

Abstract
Expression of cardiac myocyte Kv4 channels (Kv4.3 for human, Kv4.2 and Kv4.3 for rodents) is downregulated with hypertrophy in vivo leading to a decrease in the transient outward current (Ito). This effect is recapitulated in vitro with rat neonatal cardiac myocytes treated with angiotensin II (Ang II), which acts via AT1 receptors, NADPH oxidase and p38 MAP kinase to destabilize the 3′ untranslated region (3′UTR) of the Kv4.3 channel messenger RNA (mRNA). Here deletion analysis and mutagenesis identify an AU-rich element (ARE) in the Kv4.3 3′UTR that is required for Ang II-induced destabilization. Overexpression of AUF1 (ARE/poly-(U)-binding/degradation factor 1), an RNA destabilizing protein, mimics and occludes the Ang II effect, while RNA interference targeted against AUF1 blocks the Ang II effect on the Kv4.3 3′UTR. Ang II upregulates AUF1 by activating AT1 receptors, NADPH oxidase and p38 MAP kinase. Finally, pull-down assays establish that Ang II increases AUF1 binding to the ARE required for destabilization, while binding of the mRNA stabilizing protein HuR is unaffected. Hence, Ang II acts via AT1 receptors, NADPH oxidase and p38 MAP kinase to upregulate AUF1, which in turn binds to an ARE in the Kv4.3 3′UTR to destabilize the channel mRNA.
Keywords: Angiotensin, mRNA stability, Potassium channel, Heart failure, Hypertrophy, Gene expression
1. Introduction
The expression of Kv4.3 gene, which is the pore forming subunit of transient outward current (Ito) channels in human myocardium, is decreased by cardiac hypertrophy induced by congestive heart failure, pregnancy and hypertension [1–3]. This effect could contribute to reduced Ito and loss of the repolarization notch in the cardiac action potential, which in turn makes Ca2+ release in ventricular myocytes less synchronous [4]. Angiotensin II (Ang II) and hypertension are implicated in control of cardiac Kv4.3, but not KChip2, mRNA expression by in vivo rat experiments with an Angiotensin-Converting Enzyme (ACE) inhibitor, overexpression of renin and angiotensinogen, and spontaneous hypertension [3,5,6]. Furthermore, in cultured neonatal rat cardiac myocytes, AT1 receptors activated by Ang II or stretch destabilize Kv4.3 messenger RNA (mRNA) via NADPH oxidase-superoxide-p38 MAP kinase signaling [7,8]. Thus, signaling that contributes to hypertrophic regulation of Kv4.3 gene expression has been identified.
A potential link between this Ang II-activated pathway and cardiac channel mRNA stability is suggested by previous studies of AUF1, a protein that binds to AU-rich elements (AREs) in the 3′ untranslated region (3′UTR) of mRNAs to induce destabilization. Specifically, human cardiac AUF1 is chronically upregulated with congestive heart failure [9], a condition associated with decreased Kv4.3 expression [1]. Furthermore, AUF1 is upregulated by overexpression of MKK6E in cultured cardiac myocytes, which activates p38 MAP kinase [10]. The shared sensitivity of Kv4.3 and AUF1 expression to congestive heart failure and p38 MAP kinase stimulated us to test whether an AUF1 binding to a Kv4.3 ARE is involved in the Ang II effect on the channel mRNA stability.
Here we identify an ARE in the Kv4.3 3′UTR that mediates the Ang II-induced destabilization. Then AUF1 is shown to be specifically upregulated by Ang II to bind to this ARE. AUF1 upregulation requires AT1 receptor-NADPH oxidase-p38 MAP kinase signaling. Finally, AUF1 is found to be necessary and sufficient for Ang II regulation of cardiac Kv4.3 mRNA stability.
2. Materials and methods
2.1. Rat neonatal myocyte culture and treatment
Neonatal rat ventricular cardiac myocytes were isolated from collagenase-treated hearts from 1-day old Sprague–Dawley rats and cultured as described previously [7]. Transfection was also performed as described previously [8]. After culturing in the presence of serum (1 day for immunoblot experiments, 2 days for transfection experiments), the medium was replaced by serum-free MEM supplemented with human insulin (10 μg/mL), human transferrin (10 μg/mL) and bovine serum albumin (1 mg/mL). Two days later, cultures were subjected to treatment with vehicle or 100 nmol/L Ang II. For experiments utilizing signaling inhibitors, cells were preincubated with inhibitors or vehicle for 30 min before Ang II treatment.
2.2. Generation of truncated and mutant Kv4.3 3′UTR reporter constructs
All deletion constructs (PG0.4B,PG1.4B, PG2.2B, PGΔ1.8B and PGΔ3B) were generated either by restriction enzyme digestion or polymerase chain reaction (PCR) from the PG4.3B reporter construct, which contains the mouse Kv4.3 3′UTR [8]. Numbers in the name of the deletion constructs refer either to the number of kb remaining from the 3′UTR starting at its 5′ end (e.g. PG0.4B) or the number of kb deleted from the 3′UTR starting at its 5′ end (e.g. PGΔ1.8). Each digested fragment or PCR product was subcloned along with the PGL3 SV40 polyadenylation signal into the PGL3 vector utilized by XbaI and SalI sites.
Mutations of AREs in Kv1.4B were performed by 4 primer PCR. However, this approach was hindered by the low melting points of the target sequences in the DNA. Therefore, mutations were designed to generate restriction sites for evaluation and added GC content. The fidelity of mutants was verified by sequencing. Specifically, PG1.4B-m1 was made by replacing the a short sequence located 413 nucleotides downstream of the stop codon (UAUUUA, ARE #1 in Fig. 2) with GAUAUC to produce an EcoRV restriction site, while PG1.4B-m2 was made by replacing ARE #2 (UAAUUUAUU), which is 432 nucleotides downstream of the stop codon, with GUGUACAAG to generate an SspB1 site.
