

Abstract
Sepsis refers to a systemic inflammatory response syndrome resulting from a microbial infection. The inflammatory response is partly mediated by innate immune cells (such as macrophages, monocytes and neutrophils), which not only ingest and eliminate invading pathogens but also initiate an inflammatory response upon recognition of pathogen-associated molecular patterns (PAMPs). The prevailing theories of sepsis as a dysregulated inflammatory response, as manifested by excessive release of inflammatory mediators such as tumour necrosis factor and high-mobility group box 1 protein (HMGB1), are supported by extensive studies employing animal models of sepsis. Here we review emerging evidence that support extracellular HMGB1 as a late mediator of experimental sepsis, and discuss the therapeutic potential of several HMGB1-targeting agents (including neutralising antibodies and steroid-like tanshinones) in experimental sepsis.
As a consequence of cohabiting with divergent microbes (e.g. virus, bacteria and fungi), animals have to deal with various microbial infections. Epithelial barriers provide the first layer of defence by limiting the access of potential pathogens. If they are breached, the host's innate immune system mounts an immediate but nonspecific biological response – termed inflammation – at the infection site, to confine and remove invading pathogens. If the invading pathogens are effectively eliminated, inflammation resolves normally to restore immunological homeostasis (Ref. 1); however, if not, invading pathogens or pro-inflammatory mediators such as tumour necrosis factor (TNF) or other cytokines can leak into the bloodstream, triggering a systemic inflammatory response that may lead to sepsis (Fig. 1).
Figure 1.

A microbial infection can trigger a local or systemic inflammatory response. The disruption of an epithelial barrier allows invasion of microbial pathogens, which elicit an innate immune response at the site of infection. If invading pathogens are effectively eliminated by phagocytes, local inflammation resolves normally to regain immunological homeostasis. If invading pathogens are not effectively eliminated, they can leak into the bloodstream, and trigger a potentially injurious systemic inflammatory response (such as sepsis).
Sepsis refers to a systemic inflammatory response syndrome resulting from a microbial infection. As a continuum of increasing clinical severity, ‘severe sepsis’ is defined as sepsis associated with one or more acute organ dysfunctions (Ref. 2). Septic shock is severe sepsis with organ hypoperfusion and hypotension (defined as systolic blood pressure less than 90 mmHg) that are poorly responsive to fluid resuscitation. Despite recent advances in antibiotic therapy and intensive care, sepsis is still the most common cause of death in intensive care units (Ref. 2). Here, we briefly review the prevailing theories of sepsis as an uncontrolled systemic inflammatory response, and discuss potential therapeutic agents that target clinically more feasible, late-acting mediators of experimental sepsis, such as HMGB1.
Local innate immune response to mild infection
The innate immune system comprises phagocytes (such as macrophages, monocytes and neutrophils), mast cells, eosinophils, basophils and natural killer cells. It constitutes a front line of defence against most microbial infection by eliminating invading pathogens and initiating an inflammatory response.
Elimination of invading pathogens
Neutrophils and monocytes continuously patrol the body to search for invading pathogens, and infiltrate into infected/injured tissues upon detecting microbial products (Ref. 3). Neutrophils arrive at the infection site early and in high numbers, and thus usually kill more invading bacteria than other phagocytes (Ref. 4). However, neutrophils are short-lived, with an average lifespan of 1–2 days: after engulfing and killing several bacteria, neutrophils exhaust intracellular enzymes and subsequently undergo apoptotic cell death. Upon reaching extravascular tissues, monocytes can differentiate into tissue-specific macrophages. Macrophages can ingest and eliminate larger pathogens that are not handled by the neutrophils; in addition, they remove the cell debris of apoptotic neutrophils in order to resolve an inflammatory response (Ref. 5).
The recognition of pathogens by phagocytes is mediated by host bridging proteins called opsonins (such as complement or antibodies) (Ref. 6). The specific recognition of apoptotic cells is achieved through cell-surface receptors for phosphatidylserine or opsonins (such as MFG-E8) (Ref. 7). After binding to these opsonins, phagocytes engulf pathogens or damaged cells, and eliminate them through the generation of reactive oxygen species and hydrolytic enzymes.
Initiation of the innate inflammatory response
Upon recognition of molecules shared by groups of related microbes (called pathogen-associated molecular patterns; PAMPs) by pattern-recognition receptors (such as the Toll-like receptors; TLRs), innate immune cells can initiate an inflammatory response. Well-known PAMPs include bacterial endotoxin (lipopolysacharides; LPSs), peptidoglycan, and microbial unmethylated CpG-DNA (Refs 8, 9). Although there is a structural similarity among various TLRs, each TLR can recognise a specific type of PAMP. For instance, TLR2 is essential for the recognition of lipoproteins, peptidoglycan and lipoteichoic acids of most Gram-positive bacteria (Ref. 10); TLR4 recognises endotoxin of Gram-negative bacteria (Ref. 11); and TLR9 recognises microbial unmethylated CpG-DNA (Ref. 8).
Engagement of various TLRs by specific PAMPs leads to production and release of cytokines (such as TNF and the interleukins IL- 1 and IL-6) and chemokines (such as IL-8, and the ‘macrophage inflammatory proteins’ CCL3 and CCL4) (Ref. 12). Chemokines are responsible for recruiting more innate immune cells to the site of infection or injury (Ref. 13), whereas cytokines can activate these immune cells to produce more pro-inflammatory mediators (Ref. 14). Although an appropriate inflammatory response is required for host defence against infection, an uncontrolled systemic inflammatory response may contribute to the pathogenesis of lethal inflammation diseases such as sepsis.
Systemic innate immune response to severe infection
The prevailing theories of sepsis as an uncontrolled systemic inflammatory response are supported by extensive studies employing various animal models of sepsis.
Animal models of experimental sepsis
Experimental sepsis is induced in animals by three common strategies: infusion of exogenous bacterial toxin (endotoxaemia); infusion of exogenous bacteria (bacteraemia); and faecal contamination of the peritoneal cavity induced by caecal ligation and puncture (CLP). Each of these models has particular strengths and weaknesses with respect to its ability to mimic the clinical progression of human sepsis (Ref. 15).
Endotoxaemia
Endotoxaemia is induced by intraperitoneal or intravenous injection of known amounts of bacterial endotoxin to animals. It provides a model to investigate pathogenic roles of pro-inflammatory mediators in lethal systemic inflammation. Depending on the doses, endotoxin can induce transient/nonlethal or persistent/lethal haemodynamic cardiovascular responses. Thus, endotoxemia is considered as a model of septic shock rather than sepsis (Ref. 15). Other bacterial products (such as CpG-DNA) can also be used to induce septic shock in animals.
Bacteraemia
Bacteraemia is induced by intravenous or intraperitoneal infusion of exogenous viable bacteria into the host. Because many exogenous bacteria may not colonise or replicate well in the host, the doses of bacteria required to induce lethality do not mimic those inducing a typical host response to infection in the clinical setting (Ref. 15). Since various bacteria strains may induce different cytokine responses, the bacteraemia model is useful to study the host response to a particular pathogen.
Caecal ligation and puncture
Sepsis can be induced by surgical perforation of the caecum, a technique known as CLP (Ref. 15). This procedure allows bacteria spillage and faecal contamination of the peritoneal cavity, mimicking the human clinical disease of perforated appendicitis or diverticulitis. The severity of sepsis, as reflected by the eventual mortality rates, can be controlled surgically by varying the size of the needle used for caecal puncture. CLP in animals induces similar, biphasic haemodynamic cardiovascular, metabolic and immunological responses to those observed during the clinical course of human sepsis. Thus, the CLP model is considered as the most clinically relevant model for experimental sepsis.
Inflammatory mediators of experimental sepsis
In response to systemic infection, innate immune cells release large amounts of pro- and anti-inflammatory mediators that collectively determine the outcome of systemic inflammation.
Pro-inflammatory mediators
Various microbial PAMPs (e.g. LPS and CpG-DNA) stimulate innate immune cells to release a wide array of pro-inflammatory mediators, including nitric oxide (Ref. 16), TNF (Ref. 17), IL-1 (Ref. 18), leukaemia inhibitory factor (LIF) (Ref. 19), interferon (IFN)-γ (Ref. 20), and macrophage migration inhibitory factor (MIF) (Refs 21, 22, 23). Extensive studies employing animal models of sepsis suggest that various pro-inflammatory mediators, individually or in combination, contribute to the pathogenesis of lethal systemic inflammation. For instance, neutralising antibodies against bacterial products (such as endotoxin) (Ref. 24) or an early pro-inflammatory cytokine (TNF; Ref. 17) reduce lethality in an animal model of endotoxaemic/bacteraemic shock.
