correlation between oncogenic state and HSF1 dependence (Figure 7B).
Next we examined a diverse collection of breast cell lines derived from spontaneous human tumors (Figure 7C). The lines varied with regard to p53 status, carrying wild-type (MCF-7) or various mutant alleles (BT-474, MDA-MB-231, and T47D) (International Agency for Research on Cancer TP53 Database) and with regard to HER2 overexpression, estrogen sensitivity, and metastatic potential (MD Anderson Breast Cancer Cell Line Database). All were strongly affected by both of the HSF1-inhibitory hairpins; none was affected by the control hairpins.
Finally, we examined malignant cells of diverse histological origins either derived from human tumors (HeLa [cervix], PC-3 [prostate], and S462 and 90-8 [peripheral nerve sheath]) or derived by in vitro transformation (293T [kidney]). All were strongly inhibited by HSF1 knockdown (Figure 7D). Where a difference in sensitivity to the two targeting hairpins was observed, it was always the hairpin that inhibited HSF1 expression the most severely (hA6; Figure 7A) that had the stronger effect (Figures 7B and 7C). This strong correlation between the extent of HSF1 inhibition and phenotypic effects, together with the fact that similar results were obtained with independent human and mouse targeting sequences and with a mouse germline knockout, argue strongly against “off-target” factors being responsible for these effects. Note also that the hairpins had no effect on normal diploid human fibroblasts (WI-38; Figure 7D). Therefore, we conclude that, in addition to its enabling role in tumor initiation, HSF1 function helps to maintain the growth and survival of human cancer cells with diverse underlying malignant defects.
DISCUSSION
Modulation of Oncogenesis by HSF1
As master regulator of the heat shock response, HSF1 enhances organismal survival and longevity in the face of environmental challenges. In sharp contrast to its widely appreciated beneficial effects, we now report that HSF1 can act to the detriment of organisms by supporting malignant transformation. In mice, HSF1 deficiency dramatically limited spontaneous tumor formation initiated by a common, dominant-negative mutation of the p53 tumor suppressor gene. It had a similar effect on chemical-induced skin carcinogenesis associated with activating mutations of the H-Ras proto-oncogene. The reduced incidence of tumors in these animals, and their associated effects on long-term survival, cannot be due to an effect of HSF1 on mutation rates. HSF1 deficiency rendered cultured cells highly refractory to transformation initiated by mutated H-RAS and by PDGF-B overexpression. Most importantly, the depletion of HSF1 in diverse previously established human cancer cell lines strongly impaired their growth and survival while having little effect on normal cells.
Tumor cells undergo a drastic shift of intracellular milieu during transformation. Gross alterations in signal transduction, energy production, and the metabolism of nucleic acids and protein are inevitable. In addition, cells within a tumor mass are frequently exposed to harsh and rapidly changing microenvironments that include such stressors as hypoxia, acidosis, nutrient deprivation, and immune attacks from the host. To survive and prosper, like organisms living in the wild, tumor cells within a host must be able to adapt effectively. Our data indicate that HSF1, the master regulator of one of the most ancient and evolutionarily conserved adaptive mechanisms, plays a prominent role in enabling cells to accommodate such drastic alterations, survive initial oncogenic stresses, and successfully adapt to the malignant state. In this setting, HSF1 function strongly reduces life span of the host.