Fig. 2.
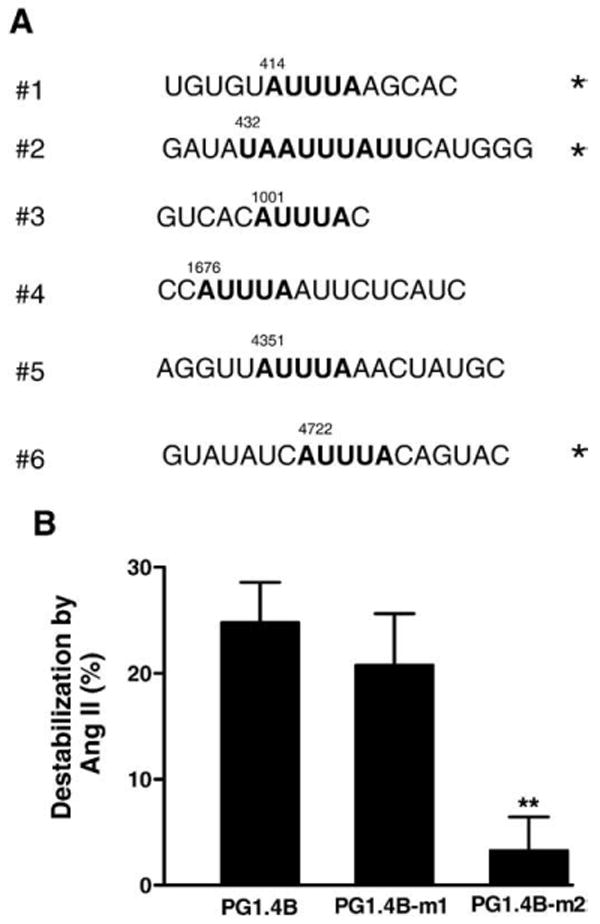
Identification of the Ang II-sensitive ARE. (A) AREs in the mouse Kv4.3 3′ untranslated region (3′UTR). Sequences conserved in human Kv4.3 are indicated with an asterisk (*). Numbers indicate number of nucleotides downstream of the stop codon (i.e., from the start of the 3′UTR). (B) The PG1.4-m2 mutant, which alters ARE #2, blocks the Ang II effect. In contrast, disrupting ARE #1 does not alter Ang II-induced destabilization. n=4. **p<0.01.
Luciferase reporter assays were carried out as described previously [8]. Each experiment included triplicate cultures to generate a single measurement. At least 3 independent experiments were performed.
2.3. Immunoblotting
Myocyte cultures were rinsed twice with ice-cold phosphate buffered saline (PBS, in mM: 137 NaCl, 2.7 KCl, 4.3 Na2HPO4, 1.47 KH2PO4). After addition of RIPA buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.25% deoxycholic acid, 1% NP-40, 1 mM EDTA and EDTA-free protease inhibitor cocktail (Roche)) on ice, cells were scraped off the dish. Following pelleting of unsolubilized debris by centrifugation at 10,000 g for 15 min, the remaining lysate was used immediately or stored at −20 °C. Protein concentration was measured by the bicinchoninic acid method according to the manufacturer's protocol (Upstate) with bovine serum albumin as a standard. Samples containing equivalent amounts of protein were loaded on 8.5% SDS-PAGE gel and, following electrophoresis, transferred onto a Polyscreen PVDF transfer membrane (Perkin Elmer). After blocking with 10% (w/v) fat-free milk powder, 0.1% Tween 20 in PBS, the membrane was incubated overnight at 4 °C with rabbit polyclonal anti-AUF1 antibody (1:1000) (Upstate) or mouse monoclonal anti-HuR antibody (1:1000) (Santa Cruz Biotechnology). The membrane was then stripped with Restore Western Blot Stripping buffer (Pierce) and reprobed with mouse monoclonal anti-Glyceraldehyde-3-Phosphate Dehydrogenase antibody (1:1000) (Upstate) for 1 h at room temperature. Membranes were washed 3 times and then incubated with goat anti-rabbit IgG-HRP antibody or goat anti-mouse IgG-HRP (Santa Cruz Biotechnology). Immunoblots were developed using high sensitivity ECL kit (Pierce) with the amplified signals detected on Kodak X-OMAT AR film. Results were then quantified on a Molecular Dynamics Personal Densitometer SI.
2.4. In vitro transcription of biotinylated RNA and pull-down assays
Digested fragments (1.4B from PG1.4B and 1.4B-m2 from PG1.4B-m2) were subcloned into pBluescript utilizing XbaI and SalI restriction sites. The resulting constructs of 1.4BpBlue and 1.4B-m2pBlue were linearized with Sal I and then transcribed in vitro with T7 polymerase and biotin RNA labeling mix (Roche). Unincorporated nucleotides were removed by ethanol precipitation. Biotinylated RNAs of wild type (1.4B) or mutant (1.4B-m2) were used as probes in pull-down assays. Probe (5 μg), cell lysate from untreated or Ang II-treated cultured cardiac myocytes (50 μg total protein), and RNA binding buffer (10 mM Hepes pH 7.6, 40 mM KCl, 3 mM MgCl2, 5% Glycerol, 1 mM DTT, 0.025 mg/mL tRNA, 2 U RNase inhibitor, and complete mini, EDTA-free protease inhibitor cocktail (Roche)) in a total volume of 200 μL were then incubated at room temperature with gentle agitation for 0.5 h. Proteins bound to the biotinylated 3′UTR were recovered by addition of 100 μL of NeutrAvidin beads (Pierce). After the incubation at room temperature for 1 h, the beads were spun down and washed 3 times with 200 μL RNA binding buffer. 4× SDS sample buffer was then added to solubilize bound proteins. After heating for 5 min at 95 °C, samples were separated by SDS-PAGE gel and subjected to immunoblotting with rabbit polyclonal anti-AUF1 antibody (1:1000) (Upstate) and, after stripping as described above, mouse monoclonal anti-HuR antibody (1:1000) (Santa Cruz).
