

Abstract
Reaction of sulfur ylide with chiral non-racemic imine afforded the desired aziridine in excellent yield. Diastereomeric ratios of >95:5 were obtained. Both enantiomeric lines of the butanediacetal-protected chiral non-racemic sulfinyl imines were examined. The sulfur ylides were generated in situ upon thermal decarboxylation of carboxylmethyl betaine functionality. The enantiomeric pairs of D-mannitol with (S)-(-)-2-methyl-2-propane sulfinamide and ascorbic acid with (R)-(-)-2-methyl-2-propane sulfinamide resulted in the highest levels of diastereocontrol when performing aziridination reactions.
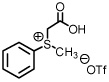
Previously communicated from our laboratory was a novel protocol for the preparation of methylphenylsulfonium methylide ii (Scheme 1).1 When prepared in situ, this sulfur ylide has been shown to effectively trap aldehydes and ketones to form terminal epoxides and imines to form terminal aziridines.2
Scheme 1.

The ease by which sulfur ylide ii is generated is one key advantage of our method over other existing technologies.3 Generation of the sulfur ylide occurs upon decarboxylation of carboxylmethyl betaine functionality. The process does not require the use of strong and often pyrophoric bases such as butyl lithium or sodium hydride, does not require expensive metal catalysts, and is compatible with ecologically benign solvent systems. However, the ‘ease by which the sulfur ylide is generated’ as stated above does come at a price. That is, while proficient when considering both scope and levels of conversion, the shortcomings of a protocol which relies on refluxing THF for complete conversion of π-acceptor to heterocycle are notable when considering asymmetry.
Our entry into stereoselective methylene transfers began with the use of chiral non-racemic sulfinyl imines (Scheme 2).2b
Scheme 2.

Reaction of thioacetate derivative 1 with a series of aryl substituted chiral non-racemic sulfinyl imines afforded the corresponding aziridines in high yield and good stereoselection. A total of seven systems were examined which consisted of both electron deficient and electron releasing aryl substituted chiral non-racemic imines. Upon in situ generation of the sulfur ylide via the thermally induced decarboxylation of carboxymethylsulfonium betaine functionality, a drop in the diastereomeric ratio was observed when going from electron deficient to electron releasing aryl substituted imines.
When working at reaction temperatures below refluxing THF, a moderate increase in stereoselection was observed. This was, however, at the expense of percent conversion and still below the level of diastereocontrol reported when working with the less stabilized dimethylsulfonium methylide under identical reaction conditions (dr = 93:7 with dimethylsulfonium methylide (84% isolated yield at room temperature) and dr = 90:10 with methylphenylsulfonium methylide ii (50% conversion at room temperature)).
In our search for alternative protocols for S-ylide generation which might be capable of sustaining highly stereoselective methylene transfers, we were intrigued by reports from Ley and co-workers using butane-2,3-diacetals as chiral building blocks.4 Butane-2,3-diacetals have not only been shown to be effective substitutes for glyceraldehyde acetonide but highly stereodiscriminating. This might be best illustrated when considering organometallic additions onto butanediacetal-protected glyceraldehyde 2 (Scheme 3).4a
Scheme 3.

As our system ideally operates in warm THF and does not involve a chiral non-racemic sulfide promoter, the combination of the above stereodirecting elements, t-butylsulfinyl imine and butane-2,3-diacetal, posed an interesting question involving stereoselective cycloadditions onto a π-acceptor. Efforts to address the match/mismatch paradigm with methylene transfers have been made and reported herein are our findings in the stereoselective assembly of heterocylic building blocks of aziridine functionality.
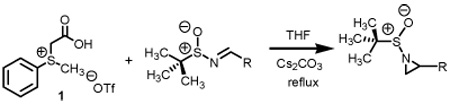
Methylphenylsulfonium methylide ii is best described as a semi-stabilized ylide. While traditional points of stabilization would focus on the alkylidene carbon, attachments onto the sulfur moiety and even solvation of the intermediate itself have also been shown to improve the half-lives of sulfur ylides.5 Accordingly, when used in excess quantities, extremely clean methylene transfers have been documented onto a host of π-acceptors. With a proven technology at hand, our attention has now shifted toward stereoselective transformations. For this study, our study began with the preparation of both enantiomeric lines of BDA protected sulfinyl imines. Scheme 4 illustrates the preparation of one of the four systems we examined.
Scheme 4.