Anti-inflammatory mediators
Microbial products also stimulate innate immune cells to produce anti-inflammatory mediators, such as prostaglandin E2 (PGE2) (Ref. 25), IL-10 (Refs 26, 27) and transforming growth factor (TGF)-β (Refs 28, 29), which counter-regulate or suppress potentially injurious pro-inflammatory mediators. Another local feedback mechanism is through spermine, a ubiquitous biogenic molecule that accumulates at sites of infection or injury and post-transcriptionally inhibits endotoxin-induced release of multiple pro-inflammatory cytokines (e.g. TNF, IL-1β, CCL3 and CCL4) from macrophages and monocytes (Refs 30, 31, 32, 33). Recent evidence suggests that the central nervous system can also attenuate the peripheral innate immune response through efferent vagus nerve signals to tissue-resident macrophages (Ref. 34). This effect is mediated by the principal neurotransmitter of the vagus nerve, acetylcholine, which inactivates macrophages via nicotinic cholinergic receptors (Ref. 34).
Current available therapies for human sepsis
Currently, available therapies for sepsis are still limited to several simple clinical interventions: (1) appropriate broad-spectrum antibiotics; (2) physiological doses of steroidal anti-inflammatory drugs (e.g. hydrocortisone); (3) adjunctive therapy with an anticoagulant agent (e.g. activated protein C); (4) early goal-directed therapies (EGDTs) to restore tissue oxygen delivery; and (5) intensive insulin therapy to regain normoglycaemia (Table 1).
Table 1.
Current available therapies for sepsis

Antibiotics
Once the infecting agents are identified, appropriate broad-spectrum antibiotics are immediately administered to patients to facilitate elimination of bacterial pathogens (Ref. 2). However, administration of antibiotics may also trigger release of bacterial products (such as endotoxin or CpG-DNA) that may further stimulate innate immune cells to release pro-inflammatory cytokines. Thus, anti-inflammatory agents may be useful to pharmacologically modulate a potentially injurious inflammatory response.
Steroidal anti-inflammatory drugs
Steroidal anti-inflammatory drugs refer to a group of steroid-like molecules that can reduce an inflammatory response. Although high-dose steroid (e.g. methylprednisolone at 30 mg/kg or dexamethasone at 0.5 mg/kg body weight/day) was harmful to septic patients (Ref. 35), one study showed low-dose steroid therapy (50 mg hydrocortisone every 6 h, plus 50 µg oral fludrocortisone for 7 days) was beneficial for septic patients with adrenal insufficiency (i.e. poor endogenous responses to steroid-inducing hormones) (Ref. 36). However, a more recent multicentre clinical trial indicated that intravenous hydrocortisone (50 mg every 6 h for 5 days) did not improve 28-day survival of patients with septic shock, regardless of whether patients were responsive or nonresponsive to steroid-inducing hormones (corticotrophin) (Ref. 37). It raises the question of whether the dose and the timing of an anti-inflammatory agent are critical for the successful management of human sepsis (Ref. 2).
Activated protein C
The systemic inflammatory response is integrally related to intravascular coagulation and endothelial activation. As a major regulator of haemostasis, thrombin has both pro- and anti-coagulant properties. The procoagulant activities of thrombin include proteolytic activation of blood-clotting factors (Va, VIIIa and XI), cleavage of fibrinogen to form a fibrin clot, and stimulation of platelet aggregation. The anticoagulant effect of thrombin is regulated by thrombomodulin, a cofactor that is expressed on the luminal surface of vascular endothelium. Following engagement of thrombin to thrombomodulin, the ability of thrombin to catalyse procoagulant reactions is inhibited, but its ability to activate a plasma anticoagulant, activated protein C, is enhanced >1000-fold, which results in inactivation of blood clotting factors. In a large clinical trial, human recombinant activated protein C (drotrecogin alfa) reduced 28-day mortality (from 30.8% in the placebo group to 24.7% in the experimental group) (Ref. 38), but was accompanied by a 1.5% increase in haemorrhagic complication risk (Ref. 38). Thus, activated protein C has been approved by the US Food and Drug Administration only for patients with severe sepsis, who are more likely to die if otherwise not treated.
In addition to activated protein C, other anticoagulation agents, such as tissue factor pathway inhibitor and antithrombin III, have also been tested in clinical sepsis trials. Although both these agents were beneficial in preclinical studies or Phase I or II clinical trials, they failed to reduce 28-day mortality rates in Phase III clinical trials (Refs 39, 40).
Early goal-directed therapies
As a supportive treatment, EGDT employs extremely tight control of several physiological parameters (such as central venous pressure, mean arterial blood pressure, central venous oxygen saturation, and haematocrit) with discrete interventions of crystalloid fluid, vasopressors and blood transfusions. In a controlled, randomised, prospective clinical trial, EGDT – combining volume resuscitation, catecholamine therapy and transfusion – effectively reduced mortality rates of patients with septic shock (from 46.5% in the placebo group to 30.5% in the experimental group) (Ref. 41). While achieving a significant reduction in mortality through relatively simple interventions, this approach is labour intensive, requiring intensive and continuous staff commitment (Ref. 42). Furthermore, a recent multicentre clinical trial indicated that low-dose vasopressin (0.01–0.03 U/min) alone did not significantly reduce the 28-day mortality rate of patients with septic shock (35.4% in the vasopressin group versus 39.3% in the noradenaline control group; P = 0.26) (Ref. 43).
Insulin
In critically ill patients, hyperglycaemia frequently occurs, and this has long been perceived as a beneficial metabolic response to stress that ensures glucose supply to insulin-insensitive organs (such as the brain and the immune system). However, this notion has recently been challenged by two landmark publications revealing that tight blood glucose control with intensive insulin therapy significantly improved morbidity and mortality in critical ill septic patients (Refs 44, 45). Nevertheless, the zeal for infusing insulin has been tempered by the announcement of unsuccessful multicentred clinical trials (Ref. 46).
HMGB1 as a novel therapeutic target for experimental sepsis
The early kinetics of systemic TNF accumulation in sepsis makes it a difficult therapeutic target in clinical settings (Ref. 17), prompting a search for other, late pro-inflammatory mediators that may offer a wider therapeutic window. Here we briefly review evidence that supports extracellular HMGB1 as a potential novel therapeutic target.
Intracellular HMGB1 as a DNA-binding protein
HMGB1 is constitutively expressed in many cell types, and a large ‘pool’ of preformed HMGB1 is stored in the nucleus as a result of the presence of two lysine-rich nuclear localisation sequences (Refs 47, 48). It contains two internal repeats of positively charged domains (‘HMG boxes’ known as the ‘A box’ and ‘B box’) in the N-terminus, and a continuous stretch of negatively charged (aspartic and glutamic acid) residues in the C-terminus. The HMG boxes enable HMGB1 to bind chromosomal DNA and fulfil its nuclear functions, including determination of nucleosomal structure and stability, and regulation of gene expression (Ref. 49).
Extracellular HMGB1 as an alarmin signal
Recently, a number of structurally diverse, multifunctional, ubiquitous host proteins – such as HMGB1 and heat shock protein 72 (HSP72) – have been categorised as ‘alarmins’ based on the following shared properties (Ref. 50) (Fig. 2).
Figure 2.

Extracellular HMGB1 functions as an alarmin signal. HMGB1 is actively secreted by innate immune cells in response to exogenous bacterial products (e.g. endotoxin or CpG-DNA) or endogenous inflammatory stimuli (e.g. TNF, IFN-γ or hydrogen peroxide), and passively released by damaged or virus-infected cells. Extracellular HMGB1 sustains an inflammatory response by stimulating migration of innate immune cells, facilitating innate recognition of bacterial products (e.g. CpG by TLR9 and endotoxin by TLR4), activating various innate immune cells, and inhibiting phagocytosis of apoptotic neutrophils. Thus, HMGB1 can function as an alarmin signal, which recruits, alerts and activates various innate immune cells, thereby sustaining a potentially injurious inflammatory response. Abbreviations: HMGB1, high-mobility group box 1 protein; IFN-γ interferon, γ; TLR, Toll-like receptor; TNF, tumour necrosis factor.
Active release and passive leakage
In response to exogenous bacterial products (such as endotoxin or CpG-DNA) (Refs 51, 52) or endogenous inflammatory stimuli (e.g. TNF, IFN-γ or hydrogen peroxide) (Refs 51, 53, 54), innate immune cells actively release HMGB1 in a dose- and time-dependent manner (Fig. 2). Lacking a leader signal sequence, HMGB1 cannot be actively secreted via the classical secretory pathway from the endoplasmic reticulum through the Golgi complex (Ref. 51). Instead, activated macrophages/monocytes acetylate HMGB1 at its nuclear localisation sequences, leading to sequestration of HMGB1 within cytoplasmic vesicles and subsequent extracellular release (Refs 48, 53, 55). In addition, HMGB1 can be released passively from damaged cells (Ref. 56) or cells infected by viruses (e.g. West Nile virus, Salmon anaemia virus, and Dengue virus) (Refs 57, 58, 59), and such HMGB1 similarly triggers an inflammatory response (Ref. 60) (Fig. 2).
Stimulation of cell migration
Accumulating evidence indicates that HMGB1 can stimulating migration of neurites (Ref. 61), smooth muscle cells (Ref. 62), tumour cells (Ref. 63), mesoangioblast stem cells (Refs 64, 65), monocytes (Ref. 66), dendritic cells (Refs 67, 68) and neutrophils (Ref. 69) (Fig. 2). It raises a possibility that extracellular HMGB1 may recruit cells to sites of infection or injury (Ref. 62), thereby functioning as a potential chemokine (Ref. 70).