2.5. AUF1 RNA interference
RNA interference used the pSILENCER-AUF15, a shRNA-generating plasmid that targets all AUF1 isoforms [11]. A control was generated by inverting a GC in the AUF1 specific sequence to CG (i.e., GTTGTAGACTGCACTCTGA was replaced with GTTGTAGACTCGACTCTGA). This scrambled sequence was synthesized and annealed into the HindIII and BamHI sites of the pSILENCER 2.0-U6 to generate pSILENCER-AUF1(s). The pSILENCER constructs, the luciferase reporter constructs (PG4.3B or PGL3) and a Renilla luciferase plasmid for normalization were transfected at 1:1:0.1 ratio into cultured cardiac myocytes using Lipofectamine 2000 (Invitrogen). Two days later, the cultures were treated with vehicle or 100 nmol/L Ang II and used for luciferase assays.
2.6. Animals and reagents
Animals were purchased from Hilltop Lab Animals Inc (Scottsdale, PA), housed in a University of Pittsburgh animal care facility, and handled according to a protocol approved by the Institutional Care and Use Committee. The shRNA expression pSILENCER plasmids were kindly provided by Dr. Myriam Gorospe (National Institute on Aging). AUF1 isoform expression plasmids [12] were kindly provided by Robert Schneider (New York University). Apocynin and SB 239063 were purchased from Aldrich Chemicals and Tocris Bioscience, respectively. Candesartan (CV-11974) was kindly provided by AstraZeneca.
2.7. Statistics
Student's t test was used with paired data. ANOVA followed by Dunnett's post-test was used for standard multiple group analysis. When ANOVA was not appropriate because of normalization, the Bonferroni test was used. Error bars represent standard error of the mean.
3. Results
3.1. Ang II acts on a Kv4.3 3′UTR AU-rich element (ARE)
Ang II treatment of cultured neonatal rat ventricular cardiac myocytes for 7 h halves the expression of native Kv4.3 mRNA and the mRNA of a luciferase reporter construct called PG4.3B, which contains the mouse Kv4.3 3′UTR [7,8]. Resultant changes in PG4.3B-induced firefly luciferase activity at this time point are smaller than the change in mRNA due to the delay from protein synthesis, but should be proportional to changes mRNA. Most importantly, the luciferase assay, which includes an internal Renilla luciferase calibration, provides a convenient and reproducible quantitative assay for studying Ang II regulation [8]. Therefore, to narrow down the Ang II-sensitive region of the Kv4.3 3′UTR, deletions were made in PG4.3B and these new constructs (Fig. 1A) were tested for sensitivity to Ang II in cardiac myocytes. The normalized decrease in luciferase activity induced by Ang II, which is plotted as % destabilization, showed that the first 1.4 kb of the Kv4.3 3′UTR mediates Ang II sensitivity (i.e., PG1.4B is statistically indistinguishable from PG4.3B) (Fig. 1B). However, the first 0.4 kb of the 3′UTR did not respond to Ang II (PG0.4B, Fig. 1B). Thus, Ang II-induced destabilization requires sequence in a 1 kb region starting 0.4 kb downstream of the stop codon.
Fig. 1.
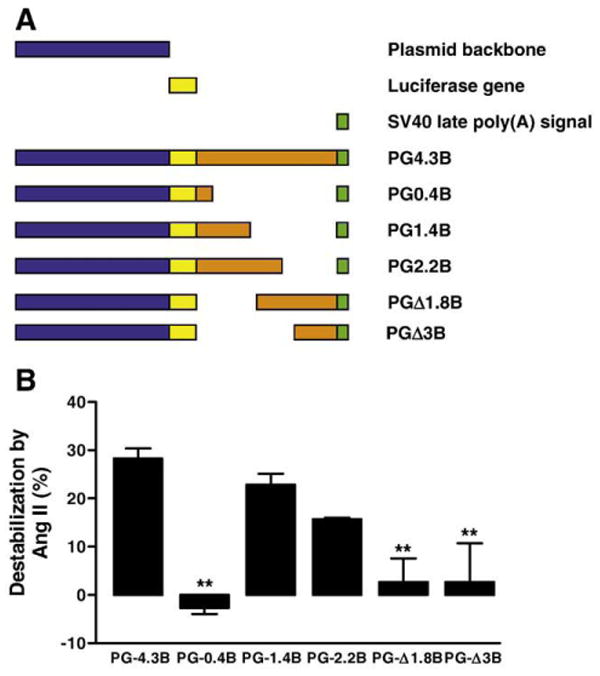
Identification of the angiotensin II-responsive ARE. (A) Luciferase reporter constructs containing various lengths of Kv4.3 3′UTR. These constructs were based on the PGL3 plasmid. Blue represents the plasmid backbone of PGL3, yellow represents the coding sequence of luciferase, orange represents Kv4.3 3′UTR sequence, and green represents the SV40 polyadenylation signal sequence. Note that different regions of the Kv4.3 3′UTR were deleted. Numbers in the name of the deletion constructs refer either to the number of kb remaining from the 3′UTR starting at its 5′ end (e.g. PG0.4B) or the number of kb deleted from the 3′UTR starting at its 5′ end (e.g. PGΔ1.8). (B) The Ang II effect on each Kv4.3 3′UTR reporter construct. Cardiac myocytes were treated with either vehicle or 100 nmol/L Ang II for 7 h. Data are from 3 to 9 independent experiments. **p<0.05.