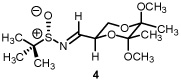
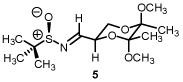
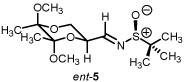
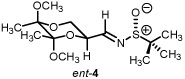
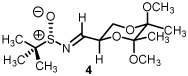
Starting from D-mannitol, butane-2,3-diacetal glyceraldehyde 3 was prepared in two steps using the experimental procedures of Ley and co-workers.4 Condensation of (S)-(-)-2-methyl-2-propane sulfinamide with aldehyde 3 resulted in the synthesis of imine 4 in excellent yield. Using (R)-(-)-2-methyl-2-propane sulfinamide and aldehyde 3, the diastereomeric isomer of 4 was prepared in again excellent yield. For ent-3, ascorbic acid started the synthetic sequence and was prepared following published reports.4 Once obtained (ent-3) identical reaction conditions were used to prepare both diastereomers (RS and SS). With all four systems at hand, we next examined methylene transfers using sulfur ylides generated by the decarboxylation of carboxymethyl betaine functionality. The data from the study is presented below in Table 1.
Table 1.
Methylene Transfers onto Butanediacetal-Protected Glyceraldehyde
Isolated chromatographically pure material.
Determined by 1H NMR (crude reaction mixture).
All reactions reported here were clean as judged by NMR analysis of the crude reaction mixture. The analysis revealed no by-products other than desired product with minor diastereomer. This was validated based upon the isolated yields obtained. Using as a baseline the data obtained when performing methylene transfers with methylphenylsulfonium methylide ii onto aryl substituted chiral non-racemic sulfinyl imines (dr ranging from 66:34 to 87:13),2b we were excited to see such high levels of diastereoselectivity when working with imine 4 (entry 1). The high isolated yield and >95:5 diastereomeric ratio was confirmed when working with ent-4 (entry 4) While entries 2 and 3 reveal slightly lower levels of stereodiscrimination, these data points support not only the trend within this enantiomeric series (5 and ent-5) but a match/mismatch paradigm when performing methylene transfers. Perhaps more significant with this study is the level of diastereocontrol observed using the reaction conditions of refluxing THF.
An increase in the diastereomeric ratio from a previous high of 87:13 when working with aryl substituted chiral non-racemic sulfinyl imines (aryl being 4-nitrophenyl) to >95:5 (entries 1 and 4, Table 1) has been achieved. Epimeric changes to either the butanediacetal-protected diol or the sulfinyl imine resulted in a drop of diastereoselectivity. While the levels of conversion were on par when examining all four butanediacetal-protected chiral non-racemic sulfinyl imines, this series using both enantiomeric lines poses an interesting question when considering the source of asymmetry. Accordingly, a series of control reactions were examined. The data from this study is presented below in Table 2.
Table 2.
Control Reactionsa

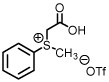
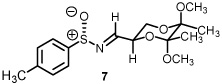
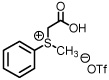
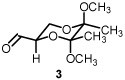
See Supporting Information for reaction details.
Isolated chromatographically pure material.
Determined by 1H NMR (crude reaction mixture).
Inextrictable crude reaction mixture (1H NMR).
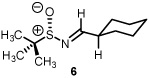
As previously reported by Stockman and co-workers, reaction of sulfur ylides (dimethylsulfonium methylide) onto cyclohexyl derivatives of t-butylsulfinyl imine 6 resulted in high levels of diastereocontrol (>95:5).6 Warmer reaction temperatures, not surprisingly, resulted in lower levels of diastereocontrol using sulfonium salt 1 and imine 6 (61:39, entry 1). Interestingly when switching from a semi-stabilized ylide (ii) to dimethylsulfonium methylide using as π-acceptor imine 4, an inextrictable crude reaction mixture was obtained (entry 2). This reaction was repeated multiple times at temperatures as low as 0°C. While starting material was consumed with each reaction, no evidence of aziridine (nor oxirane when working with aldehyde 3) was observed using dimethylsulfonium methylide based upon the 1H NMR analysis. When working with a modified sulfinyl imine 7 (4-methylphenyl, entry 3) or butane-2,3-diacetal-protected glyceraldehyde 3 itself (entry 4), lower levels of diastereoselectivity were observed.
While the present study did not establish the configuration of the new aziridine stereocenter, the data presented in both Table 1 and Table 2 clearly documents the need to have both stereodiscriminating elements present for high levels of diastereoselectivity. Even though there is ample precedent for highly discriminating additions onto either butanediacetal-protected glyceraldehyde 24 or tert-butylsulfinamides such as those present in α-chloroimines,7 our study which operates at an elevated reaction temperature when compared to the previous two mandates both when performing stereoselective methylene transfers onto butanediacetal-protected chiral non-racemic sulfinyl imines.
We do believe the stereodiscriminating event is addition onto the imine and not epimerization of the aziridine. Control reactions which examined a solution of Cs2CO3 and the aziridine using imine ent-5 did not result in a change of diastereomeric ratio even after 10 h at reflux (Table 1, entry 3). Furthermore, stopping the aziridination reaction prematurely using imine 4 had the same diastereomeric ratio as its endpoint of >95:5.
Synthesized terminal azirdines, both enantiomeric lines, starting from imines 4 and ent-4 are interesting examples in that they present a viable scaffold when considering the assembly of systems of biological and medicinal significance. Using as an example the three-carbon building block N-protected 3-amino-1,2-epoxide 8, which is an intermediate used in the assembly of many HIV protease inhibitors,8 our approach serves as a viable alternative toward the preparation of this advanced intermediate (Figure 1).
Figure 1.