Facilitation of innate recognition of microbial products
Recent studies suggested that HMGB1 can facilitate recognition of bacterial products (e.g. CpG-DNA or LPS) by innate immune cells (such as macrophages and dendritic cells) (Refs 52, 71, 72) (Fig. 2). For instance, extracellular HMGB1 can bind to biologically active microbial CpG-DNA, and facilitate its innate recognition by the intracellular TLR9 receptor, thereby augmenting CpG-DNA-induced inflammatory responses (Refs 52, 71).
Activation of innate immune cells
Extracellular HMGB1 binds to several cell-surface receptors, including the receptor for advanced glycation end products (RAGE), and pattern-recognition receptors such as TLR2 and TLR4 (Refs 73, 74). Consequently, HMGB1 activates innate immune cells (Refs 73, 74, 75, 76, 77) or endothelial cells (Refs 78, 79) to produce pro-inflammatory cytokines, chemokines and adhesion molecules (Fig. 2). Notably, the ‘A box’ of HMGB1 functions as an antagonist of HMGB1 (Refs 80, 81), whereas the ‘B box’ recapitulates the cytokine activity of full-length HMGB1 (Refs 82, 83).
In vitro, exogenous HMGB1 appears to accumulate on the macrophage cell surface within 4–6 h of HMGB1 incubation (Ref. 84), which correlates with the kinetics of HMGB1-induced release of pro-inflammatory cytokines (Ref. 85). It is not yet known whether engagement of exogenous HMGB1 to cell-surface receptors (such as TLR2, TLR4 or RAGE) induces cell-surface clustering of ligand–receptor complexes (Ref. 84), thereby activating various innate immune cells.
In the brain, exogenous HMGB1 induces the release of pro-inflammatory cytokines (Ref. 86) and excitatory amino acids (such as glutamate) (Ref. 87), induces fever (Ref. 88), and exacerbates cerebral ischaemic injury (Ref. 89). In the lung, HMGB1 induces neutrophil infiltration and acute injury (Refs 90, 91, 92). Considered together, these studies indicate that extracellular HMGB1 can function as an alarmin signal to recruit, alert and activate innate immune cells, thereby sustaining a potentially injurious inflammatory response.
Inhibition of phagocytotic elimination of apoptotic neutrophils
As mentioned above, macrophages recognise apoptotic cells through cell-surface receptors for phosphatidylserine. Interestingly, a recent study indicated that HMGB1 could interact with phosphatidylserine on the cell surface of apoptotic neutrophils, and consequently inhibit phagocytotic elimination of apoptotic neutrophils by macrophages (Ref. 93). Impaired clearance of apoptotic cells may allow excessive accumulation of late apoptotic and/or secondary necrotic cells, which may directly (Ref. 94), or indirectly (by activating phagocytes), release pro-inflammatory mediators (such as HMGB1) (Ref. 95). Thus, extracellular HMGB1 may sustain rigorous inflammatory responses by multiple mechanisms including interference with phagocytotic elimination of apoptotic neutrophils (Ref. 93) (Fig. 2).
Pathogenic role of HMGB1 in diseases
Accumulating evidence has supported a pathogenic role for extracellular HMGB1 in infection- or injury-elicited inflammatory diseases.
Experimental sepsis
In murine models of endotoxaemia and sepsis (induced by CLP), HMGB1 is first detectable in the circulation 8 h after the onset of lethal endotoxaemia and sepsis, subsequently increasing to plateau levels from 16 to 32 h (Refs 51, 80). This late appearance of circulating HMGB1 precedes and parallels the onset of animal lethality from endotoxaemia or sepsis, and distinguishes HMGB1 from TNF and other early proinflammatory cytokines (Ref. 96) (Fig. 3).
Figure 3.

Early versus late mediators of septic lethality. Vertebrates subjected to septic insult succumb at latencies of up to several days, long after serum TNF has returned to basal levels. By contrast to early pro-inflammatory cytokines, systemic HMGB1 accumulation occurs in a delayed fashion, which precedes and parallels the onset of septic lethality (Refs 96, 146). This delayed systemic accumulation makes HMGB1 a better therapeutic target with a wider therapeutic window for experimental sepsis. CLP, caecal ligation and puncture; HMGB1, high-mobility group box 1 protein; IFN-γ, interferon γ; TNF, tumour necrosis factor.
The pathogenic role of HMGB1 as a late mediator of lethal endotoxaemia was originally examined using HMGB1-specific neutralising antibodies, which conferred a dose-dependent protection against lethal endotoxaemia (Ref. 51) and endotoxin-induced acute lung injury (Ref. 90). In a more clinically relevant animal model of sepsis (induced by CLP), delayed administration of HMGB1-specific neutralising antibodies beginning 24 h after the onset of sepsis, rescued mice from lethal sepsis in a dose-dependent manner (Refs 80, 95). Similarly, anti-HMGB1 antibodies conferred protection in a rat model of sepsis (induced by CLP) (Ref. 97). In contrast, administration of exogenous HMGB1 to mice recapitulates many clinical signs of sepsis, including fever (Ref. 88), derangement of intestinal barrier function (Ref. 98), and tissue injury (Refs 90, 91). Taken together, these experimental data establish extracellular HMGB1 as a critical late mediator of experimental sepsis, with a wider therapeutic window than early pro-inflammatory cytokines (Fig. 3).
Ischaemic tissue injury
By contrast to the delayed systemic HMGB1 accumulation in experimental sepsis, HMGB1 functions as an early mediator of ischaemia–reperfusion (I–R) injury (Refs 99, 100, 101, 102). Prophylactic administration of HMGB1-specific neutralising antibody conferred significant protection against hepatic I–R injury in wild-type mice, but not in a TLR4-defective (C3H/HeJ) mutant, implicating TLR4 in HMGB1-mediated hepatic I–R injury (Ref. 99). Similarly, treatment with HMGB1 antagonist (such as HMGB1 box A) significantly reduced myocardial ischaemic injury in wild-type mice, but in this case not in RAGE-deficient mutants, indicating a potential role for RAGE in HMGB1-mediated ischaemic injury (Ref. 103).
In addition, HMGB1-specific neutralising antibodies have been proven protective against ventilator-induced acute lung injury (Ref. 104), severe acute pancreatitis (Ref. 105), and haemorrhagic shock (Ref. 106), supporting a pathogenic role for extracellular HMGB1 in various inflammatory diseases. However, HMGB1 is capable of attracting stem cells (Ref. 64), and may be important for tissue repair and regeneration. Therefore, like other cytokines, extracellular HMGB1 may have protective roles when released at low amounts (Ref. 107). It is thus important to pharmacologically modulate, rather than abrogate, systemic HMGB1 accumulation to facilitate resolution of a potentially injurious inflammatory response.
Other pro-inflammatory mediators of sepsis
In addition to HMGB1, other pro-inflammatory mediators (such as complement anaphylatoxin, C5a and MIF) also accumulate in the circulation in sepsis (Refs 21, 22, 23, 108), and contribute to the pathogenesis of sepsis. For instance, blockade of MIF with neutralising antibodies as late as 8 h after onset of experimental sepsis improved survival in mice (Ref. 22). Similarly, blockade of C5a or its cell-surface receptors (C5aR or C5L2) with specific neutralising antibodies protects animals against lethal sepsis (Refs 108, 109, 110), supporting a role for C5a in the pathogenesis of sepsis. Intriguingly, C5L2 may play an important role in the regulation of HMGB1 release, because HMGB1 release was somewhat impaired in C5L2-deficient mice following septic insult, and C5L2-deficient peritoneal macrophages following LPS stimulation (Ref. 110). Thus, many known (such as HMGB1, C5a and MIF) or as yet unidentified pro-inflammatory mediators may synergistically interact with each other and collectively contribute to the pathogenesis of sepsis.
Novel HMGB1-targeting therapeutic agents
With a limited number of effective therapies available for patients with sepsis, it is important to search for other agents capable of inhibiting clinically accessible late mediators, such as HMGB1. As discussed below, several agents have been proven protective against experimental sepsis partly through attenuating systemic HMGB1 accumulation (Table 2).
Table 2.
Potential therapeutic agents for experimental sepsis
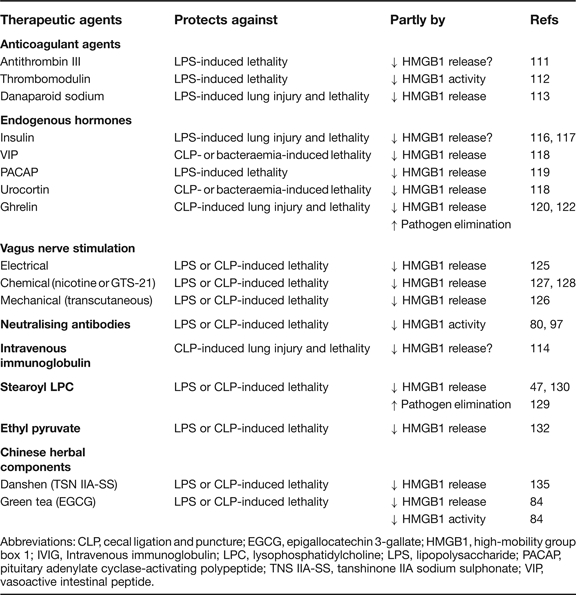
Abbreviations: CLP, cecal ligation and puncture; EGCG, epigallocatechin 3-gallate; HMGB1, high-mobility group box 1; IVIG, Intravenous immunoglobulin; LPC, lysophosphatidylcholine; LPS, lipopolysaccharide; PACAP, pituitary adenylate cyclase-activating polypeptide; TNS IIA-SS, tanshinone IIA sodium sulphonate; VIP, vasoactive intestinal peptide.