Congruent with our findings, expression of the oncogenes heregulin beta1 and Ras has recently been reported to activate HSF1. This activation protects such cells against apoptosis and enables anchorage-independent growth (Khaleque et al., 2005; Stanhill et al., 2006). During the preparation of this manuscript, another group reported that HSF1 deficiency alters the spectrum of tumors arising in p53 knockout mice (Min et al., 2007). In contrast with our findings, however, they found no significant effect either on overall tumor incidence or on tumor-free survival. Several factors may account for this major discrepancy. First, Min et al. used a different transgenic strategy to knock out Hsf1, which resulted in unanticipated recombination events and only deleted the second exon of the gene. Although wild-type HSF1 protein was not detected, a truncated Hsf1 message was produced whose biological consequences are unknown (Zhang et al., 2002). The knockout mouse strain used to generate our data expresses no Hsf1 message. Furthermore, to address potential artifacts of transgenesis, our key findings using Hsf1 knockout cells were confirmed using a variety of independent mouse and human shRNAi constructs. Second, Min et al. evaluated the effects of HSF1 deficiency in a p53 null mouse model while we looked at a clinically relevant p53 missense mutation. Missense mutations in the p53 DNA-binding domain are the most clinically relevant lesions and have been reported in more than 50 different types of human cancer. Though complete deletions are commonly associated with human malignancy for other tumor suppressor genes, they occur rarely for p53, and individuals with germline homozygous deletions are unknown, raising questions as to the relevance of homozygous p53 deletion as a cancer model (Olive et al., 2004). Perhaps most importantly, we avoided modelspecific limitations by examining a variety of mechanistically distinct oncogenic stimuli other than p53 alteration and demonstrated consistent effects in whole animals, mouse cell cultures, and diverse human cancer cell lines. Given these considerations, we believe our findings capture the role of HSF1 in oncogenesis on a broader, more biologically and clinically relevant level.
Given the global nature of the alterations in cellular physiology that occur during malignancy, rather than deeply interrogating individual pathways, we pursued a broad survey of potential mechanisms whereby HSF1 might modify tumorigenesis. We found moderate but highly significant and readily detectable effects of HSF1 on a broad array of cellular functions, all of which play a role in successful malignant transformation. First, depending on the nature of the oncogenic stimulus, HSF1 enhances cell proliferation and/or survival. Second, HSF1 modulates both of the major cancer-promoting signal transduction cascades that we tested, the RAS/MAPK and cAMP/PKA pathways, and likely affects many more. Third, HSF1 maintains efficient ribosomal biogenesis and p70 S6K activation under growth factor-limited conditions, which are particularly relevant to malignant states. Lastly, HSF1 promotes glycolysis, a key metabolic pathway for tumor growth and survival.
Our data indicate that HSF1 itself does not act as a classic oncogene or tumor suppressor. Neither enforced overexpression nor knockout directly drives transformation. Instead, HSF1 orchestrates a broad network of cellular functions that act globally to support tumorigenesis (Figure S9). Individually, any one of these effects might have only a modest influence on malignant transformation. We suggest that the profound effects of HSF1 on tumor initiation and transformation are due to their acting in concert with each other and, indeed, with yet other mechanisms still to be discovered.
As a therapeutically relevant extension to our findings on tumor initiation, we find that HSF1 also promotes cancer cell maintenance. Hsf1 knockout is not lethal, or even detrimental to normal life span, in mice. Not surprisingly, we find that HSF1 knockdown has a minimal effect on the proliferation/survival of normal primary human cells in culture. In marked contrast, compromise of HSF1 profoundly impaired a wide variety of established human malignant as well as premalignant cell lines without relationship to the diversity of their known underlying molecular genetic defects.
Coupling Cellular Physiology to Environmental Contingency by HSF1
The induction of HSPs by HSF1 can potentiate oncogenesis in a variety of ways. For example, HSP90 chaperones many signal transducers, and certain oncoproteins are particularly dependent upon it (Whitesell and Lindquist, 2005). Further, HSP70 and other chaperones interface with the apoptotic machinery with important functional consequences (Jolly and Morimoto, 2000). It is essential to realize, however, that the effects of HSF1 extend far beyond HSP induction.