Destabilization of mRNA is often conferred by 3′UTR AREs. Six AREs are present in the Kv4.3 3′UTR, but the identified region contains half of these sites: an AUUUA sequence 414 nucleotides downstream of the stop codon, a consensus nonamer of U(U/A)(U/A) UUU(U/A)(U/A)U 432 nucleotides downstream of the stop codon and a second AUUUA sequence 1001 nucleotides downstream of the stop codon (Fig. 2A). The first two AREs are conserved in the human Kv4.3 mRNA (Fig. 2A, * indicate sequences that are identical in human and mouse). However, because the function of such motifs depends on mRNA secondary structure, core consensus sequence alone is not sufficient for deducing function. Therefore, the first ARE (AUUUA) was mutated to generate the PG1.4B-m1 construct, and the second ARE (UAAUUUAUU) was mutated to generate the PG1.4B-m2 construct (see Materials and methods). Each of these mutants or the wild type control was transfected into cardiac myocytes and tested for responsiveness to Ang II. PG1.4B-m1 was indistinguishable from the wild type construct, indicating that ARE #1 is not required for the Ang II effect. However, PG1.4B-m2 did not respond to Ang II (Fig. 2B). Thus, ARE #2 is required for Ang II-induced Kv4.3 mRNA destabilization.
3.2. Upregulation of AUF1 mimics and occludes Ang II-induced Kv4.3 mRNA destabilization
If upregulation of AUF1 is responsible for the downregulation of Kv4.3 mRNA, then overexpression the AUF1 should mimic the Ang II effect. Cardiac myocytes were transfected with the reporter construct PG4.3B and plasmids encoding the p37, p42 or p45AUF1 isoforms [12,13] or an empty vector control (pcDNA3). The resulting Kv4.3 3′UTR-mediated signals were then measured with luciferase assays. In all experiments, transfection with any of these AUF1 isoform plasmids reduced Kv4.3 3′UTR-dependent luciferase activity (Fig. 3A). Furthermore, overexpression of each AUF1 isoform blocked the Ang II effect (Fig. 3B). Thus, upregulated AUF1 mimicked and occluded the Ang II destabilization of the Kv4.3 3′UTR.
Fig. 3.
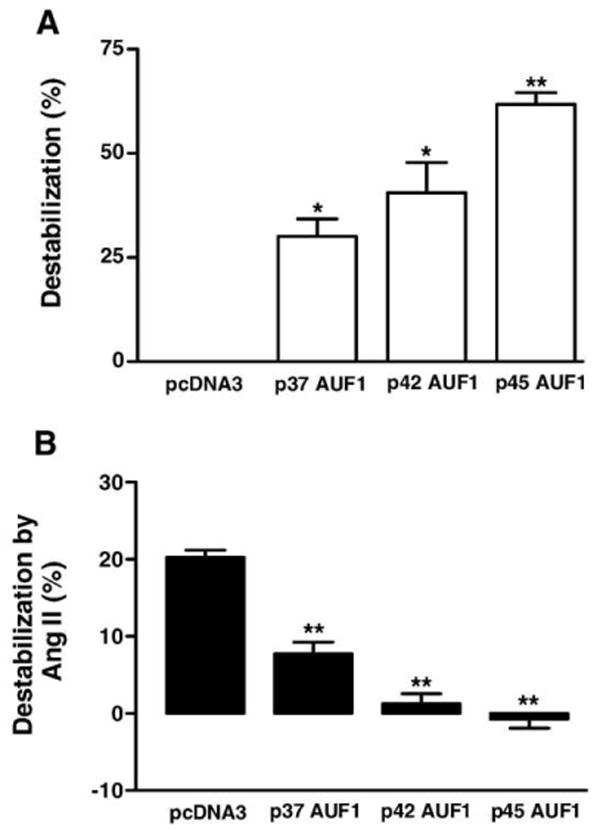
AUF1 overexpression mimics and occludes the Ang II effect. (A) pcDNA3 or individual AUF1 isoforms (p37, p42 and p45) and Kv4.3B were cotransfected into cardiac myocytes. The destabilizing effect from overexpressed isofoms was calculated based on the % difference in normalized luciferase activity with pcDNA3 and the AUF1 isoforms. (B) Ang II-induced destabilization of the Kv4.3 3′UTR was reduced by expression of AUF1 isoforms. The destabilizing effect from Ang II was calculated as the % difference between the normalized luciferase activity caused by Ang II. *p<0.05, **p<0.01, n=4.
3.3. AUF1 is necessary for the Ang II effect
RNA interference was used to test whether AUF1 is necessary for the Ang II effect. Specifically, a plasmid expressing a short hairpin RNA (shRNA) targeted against a region common to all AUF1 isoforms (pSILENCER-AUF15) [11] was transfected into cultured cardiac myocytes with the Kv4.3 3′UTR reporter vector or the pGL3 control. To test for specificity, a GC in the target sequence was scrambled to CG to generate pSILENCER-AUF1(s) (see Materials and methods). Neither of these constructs affected the signals from PGL3 (Fig. 4A), thus excluding nonspecific effects on the luciferase reporter vector. Likewise, Ang II-induced destabilization of the Kv4.3 3′UTR persisted in the presence of pSILENCER-AUF1(s) (Fig. 4B, right open bar) again showing that scrambled shRNA control did not produce nonspecific effects. Furthermore, both shRNA constructs did not affect the control signal from PG4.3B, implying that AUF1 does not functionally affect the Kv4.3 mRNA in unstimulated cells (Fig. 4A). However, pSILENCER-AUF15 inhibited the Ang II effect on the PG4.3B (Fig. 4B, right closed bar). This implies that AUF1 is necessary, as well as sufficient, for the Ang II-induced destabilization of Kv4.3 mRNA.
Fig. 4.