Anticoagulant agents
Antithrombin III
Although antithrombin III failed to reduce mortality rate in a large sepsis clinical trial (Ref. 40), a recent study suggested that antithrombin III could attenuate endotoxin-induced systemic HMGB1 accumulation, and reduced endotoxaemic lethality (Ref. 111). The mechanisms by which antithrombin III, a liver-derived anticoagulant glycoprotein, inhibits HMGB1 release remain to be investigated.
Thrombomodulin
As mentioned above, another anticoagulant molecule, thrombomodulin, can interact with thrombin to activate protein C. Interestingly, human soluble thrombomodulin (ART-123) can physically bind to HMGB1 protein, thereby inhibiting an HMGB1-mediated inflammatory response. Indeed, ART-123 conferred significant protection against lethal endotoxaemia partly by attenuating HMGB1-mediated inflammatory response (Ref. 112). It is not yet known, however, whether ART-123 confers similar protection in more clinically relevant animal models of sepsis.
Danaparoid sodium
A third anticoagulant, danaparoid sodium, also prevents blood clotting by inactivating thrombin. It is often used for individuals who cannot be given heparin because of heparin-induced thrombocytopaenia. Intriguingly, danaparoid sodium effectively protected rats against endotoxin-induced acute lung injury by attenuating systemic HMGB1 accumulation (Ref. 113).
Intravenous immunoglobulin
Intravenous immunoglobulin (IVIG) refers to IgG immunoglobulins (antibodies) pooled from the plasma of many healthy blood donors. It is usually given intravenously as a plasma protein replacement therapy to patients with various inflammatory diseases due to acute infections, autoimmune disease, or immune deficiencies. A recent study indicated that IVIG dose-dependently protected rats against sepsis-induced lung injury and lethality by attenuating systemic HMGB1 release (Ref. 114). The mechanisms by which IVIG suppresses systemic HMGB1 release remain poorly understood. Notably, it has recently been found that human IgGs can bind to HMGB1, and potentially interfere with ELISA detection of HMGB1 (Ref. 115). It is thus important to ask whether IVIG indeed attenuates systemic HMGB1 accumulation, or simply interferes with ELISA detection of HMGB1 in serum samples.
Endogenous hormones
Insulin
A recent study indicated that hyperglycaemia, induced by infusion of glucose immediately following endotoxaemia, aggravated endotoxin-induced HMGB1 release and lung injury (Ref. 116). By contrast, intensive blood glucose control by insulin conferred protection against endotoxin-induced acute lung injury, and endotoxaemic lethality (Ref. 116). It is currently unknown whether the observed protective effects are dependent on insulin's anti-inflammatory activities or its blood-glucose-modulating properties (Ref. 117).
Neuropeptides
Vasoactive intestinal peptide (VIP) is a short-lived small peptide hormone that is produced by the gut, pancreas and brain. It can induce smooth muscle relaxation, and is involved in communication between brain neurons. In animal models of sepsis induced by CLP or bacteraemia, administration of VIP attenuated systemic HMGB1 accumulation, and consequently reduced animal lethality (Ref. 118). Consistently, replenishing septic animals with recombinant HMGB1 completely reversed VIP-mediated protective effects (Ref. 118), confirming a pathogenic role for HMGB1 in experimental sepsis.
Another member of the VIP family, the pituitary adenylate cyclase-activating polypeptide (PACAP), shares 68% amino acid sequence identity with VIP. It is abundantly expressed in the central and peripheral nervous systems, and functions as a parasympathetic and sensory neurotransmitter. Interestingly, administration of PACAP peptide also significantly attenuated circulating HMGB1 levels, and similarly protected mice against lethal endotoxaemia (Ref. 119).
The neuropeptide urocortin, which belongs to the corticotropin-releasing factor family, is expressed in the brain and may be responsible for regulation of appetite. In animal models of sepsis induced by CLP or bacteraemia, administration of urocortin attenuated systemic HMGB1 accumulation and reduced animal lethality (Ref. 118), supporting a therapeutic potential for neuropeptides in experimental sepsis.
Ghrelin
Ghrelin is a stomach-derived hormone that is responsible for regulating the appetite – increasing it before eating and decreasing it afterwards. Intriguingly, plasma ghrelin levels are significantly decreased in septic animals (Ref. 120), and administration of ghrelin promoted a dose-dependent protection against sepsis-induced acute lung injury and lethality (Refs 120, 121, 122). Ghrelin may exert its protective effects through multiple mechanisms, such as by attenuating systemic HMGB1 release and by facilitating bacterial elimination (Ref. 122). Intriguingly, ghrelin may attenuate systemic accumulation of pro-inflammatory cytokines partly via the vagus nerve (Ref. 121), suggesting that pharmacological stimulation of the vagus nerve may be an effective therapy for experimental sepsis.
Vagus nerve stimulation
The vagus nerve is the structural basis for the cholinergic anti-inflammatory pathway (Ref. 123), which inhibits the innate immune response via the release of acetylcholine. Acetylcholine binds to α7 nicotinic acetylcholine receptors of various innate immune cells (Ref. 124), thereby counter-regulating potentially injurious innate immune responses. Indeed, stimulation of the vagus nerve by physical methods (e.g. electrical or mechanical) (Refs 125, 126) or chemical agents (such as the cholinergic agonists nicotine and GTS-21) (Refs 127, 128) conferred protection against lethal endotoxaemia and sepsis partly by attenuating systemic HMGB1 accumulation.
Stearoyl lysophosphatidylcholine
An endogenous phospholipid, stearoyl lysophosphatidylcholine (LPC), has recently been proven protective against experimental sepsis by stimulating neutrophils to destroy ingested bacteria in a mechanism dependent on hydrogen peroxide (Ref. 129). However, stearoyl LPC also confers protection against lethal endotoxaemia (Ref. 129), implying that it may exert protective effects through an additional, bactericidal-independent mechanism (Ref. 130). Indeed, administration of stearoyl LPC significantly attenuated circulating HMGB1 levels (Ref. 47), indicating that stearoyl LPC protects against experimental sepsis partly by facilitating elimination of invading pathogens and partly by attenuating systemic HMGB1 accumulation (Ref. 130).
Ethyl pyruvate
Ethyl pyruvate is an aliphatic ester derived from pyruvic acid, which is a final product of glycolysis and the starting substrate for the tricarboxylic acid cycle (Ref. 131). It dose-dependently inhibits LPS-induced release of early (e.g. TNF) and late (e.g. HMGB1) pro-inflammatory cytokines, and protected mice against experimental sepsis even when treatment was started as late as 12–24 h after the onset of disease (Ref. 132).
Chinese medicinal herbs
Traditional herbal medicine has formed the basis of folk remedies for various inflammatory ailments. Out of several dozen commonly used Chinese herbs (Ref. 133), we found that aqueous extracts of danggui (Angelica sinensis), green tea (Camellia sinensis), and danshen (Salvia miltorrhiza) efficiently inhibited endotoxin-induced HMGB1 release, and protected animals against experimental sepsis (Refs 84, 134, 135).
Danggui
Danggui has been traditionally used to treat gynaecological disorders (such as abnormal menstruation) (Ref. 136). Its aqueous extract dose-dependently inhibited LPS-induced HMGB1 release in macrophage and monocyte cultures, partly by interfering with HMGB1 cytoplasmic translocation (Ref. 134). Furthermore, danggui extract rescued mice from lethal sepsis even when the first dose was given at 24 h after onset of disease (Ref. 134). The active components responsible for these beneficial effects remain a subject of future investigation.
Green tea
Green tea brewed from the leaves of Camellia sinensis contains a class of biologically active polyphenols called catechins. Epigallocatechin 3-gallate (EGCG), which accounts for 50–80% of the total catechin, is effective in attenuating endotoxin-induced HMGB1 release by macrophage and monocytes (Ref. 84). In addition, EGCG dose-dependently inhibited HMGB1-induced release of TNF, IL-6 and nitric oxide in macrophage cultures (Ref. 84). Interestingly, EGCG completely abrogated accumulation/clustering of exogenous HMGB1 on the macrophage cell surface (Ref. 84), suggesting that EGCG inhibits HMGB1 cytokine activities by preventing its cell-surface accumulation/clustering.