Befitting a survival factor highly conserved for more than 1 billion years and broadly deployed to combat a wide variety of physiologically distinct toxicities, HSF1 is emerging as the coordinator of an extensive array of cellular pathways at a system-wide level. For example, heat stress activates the RAS/MAPK pathway in an HSF1-dependent manner (Mivechi and Giaccia, 1995). Given that intracellular cAMP levels rise upon heat stress and the cAMP/PKA pathway negatively regulates HSF1 (Ferguson et al., 2005; Sawaji et al., 1999), crosstalk between the cAMP/PKA pathways and other HSF1-mediated response pathways seems almost certain. In yeast, HSF1 regulates 3% of the genome and occupies the promoter regions of a wide variety of genes, including those for several key glycolytic enzymes (Hahn et al., 2004). It appears to regulate a similarly broad spectrum of genes in Drosophila and man (Westwood et al., 1991; Birch-Machin et al., 2005; Trinklein et al., 2004). Our data indicate the involvement of HSF1 in regulating translation, ribosome biogenesis, and glucose metabolism in mammalian cells. For this emerging, system-wide regulation of growth and survival pathways by HSF1, our work establishes a vital functional consequence in oncogenesis.
Balancing Aging, Longevity, and Cancer Risk
Strikingly, our data now make it clear that HSF1 plays opposite roles in the complex diseases that plague aging populations. It powerfully potentiates the development of cancers. But it has also been implicated in protection against ischemia/reperfusion injury, neurodegenerative disorders, and other broad-ranging physiological processes affecting life span, such as aging and senescence (Auluck et al., 2005; Cohen et al., 2006; Gutsmann-Conrad et al., 1998; Hsu et al., 2003; Morley and Morimoto, 2004). In aging cells and organisms, alterations in energy metabolism, signal transduction, and protein homeostasis are accompanied by blunting of the stress response, ultimately leading to compromised viability.
Such an astonishing duality for the effects of HSF1 mirrors the surprising double-edged roles recently reported for the tumor suppressors p53 and p16INK4a. On the one hand, expression of p53 or p16INK4a shortens life span by accelerating cellular senescence and limiting the regenerative capacity of stem and progenitor cells; on the other hand, their activities extend life span by suppressing the emergence of life-threatening cancers (Beausejour and Campisi, 2006; Tyner et al., 2002). Our findings establish that an even more ancient survival factor, HSF1, has a similar duality, in reverse: its normal functions extend life span but in the case of cancer have a gravely deleterious impact on organismal survival. At a fundamental level, the ability of HSF1 to enable lethal malignancies is an unfortunate legacy of its ancient role in enhancing the survival of normal cells exposed to diverse acute and chronic stresses.
Our findings also have important therapeutic implications. An expanding array of small, drug-like compounds are becoming available with potent HSF1-modulating activity in cells and whole organisms. Therapeutic induction of the HSF1-mediated stress response by noncytotoxic exposure to agents such as HSP90 inhibitors and celastroloids is being explored in hypoxic-ischemic injury and protein misfolding disorders such as Huntington’s and Parkinson’s disease (Lu et al., 2002; Wang et al., 2005). Whether the therapeutic activation of HSF1 would increase the likelihood of oncogenic transformation is unknown, but it seems imperative to determine. Conversely, based on our findings with human cell lines, inhibiting HSF1 activation could provide a multifaceted and broadly effective cancer chemopreventive as well as chemotherapeutic strategy but might accelerate neurodegenerative processes and aging. A profound dichotomy holds true for other centrally poised drug targets such as the proteasome, whose inhibition is associated with promising anticancer activity in certain malignancies (Richardson et al., 2006) but in some situations might exacerbate neurodegenerative processes (McNaught et al., 2004). Using compounds that do not cross the blood-brain barrier is just one of many possible strategies to maximize benefits and reduce risks. We have much to learn about how the ancient adaptive function of HSF1 operates in the maze-like interface between genotype and environment to modify malignancy and other complex “diseases of civilization.” At the heart of this maze lies the promise of far more effective therapeutic intervention.
EXPERIMENTAL PROCEDURES
Skin Carcinogenesis Study
Hsf1+/- mice (BALB/c × 129SvEV), a gift from Ivor J. Benjamin, were intercrossed. DMBA (Sigma) (100 μg) dissolved in acetone was applied topically to each mouse. One week later, 20 μg of TPA (Sigma) was applied to each mouse twice a week for 24 weeks. Skin tumor formation was monitored weekly, and tumor dimensions were measured by caliper. The tumor volume is calculated as [(width)2 × length] ÷ 2.