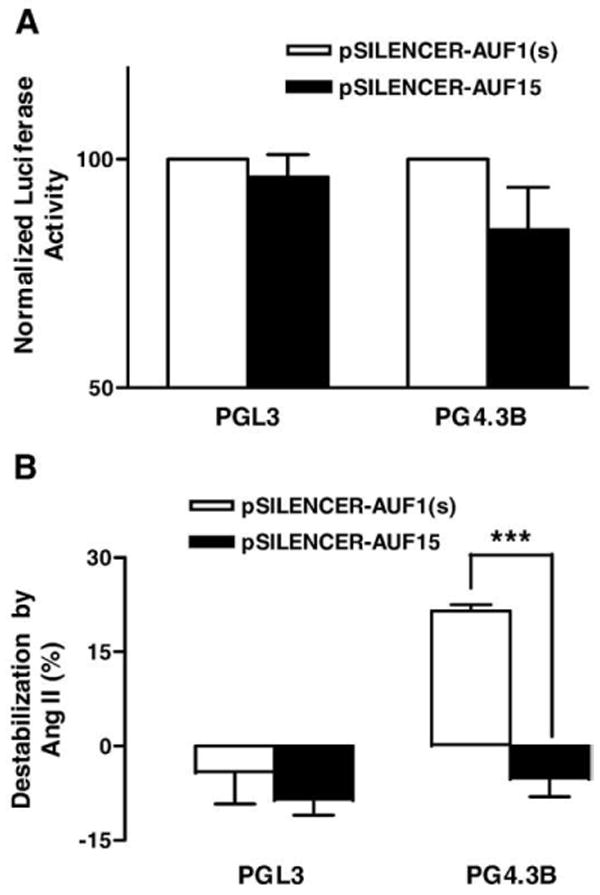
AUF1 is necessary for Ang II-induced destabilization of Kv4.3 mRNA. Cardiac myocytes expressed a short hairpin loop RNA interference construct targeted against AUF1 (pSILENCER-AUF15) or a scrambled sequence control (pSILENCER-AUF1(s)) along with PGL3 or PG4.3B luciferase reporter constructs. (A) Effect of RNA interference constructs on basal signals from the control plasmid PGL3 and the Kv4.3 3′UTR reporter plasmid. Note that both RNA interference constructs failed to produce an effect (i.e., there is no statistically significant difference between filled and open bars). (B) RNA interference directed against AUF1 specifically prevents the Ang II-induced destabilization of the Kv4.3 3′UTR. ***p<0.001, n=3.
3.4. AUF1 is induced by the AT1 receptor-NADPH oxidase-p38 pathway
Immunoblots showed that all 4 isoforms of AUF1 are present in whole cell lysates generated from cultured neonatal cardiac myocytes [13]. However, the p37 isoform of AUF1, which has the highest affinity of mRNA and is most effective at inducing destabilization [12,13], was most difficult to detect because of its low abundance (Fig. 5A). Therefore, different exposure times were used for detecting total AUF1 (Fig. 5A, left) and the p37 isoform (Fig. 5A, right; note that other isoforms yield saturating signals). Using these two assay conditions, it was found that the levels of the p37 isoform and total AUF1 each increased in response to 100 nmol/L Ang II treatment for 7 h (Fig. 5B). The differences in the Ang II effects on total and p37 AUF1 were statistically not significant (Fig. 5B, NS), implying that either the total or p37 signals could be used in follow-up studies. In contrast, the abundance of the mRNA stabilizing protein HuR, which can compete with AUF1, was not changed by Ang II (Fig. 5C). Therefore, Ang II upregulates cardiac AUF1 without affecting HuR expression.
Fig. 5.
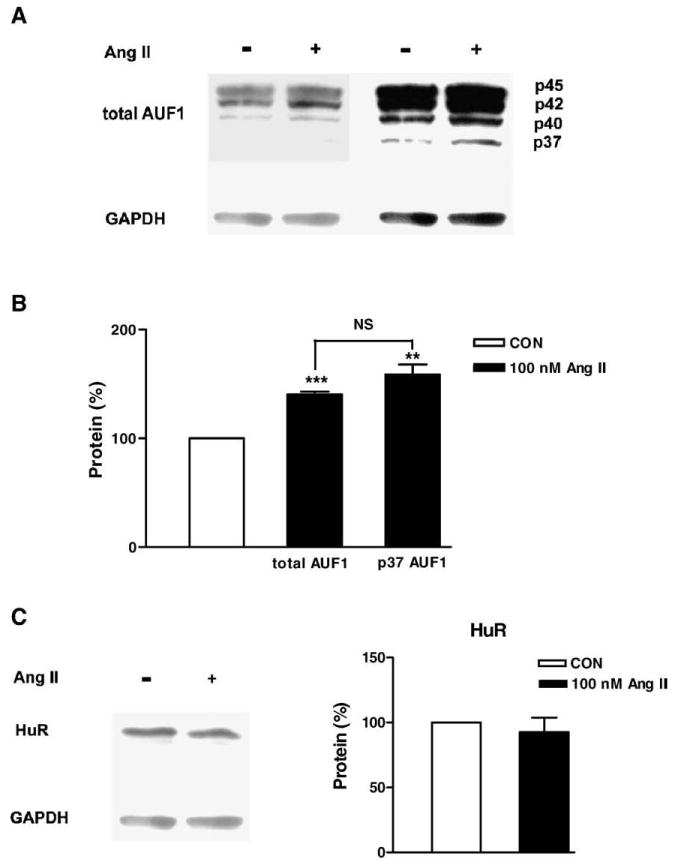
Ang II specifically increases AUF1 expression. (A) Immunoblots showing AUF1 proteins in cultured cardiac myocytes. The protein samples were collected from cardiac myocytes after treatment with vehicle or 100 nmol/L Ang II for 7 h. The blot was exposed for either 30 s for total AUF1 assays (left) or 10 min for p37 AUF1 (right). (B) Expression of total and p37 AUF1 normalized to the relative internal control GAPDH increases with Ang II. Open bars: control (CON), filled bars: Ang II. **p<0.01 and ***p<0.001 compared to control. Note that the Ang II effect is indistinguishable for total AUF1 and p37 AUF1 (NS, no significant difference for the two Ang II effects). (C) HuR protein is unaffected by Ang II. n=3–5.