In vivo, repeated administration of EGCG conferred a dose-dependent protection against lethal endotoxaemia, and rescued mice from lethal sepsis even when the first dose of EGCG was given at >24 h after onset of sepsis (Ref. 84). Consistently, delayed administration of EGCG significantly attenuated circulating levels of HMGB1, as well as surrogate markers of experimental sepsis (such as IL-6 and chemokine KC) (Refs 84, 137). Considered together, these experimental data indicate that EGCG protects mice against lethal sepsis partly by attenuating systemic HMGB1 accumulation, and partly by inhibiting HMGB1-mediated inflammatory response.
Danshen
Danshen has been widely used in China for patients with cardiovascular disorders (Refs 138, 139). Danshen contains abundant red pigments (termed tanshinone I, tanshinone IIA, and cryptotanshinone) (Fig. 4), which effectively attenuated LPS-induced HMGB1 release (Ref. 135). A water-soluble derivative (sodium sulphonate) of tanshinone IIA (TSN IIA-SS) at concentrations (100 µm) that completely abrogated LPS-induced HMGB1 release, only partially attenuated LPS-induced release of four out of 62 cytokines (IL-12p70, IL-1α, platelet factor 4 and CCL12) (Ref. 135), indicating a specificity for TSN IIA-SS in inhibiting LPS-induced HMGB1 release. Despite a structural resemblance (i.e. the presence of a fused four-ring structure) between tanshinones and steroidal anti-inflammatory drugs (such as dexamethasone and cortisone) (Fig. 4), tanshinones inhibit LPS-induced HMGB1-release in a glucocorticoid-receptor-independent mechanism (Ref. 135).
Figure 4.

Steroid-like tanshinones and water-soluble derivatives. Several steroid-like pigments (tanshinone I, tanshinone IIA, and cryptotanshinone) of the medicinal Chinese herb danshen (Salvia miltiorrhiza) are structurally similar to steroidal anti-inflammatory drugs (such as dexamethasone and cortisone) – that is, they all have a fused four-ring structure (Ref. 133). Tanshinone IIA sodium sulphonate is water-soluble, and widely used in China as a cardiovascular medicine. MW, molecular weight.
More importantly, repeated administration of TSN IIA-SS, beginning at >24 h and followed by additional doses at >48, >72 and >96 h after the onset of sepsis, dose-dependently rescued mice from lethal sepsis (Ref. 135). Notably, administration of TNS IIA-SS dose-dependently attenuated circulating HMGB1 levels in septic mice (Ref. 135), suggesting that TSN IIA-SS confers protection against experimental sepsis partly by inhibiting systemic HMGB1 accumulation.
Clinical implications
For complex systemic inflammatory diseases such as sepsis, it appears difficult to translate successful animal studies into clinical applications. For instance, although neutralising antibodies against endotoxin (Ref. 24) or cytokines (e.g. TNF) (Refs 17, 140) are protective in animal models of endotoxaemia or bacteraemia, these agents failed in sepsis clinical trials (Refs 141, 142, 143). This failure partly reflects the complexity of the underlying pathogenic mechanisms of sepsis and the heterogeneity of the patient population (Refs 2, 144). It may also be attributable to pitfalls in the selection of (1) feasible therapeutic targets or drugs, (2) optimal doses and timing of drugs, and (3) nonrealistic clinical outcome measures (such as mortality rates) (Refs 2, 144).
Nevertheless, the investigation of pathogenic cytokines in animal models of diseases has led to the development of anti-TNF therapy for patients with debilitating chronic inflammatory diseases, such as rheumatoid arthritis (Ref. 145). Consequently, a chimaeric anti-TNF monoclonal antibody (infliximab) and a soluble TNF-receptor–Fc fusion protein (sTNF-R–Fc; etanercept) have been approved by regulatory authorities in the USA and Europe for treating rheumatoid arthritis. Since pro-inflammatory cytokines are indeed pathogenic in human inflammatory diseases (such as rheumatoid arthritis), it is necessary to continue the search for clinically feasible therapeutic targets and drugs for other inflammatory diseases (such as sepsis).
Will HMGB1 ever become a clinically feasible therapeutic target for human sepsis? We cannot answer this question until HMGB1-neutralising antibodies have been tested for efficacy in large clinical trials. Although HMGB1 appears to be a feasible therapeutic target for experimental sepsis (Refs 96, 146, 147), its levels in unfractionated crude serum of septic patients did not correlated well with disease severity (Refs 148, 149). Upon separation of serum proteins by ultrafiltration through membrane with a defined molecular weight cut-off (100 kDa), a 30 kDa HMGB1 band was detected (by western blotting analysis) in both low (<100 kDa) and high (>100 kDa) molecular weight serum fractions of many septic patients. Furthermore, HMGB1 levels in the low (<100 kDa) serum fraction were significantly higher in septic patients who died of sepsis than those who survived (Ref. 51). This observation suggested a possibility that HMGB1 may interact with other serum components to form large (>100 kDa) complexes.
Indeed, many exogenous bacterial products (such as endotoxin or CpG-DNA) (Refs 52, 71, 72) or endogenous proteins (such as human thrombomodulin, IgG1 or IL-1) (Refs 115, 150) may physically interact with HMGB1 to form various complexes. It is not yet known how these and as yet unidentified HMGB1-binding molecules affect the biological activities, or immunodetection, of HMGB1 in septic patients (Refs 115, 148). In addition, chemical modifications may similarly affect the biological activities of HMGB1. For instance, a recent study indicated that reactive oxygen species (ROS) may oxidise HMGB1 to form an intramolecular disulphide bond between the thiol group of Cys106 and Cys23 or Cys45, and consequently abolish HMGB1-mediated immunostimulatory activities (Ref. 151). Because Cys106 is located within the 18-amino-acid cytokine domain of HMGB1 B box, it will be important to investigate whether oxidisation similarly affects biological activities of HMGB1 in future studies.
Will any specific HMGB1 inhibitor ever become a therapeutic agent for human sepsis? One of the most selective HMGB1 inhibitors, TSN IIA-SS, has already been used in China as a medicine for patients with cardiovascular disorders (Ref. 138). Even in septic animals, TSN IIA-SS reduced total peripheral vascular resistance, and yet increased cardiac stroke volume and cardiac output (Ref. 135). Because HMGB1 may function as a myocardial depressant factor by reducing contractility of cardiac myocytes (Ref. 152), it is plausible that TSN IIA-SS improves cardiovascular function partly by attenuating HMGB1 release. The dual effects of TSN IIA-SS in attenuating late inflammatory response and improving cardiovascular function make it a promising therapeutic agent for sepsis.
Conclusions and perspectives
The ubiquitous nuclear protein HMGB1 is released by activated macrophages/monocytes, and functions as a late mediator of experimental sepsis. First, circulating HMGB1 levels are elevated in a delayed fashion in endotoxaemic and septic animals. Second, administration of exogenous HMGB1 to mice induces fever, derangement of intestinal barrier function, and tissue injury. Third, administration of anti-HMGB1 antibodies or inhibitors rescues mice from lethal experimental sepsis even when the first dose is given 24 h after onset of sepsis. Taken together, these data establish HMGB1 as a late mediator of experimental sepsis with a wider therapeutic window than early mediators such as TNF.
HMGB1-specific neutralising antibodies and small-molecule inhibitors (such as tanshinone IIA derivative) have been proven protective in animal models of experimental sepsis. Currently, the intricate mechanisms by which various agents attenuate systemic HMGB1 release and protect against experimental sepsis remain poorly understood. In addition, it is not yet known whether a better protection could be achieved by combinational therapy with several anti-HMGB1 agents. It is thus important to further explore the therapeutic potential of these HMGB1-inhibiting agents in future studies.
Acknowledgements and funding
We are grateful to the peer reviewers for their critical and constructive comments. Work in the authors' laboratories was supported by grants from the National Institutes of Health, National Institute of General Medical Science (R01GM063075 and R01GM070817 to H.W.).