Tumorigenesis Study of p53R172H Mice
p53R172H/+ mice (129S4/SvJae), a gift from Tyler Jacks, were crossed with Hsf1+/- mice. Moribund mice or mice with severely compromised health conditions were euthanized, and all major tissues were harvested and fixed in 10% formalin (soft tissues) or Bouin’s fixative (bones). Tumors were identified and diagnosed by an experienced veterinary pathologist (A.B.R.). Mice harboring no tumors were considered as censored subjects.
Focus Formation Assay and Image Analysis
Cells (5 × 104 or 1 × 105) were incubated with viral supernatants overnight. After 2 or 3 weeks, cells were fixed in cold 100% methanol. Foci were visualized by staining cells with 0.1% toluidine blue solution followed by destaining with 1% acetic acid. Images were documented with a FluorChem Imaging System (Alpha Innotech). The numbers and sizes of foci were further measured using CellProfiler software.
Lentiviral shRNA Knockdown Experiment
Lentiviral pLKO.1-puro shRNA constructs were obtained from the RNAi Consortium. Specific hairpin sequences and detailed viral production procedures can be found in the Supplemental Data.
Cell Proliferation Assay
Cell number and mass were quantitated by DNA (Hoechst 33342) and protein (sulforhodamine B) staining, respectively, using standard procedures. Detailed procedures are described in the Supplemental Data.
Cell Counting and Viability Assay
Cell number and viability were quantitated by microcapillary flow cytometer (Guava EasyCyte System, Guava Technologies) using the ViaCount reagent (Guava Technologies).
In-Cell Western
Cells were fixed with 10% formalin/PBS solution for 20 min. Wells were blocked with Odyssey blocking buffer (LI-COR Biosciences) for 2 hr at RT and incubated with phospho-p44/42 MAPK (Thr202/Tyr204) Ab (4377, Cell Signaling Technology) at 4°C overnight. After washing, cells were stained with IRDye 800CW Donkey Anti-Rabbit (LI-COR Biosciences) and TO-PRO-3 iodide (Invitrogen) for 1 hr at RT. After washing, microplates were scanned using the Odyssey Infrared imaging system (LI-COR Biosciences).
PKA Activity Assay
PKA activity was measured with the PepTag Non-Radioactive cAMP-Dependent Protein Kinase Assay kit (Promega).
Glucose Uptake Analysis
After incubation with medium containing 100 mM 2-NBDG (Invitrogen) overnight, cells were washed with PBS and detached. The relative fluorescence intensity of cells was measured using a Guava EasyCyte cytometer, and histograms were plotted using WinMDI software.
Cell Survival Assay
Relative cell growth/survival was measured using the CellTiter-Blue Cell Viability Assay Reagent (Promega).
Statistical Analysis
All statistical analyses were performed using Prism 4 (GraphPad Software).
Supplementary Material
ACKNOWLEDGMENTS
We thank I. Benjamin, T. Jacks, K. Cichowski, and T. Ince for mice and cell lines; R. Weinberg and H. Varmus for reagents; and the RNAi Consortium for shRNAi constructs. A. Steele and M. Topolszki provided excellent technical help, and R. Bronson provided expert pathological consultation. We thank T. DiCesare for graphic support and members of the Lindquist laboratory for advice and discussions. S.L. is a senior investigator of the Howard Hughes Medical Institute. This work was supported in part by the Mathers Foundation and fellowships from the Children’s Tumor Foundation (C.D.) and Radcliffe Institute for Advanced Study (L.W.).