If Ang II acts via AUF1 upregulation, then this effect should require the signal transduction that mediates destabilization of the Kv4.3 mRNA. Ang II destabilizes Kv4.3 mRNA by activating AT1 receptors, which in turn stimulate NADPH oxidase and p38 MAP kinase [8]. To address whether this signaling mediates AUF1 upregulation, cultured cardiac myocytes were first treated with the AT1 receptor antagonist candesartan and total AUF1 protein was quantified. Immunoblot analysis showed that 100 nmol/L candesartan, which inhibits the destabilization of Kv4.3 mRNA by Ang II [8], also blocked AUF1 protein upregulation (Fig. 6A). Likewise, the NADPH oxidase inhibitor apocynin and the p38 kinase inhibitor SB239063 at doses that abolished the Ang II-induced destabilization of Kv4.3 mRNA [8] inhibited Ang II-induced AUF1 upregulation (Fig. 6B). The shared requirement for the AT1 receptor-NADPH oxidase-p38 MAP kinase signaling is consistent with the hypothesis that upregulation of AUF1 is responsible for Kv4.3 mRNA destabilization by Ang II.
Fig. 6.
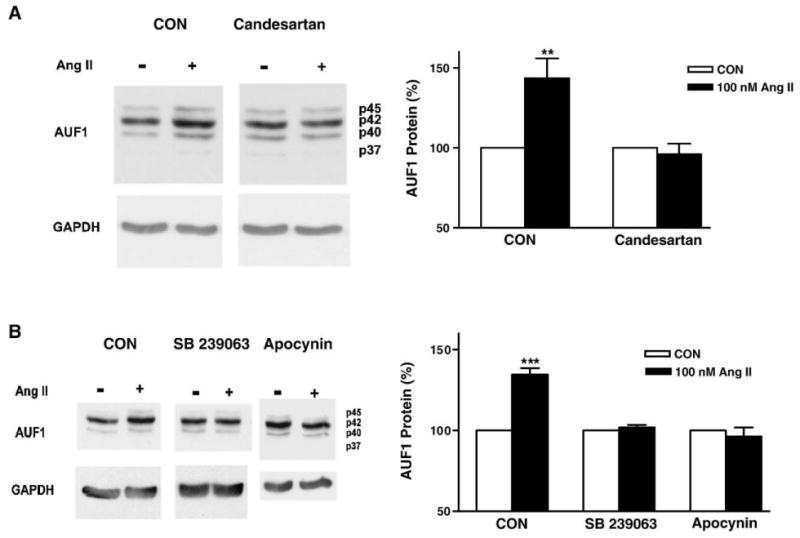
AT1 receptors, NADPH oxidase and p38 MAP kinase mediate the upregulation of AUF1 by Ang II. (A) Protein samples were collected from cardiac myocytes with or without incubation with 100 nM candesartan (an AT1 receptor antagonist) during treatment with vehicle or 100 nmol/L Ang II for 7 h. (B) NADPH oxidase and p38 MAP kinase are required for the Ang II-induced upregulation of AUF1. Protein samples were collected from cardiac myocytes with or without incubation with 100 μM apocynin or 20 μM SB 239063 during treatment with vehicle or 100 nmol/L Ang II for 7 h. Bar graphs show total AUF1 protein normalized to GAPDH. Open bars: control (CON), filled bars: Ang II. **p<0.01, ***p<0.001; n=3 in all panels.
3.5. Ang II-upregulated AUF1 binds to the Ang II-sensitive ARE
To investigate whether the Kv4.3 3′UTR interacts directly with AUF1, we performed RNA pull-down experiments. Specifically, the first 1.4 kb of the Kv4.3 3′UTR was transcribed in vitro to generate a biotinylated probe to bind interacting proteins from a cultured cardiac myocyte lysate. Bound proteins were recovered with avidin-conjugated beads and then assayed by immunoblot. This analysis showed that the Kv4.3 3′UTR interacts with both HuR and AUF1. Furthermore, Ang II increased the interaction with p37 AUF1 nearly two-fold (Figs. 7A,B). In contrast, Ang II did not significantly affect the binding of the other AUF1 isoforms (data not shown) and HuR (Figs. 7A,C). Thus, the increase in p37 AUF1 protein expression (Figs. 5A,B) is associated with elevated p37 AUF1 binding to the channel 3′UTR.
Fig. 7.