References
- 1.Serhan C.N.. Savill J.. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. and . [DOI] [PubMed] [Google Scholar]
- 2.Dellinger R.P.. et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36:296–327. doi: 10.1097/01.CCM.0000298158.12101.41. [DOI] [PubMed] [Google Scholar]
- 3.Luster A.D., Alon R.. von Andrian U.H.. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. , and . [DOI] [PubMed] [Google Scholar]
- 4.Scapini P.. et al. The neutrophil as a cellular source of chemokines. Immunol Rev. 2000;177:195–203. doi: 10.1034/j.1600-065x.2000.17706.x. [DOI] [PubMed] [Google Scholar]
- 5.Liles W.C.. Immunomodulatory approaches to augment phagocyte-mediated host defense for treatment of infectious diseases. Semin Respir Infect. 2001;16:11–17. doi: 10.1053/srin.2001.22724. [DOI] [PubMed] [Google Scholar]
- 6.Mosser D.M.. Receptors on phagocytic cells involved in microbial recognition. Immunol Ser. 1999;60:99–114. [PubMed] [Google Scholar]
- 7.Wu Y., Tibrewal N.. Birge R.B.. Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol. 2006;16:189–197. doi: 10.1016/j.tcb.2006.02.003. and . [DOI] [PubMed] [Google Scholar]
- 8.Hemmi H.. et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 9.Krieg A.M.. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 10.Brightbill H.D.. et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999;285:732–736. doi: 10.1126/science.285.5428.732. [DOI] [PubMed] [Google Scholar]
- 11.Poltorak A.. et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 12.Akira S.. Takeda K.. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. and . [DOI] [PubMed] [Google Scholar]
- 13.Baggiolini M.. Loetscher P.. Chemokines in inflammation and immunity. Immunol Today. 2000;21:418–420. doi: 10.1016/s0167-5699(00)01672-8. and . [DOI] [PubMed] [Google Scholar]
- 14.Balkwill F.. Cytokines-soluble factors in immune responses. Curr Opin Immunol. 1988;1:241–249. doi: 10.1016/0952-7915(88)90008-8. [DOI] [PubMed] [Google Scholar]
- 15.Wichterman K.A., Baue A.E.. Chaudry I.H.. Sepsis and septic shock–a review of laboratory models and a proposal. J Surg Res. 1980;29:189–201. doi: 10.1016/0022-4804(80)90037-2. and . [DOI] [PubMed] [Google Scholar]
- 16.Lowenstein C.J., Dinerman J.L.. Snyder S.H.. Nitric oxide: a physiologic messenger. Ann Intern Med. 1994;120:227–237. doi: 10.7326/0003-4819-120-3-199402010-00009. and . [DOI] [PubMed] [Google Scholar]
- 17.Tracey K.J.. et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330:662–664. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- 18.Dinarello C.A.. Thompson R.C.. Blocking IL-1: interleukin 1 receptor antagonist in vivo and in vitro. Immunol Today. 1991;12:404–410. doi: 10.1016/0167-5699(91)90142-G. and . [DOI] [PubMed] [Google Scholar]
- 19.Block M.I.. et al. Passive immunization of mice against D factor blocks lethality and cytokine release during endotoxemia. J Exp Med. 1993;178:1085–1090. doi: 10.1084/jem.178.3.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinzel F.P.. The role of IFN-gamma in the pathology of experimental endotoxemia. J Immunol. 1990;145:2920–2924. [PubMed] [Google Scholar]
- 21.Bernhagen J.. et al. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365:756–759. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 22.Calandra T.. et al. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med. 2000;6:164–170. doi: 10.1038/72262. [DOI] [PubMed] [Google Scholar]
- 23.Bozza M.. et al. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med. 1999;189:341–346. doi: 10.1084/jem.189.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis C.E.. et al. Prevention of death from endotoxin with antisera. I. The risk of fatal anaphylaxis to endotoxin. J Immunol. 1969;102:563–572. [PubMed] [Google Scholar]
- 25.Knudsen P.J.. et al. Prostaglandins posttranscriptionally inhibit monocyte expression of interleukin 1 activity by increasing intracellular cyclic adenosine monophosphate. J Immunol. 1986;137:3189–3194. [PubMed] [Google Scholar]
- 26.Oswald I.P.. et al. Interleukin 10 inhibits macrophage microbicidal activity by blocking the endogenous production of tumor necrosis factor alpha required as a costimulatory factor for interferon gamma-induced activation. Proc Natl Acad Sci U S A. 1992;89:8676–8680. doi: 10.1073/pnas.89.18.8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bogdan C., Vodovotz Y.. Nathan C.. Macrophage deactivation by interleukin 10. J Exp Med. 1991;174:1549–1555. doi: 10.1084/jem.174.6.1549. and . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsunawaki S.. et al. Deactivation of macrophages by transforming growth factor-beta. Nature. 1988;334:260–262. doi: 10.1038/334260a0. [DOI] [PubMed] [Google Scholar]
- 29.Miller-Graziano C.L.. et al. Role of elevated monocyte transforming growth factor beta (TGF beta) production in posttrauma immunosuppression. J Clin Immunol. 1991;11:95–102. doi: 10.1007/BF00917745. [DOI] [PubMed] [Google Scholar]
- 30.Zhang M.. et al. Spermine inhibits proinflammatory cytokine synthesis in human mononuclear cells: a counterregulatory mechanism that restrains the immune response. J Exp Med. 1997;185:1759–1768. doi: 10.1084/jem.185.10.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang M.. et al. Spermine inhibition of monocyte activation and inflammation. Mol Med. 1999;5:595–605. [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H.. et al. Fetuin (alpha2-HS-glycoprotein) opsonizes cationic macrophagedeactivating molecules. Proc Natl Acad Sci U S A. 1998;95:14429–14434. doi: 10.1073/pnas.95.24.14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang M., Wang H.. Tracey K.J.. Regulation of macrophage activation and inflammation by spermine: a new chapter in an old story. Crit Care Med. 2000;28:N60–N66. doi: 10.1097/00003246-200004001-00007. and . [DOI] [PubMed] [Google Scholar]
- 34.Borovikova L.V.. et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 35.Lefering R.. Neugebauer E.A.. Steroid controversy in sepsis and septic shock: a meta-analysis. Crit Care Med. 1995;23:1294–1303. doi: 10.1097/00003246-199507000-00021. and . [DOI] [PubMed] [Google Scholar]
- 36.Annane D.. et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862–871. doi: 10.1001/jama.288.7.862. [DOI] [PubMed] [Google Scholar]
- 37.Sprung C.L.. et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358:111–124. doi: 10.1056/NEJMoa071366. [DOI] [PubMed] [Google Scholar]
- 38.Bernard G.R.. et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 39.Warren B.L.. et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001;286:1869–1878. doi: 10.1001/jama.286.15.1869. [DOI] [PubMed] [Google Scholar]
- 40.Abraham E.. et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003;290:238–247. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]
- 41.Rivers E.. et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368–1377. doi: 10.1056/NEJMoa010307. [DOI] [PubMed] [Google Scholar]
- 42.Sama A.E.. et al. Bench to bedside: HMGB1-a novel proinflammatory cytokine and potential therapeutic target for septic patients in the emergency department. Acad Emerg Med. 2004;11:867–873. doi: 10.1197/j.aem.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 43.Russell J.A.. et al. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med. 2008;358:877–887. doi: 10.1056/NEJMoa067373. [DOI] [PubMed] [Google Scholar]
- 44.van den Berghe G.. et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359–1367. doi: 10.1056/NEJMoa011300. [DOI] [PubMed] [Google Scholar]
- 45.van den Berghe G.. et al. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:449–461. doi: 10.1056/NEJMoa052521. [DOI] [PubMed] [Google Scholar]
- 46.Brunkhorst F.M.. et al. Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med. 2008;358:125–139. doi: 10.1056/NEJMoa070716. [DOI] [PubMed] [Google Scholar]
- 47.Chen G.. et al. Suppression of HMGB1 release by stearoyl lysophosphatidylcholine:an additional mechanism for its therapeutic effects in experimental sepsis. J Lipid Res. 2005;46:623–627. doi: 10.1194/jlr.C400018-JLR200. [DOI] [PubMed] [Google Scholar]
- 48.Bonaldi T.. et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bustin M.. At the crossroads of necrosis and apoptosis: signaling to multiple cellular targets by HMGB1. Sci STKE. 2002;2002:E39. doi: 10.1126/stke.2002.151.pe39. [DOI] [PubMed] [Google Scholar]
- 50.Oppenheim J.J.. Yang D.. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. 2005;17:359–365. doi: 10.1016/j.coi.2005.06.002. and . [DOI] [PubMed] [Google Scholar]
- 51.Wang H.. et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 52.Ivanov S.. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood. 2007;110:1970–1981. doi: 10.1182/blood-2006-09-044776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rendon-Mitchell B.. et al. IFN-gamma Induces High Mobility Group Box 1 Protein Release Partly Through a TNF-Dependent Mechanism. J Immunol. 2003;170:3890–3897. doi: 10.4049/jimmunol.170.7.3890. [DOI] [PubMed] [Google Scholar]
- 54.Tang D.. et al. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J Leukoc Biol. 2007;81:741–747. doi: 10.1189/jlb.0806540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gardella S.. et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:955–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scaffidi P., Misteli T.. Bianchi M.E.. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. and . [DOI] [PubMed] [Google Scholar]