REFERENCES
- Auluck PK, Meulener MC, Bonini N. Mechanisms of suppression of {alpha}-synuclein neurotoxicity by geldanamycin in Drosophila. J. Biol. Chem. 2005;280:2873–2878. doi: 10.1074/jbc.M412106200. [DOI] [PubMed] [Google Scholar]
- Balmain A, Ramsden M, Bowden GT, Smith J. Activation of the mouse cellular Harvey-ras gene in chemically induced benign skin papillomas. Nature. 1984;307:658–660. doi: 10.1038/307658a0. [DOI] [PubMed] [Google Scholar]
- Beausejour CM, Campisi J. Ageing: balancing regeneration and cancer. Nature. 2006;443:404–405. doi: 10.1038/nature05221. [DOI] [PubMed] [Google Scholar]
- Birch-Machin I, Gao S, Huen D, McGirr R, White RA, Russell S. Genomic analysis of heat-shock factor targets in Drosophila. Genome Biol. 2005;6:R63. doi: 10.1186/gb-2005-6-7-r63. Published online June 10, 2005. 10.1186/gb-2005-6-7-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, Rambeck WA, White RC, Bassham JA. Glycerol phosphate shuttle in virus-transformed cells in culture. Science. 1976;191:856–858. doi: 10.1126/science.175441. [DOI] [PubMed] [Google Scholar]
- Bossis I, Voutetakis A, Bei T, Sandrini F, Griffin KJ, Stratakis CA. Protein kinase A and its role in human neoplasia: the Carney complex paradigm. Endocr. Relat. Cancer. 2004;11:265–280. doi: 10.1677/erc.0.0110265. [DOI] [PubMed] [Google Scholar]
- Christians ES, Yan LJ, Benjamin I. Heat shock factor 1 and heat shock proteins: critical partners in protection against acute cell injury. Crit. Care Med. 2002;30:S43–S50. [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, Weinberg RA. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- Ferguson SB, Anderson ES, Harshaw RB, Thate T, Craig NL, Nelson HC. Protein kinase A regulates constitutive expression of small heat-shock genes in an Msn2/4p-independent and Hsf1p-dependent manner in Saccharomyces cerevisiae. Genetics. 2005;169:1203–1214. doi: 10.1534/genetics.104.034256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- GutsmannConrad A, Heydari AR, You S, Richardson A. The expression of heat shock protein 70 decreases with cellular senescence in vitro and in cells derived from young and old human subjects. Exp. Cell Res. 1998;241:404–413. doi: 10.1006/excr.1998.4069. [DOI] [PubMed] [Google Scholar]
- Hahn JS, Hu Z, Thiele DJ, Iyer VR. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell. Biol. 2004;24:5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Jedlicka P, Mortin MA, Wu C. Multiple functions of Drosophila heat shock transcription factor in vivo. EMBO J. 1997;16:2452–2462. doi: 10.1093/emboj/16.9.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly C, Morimoto RI. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 2000;92:1564–1572. doi: 10.1093/jnci/92.19.1564. [DOI] [PubMed] [Google Scholar]
- Khaleque MA, Bharti A, Sawyer D, Gong J, Benjamin IJ, Stevenson MA, Calderwood SK. Induction of heat shock proteins by heregulin beta1 leads to protection from apoptosis and anchorage-independent growth. Oncogene. 2005;24:6564–6573. doi: 10.1038/sj.onc.1208798. [DOI] [PubMed] [Google Scholar]
- Lu A, Ran R, Parmentier-Batteur S, Nee A, Sharp FR. Geldanamycin induces heat shock proteins in brain and protects against focal cerebral ischemia. J. Neurochem. 2002;81:355–364. doi: 10.1046/j.1471-4159.2002.00835.x. [DOI] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- McMillan DR, Xiao X, Shao L, Graves K, Benjamin IJ. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J. Biol. Chem. 1998;273:7523–7528. doi: 10.1074/jbc.273.13.7523. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Perl DP, Brownell AL, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann. Neurol. 2004;56:149–162. doi: 10.1002/ana.20186. [DOI] [PubMed] [Google Scholar]