Potential applications.
The chemistry recently reported by Hodgson and co-workers best illustrates the potential in that regioselective ring-opening of the terminal aziridine bearing sulfinyl functionality using PhMgBr in the presence of CuI would allow for the installation of the benzyl functionality found in both amprenivir and saquanivir.9 Switching PhMgBr with thiophenol, an advanced intermediate used in the preparation of nelfinavir can be realized. That is, the final steps toward the preparation of N-protected 3-amino-1,2-epoxide 8 would include deprotection of the aminodiol using HCl in dioxane,4,9 BOC-protection of the amine, and functional group interconversion of the diol to epoxy functionality.8a,10
In summary, a number of groups have demonstrated and refined ingenious approaches toward the preparation of homochiral carbo- and heterocyclic rings using sulfur ylide technology. While our approach has several shortcomings in that the decarboxylative process does not operate at or well below ambient reaction temperatures, high levels of diastereocontrol can be achieved. Diastereomeric ratios of >95:5 were obtained when performing methylene transfers onto imines originating from the D-mannitol and (S)-(-)-2-methyl-2-propane sulfinamide or ascorbic acid and (R)-(-)-2-methyl-2-propane sulfinamide. Currently we are active in examining the source of stereocontrol through computational studies and applying this technology in the assembly of materials of biological and medicinal importance. Results from these studies will be reported in due course.
Acknowledgments
This work was supported in part by NIGMS (NIH NIGMS 1R15GM085936), NSF (CHE 0514004), and the Camille and Henry Dreyfus Foundation (TH-06-008).
Footnotes
Electronic Supplementary Information (ESI) available: Experimental and spectral data in the preparation of compounds 4 and 5, the corresponding aziridines and control reactions.. See DOI: 10.1039/b000000x/
Notes and references
- 1.Forbes DC, Amin SR, Bean CJ, Standen MC. J. Org. Chem. 2006;71:8287. doi: 10.1021/jo061370u. [DOI] [PubMed] [Google Scholar]
- 2.(a) Forbes DC, Bettigeri SV, Patrawala SA, Pischek SC, Standen MC. Tetrahedron. 2009;65:70. doi: 10.1016/j.tet.2008.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Forbes DC, Bettigeri SV, Amin SR, Bean CJ, Law AM, Stockman RA. Synth. Commun. doi: 10.1080/00397910802654898. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forbes DC, Standen MC, Lewis DL. Org. Lett. 2003;5:2283. doi: 10.1021/ol034612a. [DOI] [PubMed] [Google Scholar]
- 4.(a) Michel P, Ley SV. Angew. Chem. Int. Ed. 2002;41:3898. doi: 10.1002/1521-3773(20021018)41:20<3898::AID-ANIE3898>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]; (b) Michel P, Ley SV. Synthesis. 2003;10:1598. [Google Scholar]; (c) Michel P, Ley SV. Synthesis. 2004;1:147. [Google Scholar]
- 5.(a) Cheng J-P, Liu B, Zhang X-M. J. Org. Chem. 1998;63:7574. doi: 10.1021/jo981129i. [DOI] [PubMed] [Google Scholar]; (b) Johnson AW, Amel RT. Can. J. Chem. 1968;46:461. [Google Scholar]
- 6.Morton D, Pearson D, Field RA, Stockman RA. Synlett. 2003:1985. [Google Scholar]
- 7.Hodgson DM, Kloesges J, Evans B. Org. Lett. 2008;10:2781. doi: 10.1021/ol800961a. [DOI] [PubMed] [Google Scholar]
- 8.(a) Izawa K, Onishi T. Chem. Rev. 2006;106:2811. doi: 10.1021/cr050997u. [DOI] [PubMed] [Google Scholar]; (b) Honda Y, Katayama S, Kojima M, Suzuki T, Kishibata N, Izawa K. Org. Biomol. Chem. 2004;2:2061. doi: 10.1039/b404071f. [DOI] [PubMed] [Google Scholar]; (c) Wang D, Schwinden MD, Radesca L, Patel B, Kronenthal D, Huang M-H, Nugent W. J. Org. Chem. 2004;69:1629. doi: 10.1021/jo035733r. [DOI] [PubMed] [Google Scholar]; (d) Evans BE, Rittle KE, Homnick CF, Springer JP, Hirshfield J, Veber DF. J. Org. Chem. 1985;50:4615. [Google Scholar]; (e) Beaulieu PL, Wernic D, Duceppe J-S, Guindon Y. Tetrahedron Lett. 1995;36:3317. [Google Scholar]; (f) Reetz MT, Binder J. Tetrahedron Lett. 1989;30:5425. [Google Scholar]; (g) Rotella DP. Tetrahdron Lett. 1995;36:5453. [Google Scholar]
- 9.Hodgson DM, Hughes SP, Thompson AL, Heightman TD. Org. Lett. 2008;10:3453. doi: 10.1021/ol801224g. [DOI] [PubMed] [Google Scholar]
- 10.Kim BM, Bae SJ, So SM, Yoo HT, Chang SK, Lee JH, Kang J. Org. Lett. 2001;3:2349. doi: 10.1021/ol016147s. [DOI] [PubMed] [Google Scholar]