- 57.Chu J.J.. Ng M.L.. The mechanism of cell death during West Nile virus infection is dependent on initial infectious dose. J Gen Virol. 2003;84:3305–3314. doi: 10.1099/vir.0.19447-0. and . [DOI] [PubMed] [Google Scholar]
- 58.Joseph T.. et al. Mechanism of cell death during infectious salmon anemia virus infection is cell type-specific. J Gen Virol. 2004;85:3027–3036. doi: 10.1099/vir.0.80091-0. [DOI] [PubMed] [Google Scholar]
- 59.Chen L.C.. et al. Dengue virus infection induces passive release of high mobility group box 1 protein by epithelial cells. J Infect. 2008;56:143–150. doi: 10.1016/j.jinf.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 60.Wang H.. et al. Potential role of high mobility group box 1 in viral infectious diseases. Viral Immunol. 2006;19:3–9. doi: 10.1089/vim.2006.19.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fages C.. et al. Regulation of cell migration by amphoterin. J Cell Sci. 2000;113((Pt 4)):611–620. doi: 10.1242/jcs.113.4.611. [DOI] [PubMed] [Google Scholar]
- 62.Degryse B.. et al. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. 2001;152:1197–1206. doi: 10.1083/jcb.152.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huttunen H.J.. et al. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 2002;62:4805–4811. [PubMed] [Google Scholar]
- 64.Palumbo R.. et al. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol. 2004;164:441–449. doi: 10.1083/jcb.200304135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Palumbo R.. et al. Cells migrating to sites of tissue damage in response to the danger signal HMGB1 require NF-kappaB activation. J Cell Biol. 2007;179:33–40. doi: 10.1083/jcb.200704015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rouhiainen A.. et al. Regulation of monocyte migration by amphoterin (HMGB1) Blood. 2004;104:1174–1182. doi: 10.1182/blood-2003-10-3536. [DOI] [PubMed] [Google Scholar]
- 67.Yang D.. et al. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J Leukoc Biol. 2007;81:59–66. doi: 10.1189/jlb.0306180. [DOI] [PubMed] [Google Scholar]
- 68.Dumitriu I.E.. et al. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukoc Biol. 2007;81:84–91. doi: 10.1189/jlb.0306171. [DOI] [PubMed] [Google Scholar]
- 69.Orlova V.V.. et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007;26:1129–1139. doi: 10.1038/sj.emboj.7601552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Degryse B.. de Virgilio M.. The nuclear protein HMGB1, a new kind of chemokine? FEBS Lett. 2003;553:11–17. doi: 10.1016/s0014-5793(03)01027-5. and . [DOI] [PubMed] [Google Scholar]
- 71.Tian J.. et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 72.Silva E.. et al. HMGB1 and LPS induce distinct patterns of gene expression and activation in neutrophils from patients with sepsis-induced acute lung injury. Intensive Care Med. 2007;33:1829–1839. doi: 10.1007/s00134-007-0748-2. [DOI] [PubMed] [Google Scholar]
- 73.Park J.S.. et al. Involvement of TLR 2 and TLR 4 in cellular activation by high mobility group box 1 protein (HMGB1) J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 74.Yu M.. et al. HMGB1 signals through Toll-like Receptor (TLR) 4 and TLR2. Shock. 2006;26:174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 75.Park J.S.. et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol 290, C917-C924. 2006 doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 76.Kokkola R.. et al. RAGE is the Major Receptor for the Proinflammatory Activity of HMGB1 in Rodent Macrophages. Scand J Immunol. 2005;61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- 77.Pedrazzi M.. et al. Selective proinflammatory activation of astrocytes by high-mobility group box 1 protein signaling. J Immunol. 2007;179:8525–8532. doi: 10.4049/jimmunol.179.12.8525. [DOI] [PubMed] [Google Scholar]
- 78.Fiuza C.. et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 79.Treutiger C.J.. et al. High mobility group 1 B-box mediates activation of human endothelium. J Intern Med. 2003;254:375–385. doi: 10.1046/j.1365-2796.2003.01204.x. [DOI] [PubMed] [Google Scholar]
- 80.Yang H.. et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kokkola R.. et al. Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. 2003;48:2052–2058. doi: 10.1002/art.11161. [DOI] [PubMed] [Google Scholar]
- 82.Li J.. et al. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med. 2003;9:37–45. [PMC free article] [PubMed] [Google Scholar]
- 83.Messmer D.. et al. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J Immunol. 2004;173:307–313. doi: 10.4049/jimmunol.173.1.307. [DOI] [PubMed] [Google Scholar]
- 84.Li W.. et al. A major ingredient of green tea rescues mice from lethal sepsis partly by inhibiting HMGB1. PLoS ONE 2, e1153. 2007 doi: 10.1371/journal.pone.0001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Andersson U.. et al. High Mobility Group 1 Protein (HMG-1) Stimulates Proinflammatory Cytokine Synthesis in Human Monocytes. J Exp Med. 2000;192:565–570. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Agnello D.. et al. HMGB1, a DNA-binding protein with cytokine activity, induces brain TNF and IL-6 production, and mediates anorexia and taste aversion. Cytokine. 2002;18:231–236. doi: 10.1006/cyto.2002.0890. [DOI] [PubMed] [Google Scholar]
- 87.Pedrazzi M.. et al. Stimulation of excitatory amino acid release from adult mouse brain glia subcellular particles by high mobility group box 1 protein. J. Neurochem. 2006;99:827–838. doi: 10.1111/j.1471-4159.2006.04120.x. [DOI] [PubMed] [Google Scholar]
- 88.O'Connor K.A.. et al. Further characterization of high mobility group box 1 (HMGB1) as a proinflammatory cytokine: central nervous system effects. Cytokine. 2003;24:254–265. doi: 10.1016/j.cyto.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 89.Liu K.. et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007;21:3904–3916. doi: 10.1096/fj.07-8770com. [DOI] [PubMed] [Google Scholar]
- 90.Abraham E.. et al. HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165:2950–2954. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- 91.Ueno H.. et al. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am J Respir Crit Care Med. 2004;170:1310–1316. doi: 10.1164/rccm.200402-188OC. [DOI] [PubMed] [Google Scholar]
- 92.Lin X.. et al. {alpha}-Chemokine receptor blockade reduces high mobility group box 1 protein-induced lung inflammation and injury and improves survival in sepsis. Am J Physiol Lung Cell Mol Physiol. 2005;289:L583–L590. doi: 10.1152/ajplung.00091.2005. [DOI] [PubMed] [Google Scholar]
- 93.Liu G.. et al. High mobility group protein-1 Inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J Immunol. 2008;181:4240–4246. doi: 10.4049/jimmunol.181.6.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bell C.W.. et al. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol. 2006;291:C1318–1325. doi: 10.1152/ajpcell.00616.2005. [DOI] [PubMed] [Google Scholar]
- 95.Qin S.. et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. 2006;203:1637–1642. doi: 10.1084/jem.20052203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang H.. et al. HMGB1 as a Late Mediator of Lethal Systemic Inflammation. Am J Respir Crit Care Med. 2001;164:1768–1773. doi: 10.1164/ajrccm.164.10.2106117. [DOI] [PubMed] [Google Scholar]
- 97.Suda K.. et al. Anti-high-mobility group box chromosomal protein 1 antibodies improve survival of rats with sepsis. World J Surg. 2006;30:1755–1762. doi: 10.1007/s00268-005-0369-2. [DOI] [PubMed] [Google Scholar]
- 98.Sappington P.L.. et al. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology. 2002;123:790–802. doi: 10.1053/gast.2002.35391. [DOI] [PubMed] [Google Scholar]
- 99.Tsung A.. et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Oozawa A.. et al. Effects of HMGB1 on ischemia-reperfusion injury in the rat heart. Circ J. 2008;72:1178–1184. doi: 10.1253/circj.72.1178. [DOI] [PubMed] [Google Scholar]
- 101.Wu H.. et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu K.. et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007;21:3904–3916. doi: 10.1096/fj.07-8770com. [DOI] [PubMed] [Google Scholar]
- 103.Andrassy M.. et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation. 2008;117:3216–3226. doi: 10.1161/CIRCULATIONAHA.108.769331. [DOI] [PubMed] [Google Scholar]
- 104.Ogawa E.N.. et al. Contribution of high-mobility group box-1 to the development of ventilator-induced lung injury. Am J Respir Crit Care Med. 2006;174:400–407. doi: 10.1164/rccm.200605-699OC. [DOI] [PubMed] [Google Scholar]
- 105.Sawa H.. et al. Blockade of high mobility group box-1 protein attenuates experimental severe acute pancreatitis. World J Gastroenterol. 2006;12:7666–7670. doi: 10.3748/wjg.v12.i47.7666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang R.. et al. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med. 2006;12:105–114. doi: 10.2119/2006-00010.Yang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li W., Sama A.E.. Wang H.. Role of HMGB1 in cardiovascular diseases. Curr Opin Pharmacol. 2006;6:130–135. doi: 10.1016/j.coph.2005.10.010. and . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Czermak B.J.. et al. Protective effects of C5a blockade in sepsis. Nat Med. 1999;5:788–792. doi: 10.1038/10512. [DOI] [PubMed] [Google Scholar]