- Min JN, Huang L, Zimonjic DB, Moskophidis D, Mivechi NF. Selective suppression of lymphomas by functional loss of Hsf1 in a p53-deficient mouse model for spontaneous tumors. Oncogene. 2007;26:5086–5097. doi: 10.1038/sj.onc.1210317. Published online February 19, 2007. 10.1038/ sj.onc.1210317. [DOI] [PubMed] [Google Scholar]
- Mivechi NF, Giaccia AJ. Mitogen-activated protein kinase acts as a negative regulator of the heat shock response in NIH3T3 cells. Cancer Res. 1995;55:5512–5519. [PubMed] [Google Scholar]
- Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol. Biol. Cell. 2004;15:657–664. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Mitsiades C, Hideshima T, Anderson KC. Bortezomib: proteasome inhibition as an effective anticancer therapy. Annu. Rev. Med. 2006;57:33–47. doi: 10.1146/annurev.med.57.042905.122625. [DOI] [PubMed] [Google Scholar]
- Sawaji Y, Sato T, Seiki M, Ito A. Transient increase of intracellular cAMP by heat shock initiates the suppression of MT1-MMP production in tumor cells. Ann. NY Acad. Sci. 1999;878:707–709. doi: 10.1111/j.1749-6632.1999.tb07768.x. [DOI] [PubMed] [Google Scholar]
- Sebti SM, Adjei AA. Farnesyltransferase inhibitors. Semin. Oncol. 2004;31:28–39. doi: 10.1053/j.seminoncol.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Stanhill A, Levin V, Hendel A, Shachar I, Kazanov D, Arber N, Kaminski N, Engelberg D. Ha-ras(val12) induces HSP70b transcription via the HSE/HSF1 system, but HSP70b expression is suppressed in Ha-ras(val12)-transformed cells. Oncogene. 2006;25:1485–1495. doi: 10.1038/sj.onc.1209193. [DOI] [PubMed] [Google Scholar]
- Tang D, Khaleque MA, Jones EL, Theriault JR, Li C, Wong WH, Stevenson MA, Calderwood SK. Expression of heat shock proteins and heat shock protein messenger ribonucleic acid in human prostate carcinoma in vitro and in tumors in vivo. Cell Stress Chaperones. 2005;10:46–58. doi: 10.1379/CSC-44R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinklein ND, Murray JI, Hartman SJ, Botstein D, Myers RM. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol. Biol. Cell. 2004;15:1254–1261. doi: 10.1091/mbc.E03-10-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- Wang J, Gines S, MacDonald ME, Gusella JF. Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci. 2005;6:1. doi: 10.1186/1471-2202-6-1. Published online January 13, 2005. 10.1186/1471-2202-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide SD, Morimoto RI. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J. Biol. Chem. 2005;280:33097–33100. doi: 10.1074/jbc.R500010200. [DOI] [PubMed] [Google Scholar]
- Westwood JT, Clos J, Wu C. Stress-induced oligomerization and chromosomal relocalization of heat-shock factor. Nature. 1991;353:822–827. doi: 10.1038/353822a0. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Xiao X, Zuo X, Davis AA, McMillan DR, Curry BB, Richardson JA, Benjamin IJ. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J. 1999;18:5943–5952. doi: 10.1093/emboj/18.21.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin C, Knudson CM, Korsmeyer SJ, Van Dyke T. Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature. 1997;385:637–640. doi: 10.1038/385637a0. [DOI] [PubMed] [Google Scholar]
- Yuspa SH. The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis—thirty-third G. H. A. Clowes Memorial Award Lecture. Cancer Res. 1994;54:1178–1189. [PubMed] [Google Scholar]
- Zhang Y, Huang L, Zhang J, Moskophidis D, Mivechi NF. Targeted disruption of hsf1 leads to lack of thermotolerance and defines tissue-specific regulation for stress-inducible Hsp molecular chaperones. J. Cell. Biochem. 2002;86:376–393. doi: 10.1002/jcb.10232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.