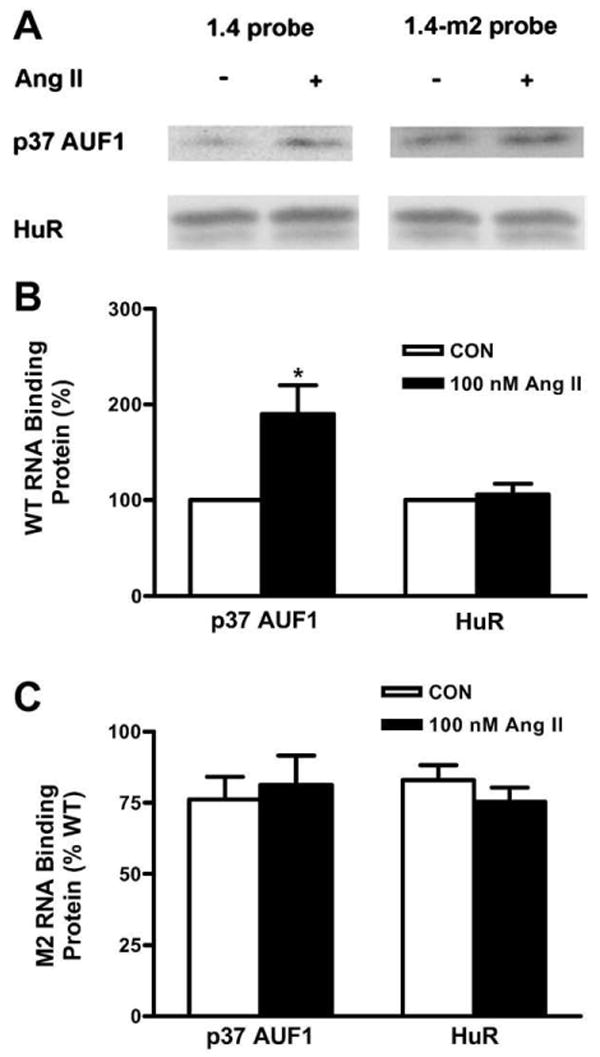
Ang II increases p37 AUF1 interaction with the Ang II-sensitive ARE. (A) Left, pull-down immunoblots show that Ang II increases p37 AUF1 binding to the wild type Kv4.3 3′UTR probe (left), but not to the mutant probe (right). In contrast, HuR binding is unaffected by Ang II or the mutant. (B) Quantification of binding to a wild type (WT) construct (*p<0.05). (C) Same as B, but m2 mutation in the Ang II-sensitive ARE was used in the pull-down assay. n=5.
If this increase in p37 AUF1 binding reflects involvement in the Ang II effect, then the 3′UTR ARE mutation that disrupts Ang II-induced destabilization of Kv4.3 mRNA (Fig. 2C) should also inhibit AUF1 binding to the mRNA. In fact, pull-down experiments demonstrated that the increased p37 AUF1 RNA binding induced by Ang II was not evident with the PG1.4B-m2 mutant (Figs. 7A,C). In contrast to the disruption of Ang II-induced AUF1 binding, HuR binding was unaffected. Therefore, Ang II induces AUF1 binding to the ARE responsible for Kv4.3 mRNA destabilization.
4. Discussion
While changes in cardiac channel gene expression have often been attributed to regulated transcription, Ang II destabilizes cardiac Kv4.3 channel mRNA [7,8]. This response is mediated by AT1 receptor-NADPH oxidase-p38 MAP kinase signaling [8]. Here an ARE was identified in the 3′UTR of the Kv4.3 mRNA that is required for this effect. Furthermore, Ang II was shown to upregulate AUF1, which results in increased binding of the high affinity p37 AUF1 isoform to this ARE. Also, AUF1 was demonstrated to be necessary and sufficient for Kv4.3 mRNA destabilization by Ang II. Finally, AUF1 upregulation was shown to require the same signaling (AT1 receptors, NADPH oxidase, p38 MAP kinase) as destabilization of the channel mRNA. Therefore, the simplest conclusion from these results is that Ang II induces AUF1, which then binds to a specific ARE in the 3′UTR of the channel mRNA to induce destabilization. The identified ARE is conserved in the human Kv4.3 gene (Fig. 2A). Hence, AUF1 binding to this ARE may be relevant in congestive heart failure in humans, which is associated with upregulation of AUF1 and downregulation of Kv4.3 [1,9].
In addition to supporing the above conclusion, the results also reveal some complexities. First, multiple AUF1 isoforms are upregulated by Ang II and, upon overexpression, can destabilize the Kv4.3 3′UTR. However, pull-down assays revealed that Ang II only increased p37 AUF1 association with the Kv4.3 ARE. These results could reflect the higher affinity for ARE binding of the p37 isoform, diverse subcellular localization for individual isoforms, confounding effects of overexpression, and limitations in the binding assay used here [12,14–16]. Second, although basal AUF1 expression was detected, RNA interference experiments indicated that AUF1 became functionally relevant only after stimulation with Ang II. One potential explanation for this finding is that phosphorylation can regulate AUF1 mRNA binding and function [14,17,18]. Thus, coordinated upregulation and posttranslational modification may be involved in the Ang II effect of Kv4.3 mRNA stability. It is also possible that Ang II affects other proteins that either assist or compete with AUF1 [16,19]. Addressing the above issues to provide a more detailed understanding of how AUF1 binding to a specific Kv4.3 3′UTR ARE mediates Ang II-induced destabilization of Kv4.3 mRNA will require further experiments.
The results presented here raise the question of whether other cardiac mRNAs are subject to regulation by this mechanism. AUF1 is induced by β agonists to destabilize β-adrenergic receptor mRNA [9]. AUF1 has also been implicated in the control of mRNA stability for SERCA2, which pumps Ca2+ into the sarcoplasmic reticulum [18,19]. Other important proteins are also downregulated with cardiac hypertrophy and Ang II signaling. For example, a decrease in connexin 43 expression likely contributes to arrhythmias and death in renin–angiotensinogen-overexpressing rats [5]. The connexin 43 3′UTR contains sequences for controlling mRNA stability [20], suggesting a potential involvement of AUF1. Therefore, AUF1 might affect expression of numerous cardiac genes in addition to Kv4.3. AUF1-mediated mRNA destabilization may be especially important in heart pathology because this mechanism reduces expression of specific proteins despite the generalized increase in translation associated with cardiac hypertrophy.
Acknowledgments
We thank Drs. M. Gorospe (NIH) and R. J. Schneider (NYU) for AUF1 RNA interference and expression constructs, respectively. We also thank Dr. C. Milcarek (University of Pittsburgh) for advice. This study was supported by NIH grant R01 HL80632.