- 109.Riedemann N.C.. et al. Increased C5a receptor expression in sepsis. J Clin Invest. 2002;110:101–108. doi: 10.1172/JCI15409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rittirsch D.. et al. Functional roles for C5a receptors in sepsis. Nat Med. 2008;14:551–557. doi: 10.1038/nm1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hagiwara S.. et al. High dose antithrombin III inhibits HMGB1 and improves endotoxin-induced acute lung injury in rats. Intensive Care Med. 2008;34:361–367. doi: 10.1007/s00134-007-0887-5. [DOI] [PubMed] [Google Scholar]
- 112.Abeyama K.. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. 2005;115:1267–1274. doi: 10.1172/JCI22782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hagiwara S.. et al. Danaparoid sodium inhibits systemic inflammation and prevents endotoxin-induced acute lung injury in rats. Crit Care. 2008;12:R43. doi: 10.1186/cc6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hagiwara S.. et al. High-dose intravenous immunoglobulin G improves systemic inflammation in a rat model of CLP-induced sepsis. Intensive Care Med. 2008;34:1812–1819. doi: 10.1007/s00134-008-1161-1. [DOI] [PubMed] [Google Scholar]
- 115.Urbonaviciute V.. et al. Factors masking HMGB1 in human serum and plasma. J Leukoc Biol. 2007;81:67–74. doi: 10.1189/jlb.0306196. [DOI] [PubMed] [Google Scholar]
- 116.Hagiwara S.. et al. Effects of hyperglycemia and insulin therapy on high mobility group box 1 in endotoxin-induced acute lung injury in a rat model. Crit Care Med. 2008;36:2407–2413. doi: 10.1097/CCM.0b013e318180b3ba. [DOI] [PubMed] [Google Scholar]
- 117.Wang H.. et al. Hyperglycemia aggravates endotoxin-induced high mobility group box 1 protein release: yet another reason not to be too sweet. Crit Care Med. 2008;36:2475–2476. doi: 10.1097/CCM.0b013e318181159c. [DOI] [PubMed] [Google Scholar]
- 118.Chorny A.. Delgado M.. Neuropeptides rescue mice from lethal sepsis by down-regulating secretion of the late-acting inflammatory mediator high mobility group box 1. Am J Pathol. 2008;172:1297–1307. doi: 10.2353/ajpath.2008.070969. and . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tang Y.. et al. PACAP inhibit the release and cytokine activity of HMGB1 and improve the survival during lethal endotoxemia. Int Immunopharmacol. 2008;8:1646–1651. doi: 10.1016/j.intimp.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 120.Wu R.. et al. Ghrelin attenuates sepsis-induced acute lung injury and mortality in rats. Am J Respir Crit Care Med. 2007;176:805–813. doi: 10.1164/rccm.200604-511OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wu R.. et al. Ghrelin down-regulates proinflammatory cytokines in sepsis through activation of the vagus nerve. Ann Surg. 2007;245:480–486. doi: 10.1097/01.sla.0000251614.42290.ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chorny A.. et al. Ghrelin protects against experimental sepsis by inhibiting high-mobility group box 1 release and by killing bacteria. J Immunol. 2008;180:8369–8377. doi: 10.4049/jimmunol.180.12.8369. [DOI] [PubMed] [Google Scholar]
- 123.Tracey K.J.. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117:289–296. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang H.. et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 125.Huston J.M.. et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med. 2006;203:1623–1628. doi: 10.1084/jem.20052362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Huston J.M.. et al. Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit Care Med. 2007;35:2762–2768. doi: 10.1097/01.CCM.0000288102.15975.BA. [DOI] [PubMed] [Google Scholar]
- 127.Wang H.. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med. 2004;10:1216–1221. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- 128.Pavlov V.A.. et al. Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit Care Med. 2007;35:1139–1144. doi: 10.1097/01.CCM.0000259381.56526.96. [DOI] [PubMed] [Google Scholar]
- 129.Yan J.J.. et al. Therapeutic effects of lysophosphatidylcholine in experimental sepsis. Nat Med. 2004;10:161–167. doi: 10.1038/nm989. [DOI] [PubMed] [Google Scholar]
- 130.Wang H., Czura C.J.. Tracey K.J.. Lipid unites disparate syndromes of sepsis. Nat Med. 2004;10:124–125. doi: 10.1038/nm0204-124. and . [DOI] [PubMed] [Google Scholar]
- 131.Fink M.P.. Ethyl pyruvate: a novel treatment for sepsis. Curr Drug Targets. 2007;8:515–518. [PubMed] [Google Scholar]
- 132.Ulloa L.. et al. Ethyl pyruvate prevents lethality in mice with established lethal sepsis and systemic inflammation. Proc Natl Acad Sci U S A. 2002;99:12351–12356. doi: 10.1073/pnas.192222999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhu S.. et al. Caging a beast in the inflammation arena: use of Chinese medicinal herbs to inhibit a late mediator of lethal sepsis, HMGB1. Int J Clin Exp Med. 2008;1:64–79. [PMC free article] [PubMed] [Google Scholar]
- 134.Wang H.. et al. The aqueous extract of a popular herbal nutrient supplement, Angelica sinensis, protects mice against lethal endotoxemia and sepsis. J Nutr. 2006;136:360–365. doi: 10.1093/jn/136.2.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li W.. et al. A cardiovascular drug rescues mice from lethal sepsis by selectively attenuating a late-acting proinflammatory mediator, high mobility group box 1. J Immunol. 2007;178:3856–3864. doi: 10.4049/jimmunol.178.6.3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Piersen C.E.. Phytoestrogens in botanical dietary supplements: implications for cancer. Integr Cancer Ther. 2003;2:120–138. doi: 10.1177/1534735403002002004. [DOI] [PubMed] [Google Scholar]
- 137.Osuchowski M.F.. et al. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J Immunol. 2006;177:1967–1974. doi: 10.4049/jimmunol.177.3.1967. [DOI] [PubMed] [Google Scholar]
- 138.Ji X.Y., Tan B.K.. Zhu Y.Z.. Salvia miltiorrhiza and ischemic diseases. Acta Pharmacol Sin. 2000;21:1089–1094. and . [PubMed] [Google Scholar]
- 139.Cheng T.O.. Cardiovascular effects of Danshen. Int J Cardiol. 2007;121:9–22. doi: 10.1016/j.ijcard.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 140.Beutler B., Milsark I.W.. Cerami A.C.. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229:869–871. doi: 10.1126/science.3895437. and . [DOI] [PubMed] [Google Scholar]
- 141.Ziegler E.J.. et al. Treatment of gram-negative bacteremia and septic shock with HA-1A human monoclonal antibody against endotoxin. A randomized, double-blind, placebo-controlled trial. The HA-1A Sepsis Study Group. N Engl J Med. 1991;324:429–436. doi: 10.1056/NEJM199102143240701. [DOI] [PubMed] [Google Scholar]
- 142.Ziegler E.J.. et al. Treatment of gram-negative bacteremia and shock with human antiserum to a mutant Escherichia coli. N Engl J Med. 1982;307:1225–1230. doi: 10.1056/NEJM198211113072001. [DOI] [PubMed] [Google Scholar]
- 143.Abraham E.. et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAb Sepsis Study Group. JAMA. 1995;273:934–941. [PubMed] [Google Scholar]
- 144.Cohen J.. Adjunctive therapy in sepsis: a critical analysis of the clinical trial programme. Br Med Bull. 1999;55:212–225. doi: 10.1258/0007142991902222. [DOI] [PubMed] [Google Scholar]
- 145.Feldmann M.. Maini R.N.. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163–196. doi: 10.1146/annurev.immunol.19.1.163. and . [DOI] [PubMed] [Google Scholar]
- 146.Wang H., Yang H.. Tracey K.J.. Extracellular role of HMGB1 in inflammation and sepsis. J Intern Med. 2004;255:320–331. doi: 10.1111/j.1365-2796.2003.01302.x. and . [DOI] [PubMed] [Google Scholar]
- 147.Lotze M.T.. Tracey K.J.. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. and . [DOI] [PubMed] [Google Scholar]
- 148.Sunden-Cullberg J.. et al. Persistent elevation of high mobility group box-1 protein (HMGB1) in patients with severe sepsis and septic shock. Crit Care Med. 2005;33:564–573. doi: 10.1097/01.ccm.0000155991.88802.4d. [DOI] [PubMed] [Google Scholar]
- 149.Angus D.C.. et al. Circulating high-mobility group box 1 (HMGB1) concentrations are elevated in both uncomplicated pneumonia and pneumonia with severe sepsis. Crit Care Med. 2007;35:1061–1067. doi: 10.1097/01.CCM.0000259534.68873.2A. [DOI] [PubMed] [Google Scholar]
- 150.Sha Y.. et al. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol. 2008;180:2531–2537. doi: 10.4049/jimmunol.180.4.2531. [DOI] [PubMed] [Google Scholar]
- 151.Kazama H.. et al. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32. doi: 10.1016/j.immuni.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tzeng H.P.. et al. Negative inotropic effects of high-mobility group box 1 protein in isolated contracting cardiac myocytes. Am J Physiol Heart Circ Physiol 294, H1490-H1496 antiserum to a mutant Escherichia coli. N Engl J Med. 2008;307:1225–1230. doi: 10.1152/ajpheart.00910.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Further reading, resources and contacts
- Chadwick D.J. Goode J. New Therapies. John Wiley & Sons; New York, USA: 2007. Novartis Foundation Symposium 280: Sepsis – New Insights. and . , eds ( [Google Scholar]; Sepsis.com provides more information about the signs, diagnosis and potential treatment for severe sepsis:
- http://www.sepsis.com http://www.sepsis.com; Surviving sepsis.org promotes the Surviving Sepsis Campaign (SSC), an initiative to improve the management, diagnosis and treatment of sepsis:
- http://www.survivingsepsis.org http://www.survivingsepsis.org; MDidea.com provides a detailed description about potential use of Danshen and components (Tanshinone IIA) in the treatment of various inflammatory ailments:
- http://www.mdidea.com/products/herbextract/danshen/data.html http://www.mdidea.com/products/herbextract/danshen/data.html