References
- 1.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–83. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 2.Eghbali M, Deva R, Alioua A, Minosyan TY, Ruan H, Wang Y, Toro L, Stefani E. Molecular and functional signature of heart hypertrophy during pregnancy. Circ Res. 2005;96:1208–16. doi: 10.1161/01.RES.0000170652.71414.16. [DOI] [PubMed] [Google Scholar]
- 3.Takimoto K, Li D, Hershman KM, Li P, Jackson EK, Levitan ES. Decreased expression of Kv4.2 and novel Kv4.3 K+ channel subunit mRNAs in ventricles of renovascular hypertensive rats. Circ Res. 1997;81:533–9. doi: 10.1161/01.res.81.4.533. [DOI] [PubMed] [Google Scholar]
- 4.Harris DM, Mills GD, Chen X, Kubo H, Berretta RM, Votaw VS, Santana LF, Houser SR. Alterations in early action potential repolarization causes localized failure of sarcoplasmic reticulum Ca2+ release. Circ Res. 2005;96:543–50. doi: 10.1161/01.RES.0000158966.58380.37. [DOI] [PubMed] [Google Scholar]
- 5.Fischer, et al. Angiotensin II-induced sudden arrhythmic death and electrical remodeling. Am J Physiol Heart Circ Physiol. 2007;293:H1242–1253. doi: 10.1152/ajpheart.01400.2006. [DOI] [PubMed] [Google Scholar]
- 6.Goltz D, Schultz JH, Stucke C, Wagner M, Bassalaý P, Schwoerer AP, Ehmke H, Volk T. Diminished Kv4.2/3 but not KChIP2 levels reduce the cardiac transient outward K+ current in spontaneously hypertensive rats. Cardiovasc Res. 2007 Apr 1;74:85–95. doi: 10.1016/j.cardiores.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Zhang TT, Takimoto K, Stewart AF, Zhu C, Levitan ES. Independent regulation of cardiac Kv4.3 potassium channel expression by angiotensin II and phenylephrine. Circ Res. 2001;88:476–82. doi: 10.1161/01.res.88.5.476. [DOI] [PubMed] [Google Scholar]
- 8.Zhou C, Ziegler C, Birder LA, Stewart AF, Levitan ES. Angiotensin II and stretch activate NADPH oxidase to destabilize cardiac Kv4.3 channel mRNA. Circ Res. 2006;98:1040–7. doi: 10.1161/01.RES.0000218989.52072.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pende A, Tremmel KD, DeMaria CT, Blaxall BC, Minobe WA, Sherman JA, Bisognano JD, Bristow MR, Brewer G, Port J. Regulation of the mRNA-binding protein AUF1 by activation of the beta-adrenergic receptor signal transduction pathway. J Biol Chem. 1996;271:8493–501. doi: 10.1074/jbc.271.14.8493. [DOI] [PubMed] [Google Scholar]
- 10.Glaser ND, Lukyanenko YO, Wang Y, Wilson GM, Rogers TB. JNK activation decreases PP2A regulatory subunit B56α expression and mRNA stability and increases AUF1 expression in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2006;291:H1183–92. doi: 10.1152/ajpheart.01162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004;23(15):3092–102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarkar B, Xi Q, He C, Schneider RJ. Selective degradation of AU-rich mRNAs promoted by the p37 AUF1 protein isoform. Mol Cell Biol. 2003;23:6685–93. doi: 10.1128/MCB.23.18.6685-6693.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner BJ, DeMaria CT, Sun Y, Wilson GM, Brewer G. Structure and genomic organization of the human AUF1 gene: alternative pre-mRNA splicing generates four protein isoforms. Genomics. 1998;48:195–202. doi: 10.1006/geno.1997.5142. [DOI] [PubMed] [Google Scholar]
- 14.Wilson GM, Lu J, Sutphen K, Sun Y, Huynh Y, Brewer G. Regulation of A+U-rich element-directed mRNA turnover involving reversible phosphorylation of AUF1. J Biol Chem. 2003;278:33029–38. doi: 10.1074/jbc.M305772200. [DOI] [PubMed] [Google Scholar]
- 15.Sarkar B, Lu JY, Schneider RJ. Nuclear import and export functions in the different isoforms of the AUF1/heterogeneous nuclear ribonucleoprotein protein family. J Biol Chem. 2003;278:20700–7. doi: 10.1074/jbc.M301176200. [DOI] [PubMed] [Google Scholar]
- 16.Loflin P, Chen CY, Shyu AB. Unraveling a cytoplasmic role for hnRNP D in the in vivo mRNA destabilization directed by the AU-rich element. Genes Dev. 1999;13:1884–97. doi: 10.1101/gad.13.14.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson GM, Lu J, Sutphen K, Suarez Y, Sinha S, Brewer B, Villanueva-Feliciano EC, Ysla RM, Charles S, Brewer G. Phosphorylation of p40AUF1 regulates binding to A+U-rich mRNA-destabilizing elements and protein-induced changes in ribonucleoprotein structure. J Biol Chem. 2003;278:33039–48. doi: 10.1074/jbc.M305775200. [DOI] [PubMed] [Google Scholar]
- 18.Blum JL, Samarel AM, Mestril R. Phosphorylation and binding of AUF1 to the 3′-untranslated region of cardiomyocyte SERCA2a mRNA. Am J Physiol Heart Circ Physiol. 2005;289:H2543–2550. doi: 10.1152/ajpheart.00545.2005. [DOI] [PubMed] [Google Scholar]
- 19.Misquitta CM, Chen T, Grover AK. Control of protein expression through mRNA stability in calcium signalling. Cell Calcium. 2006;40:329–46. doi: 10.1016/j.ceca.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 20.Clairmont A, Sies H. Evidence for a posttranscriptional effect of retinoic acid on connexin43 gene expression via the 3′-untranslated region. FEBS Lett. 1997;419:268–70. doi: 10.1016/s0014-5793(97)01468-3. [DOI] [PubMed] [Google Scholar]