

Abstract
We have shown previously that SNM1A co-localizes with 53BP1 at sites of double-strand breaks (DSBs) induced by IR, and that these proteins interact with or without DNA damage. However, the role of SNM1A in the DNA damage response has not been elucidated. Here, we show that SNM1A is required for an efficient G1 checkpoint arrest after IR exposure. Interestingly, the localization of SNM1A to sites of DSBs does not require either 53BP1 or H2AX, nor does the localization of 53BP1 require SNM1A. However, the localization of SNM1A does require ATM. Furthermore, SNM1A is shown to be a phosphorylation substrate of ATM in vitro, and to interact with ATM in vivo particularly after exposure of cells to IR. In addition, in the absence of SNM1A the activation of the downstream ATM target p53 is reduced. These findings suggest that SNM1A acts with ATM to promote the G1 cell cycle checkpoint.
Keywords: SNM1A, G1 checkpoint, Ionizing radiation, ATM
Introduction
SNM1A is a member of a small gene family that is characterized by a metallo-β-lactamase fold and an appended β-CASP domain that together are proposed to function as a DNA nuclease [1; 2; 3; 4]. In budding yeast the single SNM1 gene is involved in mediating resistance to interstrand cross-linking drugs such as nitrogen mustard or mitomycin C (MMC), but not to other forms of DNA damage such as that induced by IR or UV [5; 6]. scSNM1 appears to function to repair double-strand breaks (DSBs) that are created when a replication fork encounters an interstrand cross-link (ICL), although, its precise function remains unidentified [7]. In mammalian cells five homologs have been identified and include SNM1A, SNM1B/Apollo, Artemis, ELAC2, and CPSF73 [8; 9; 10; 11]. Each of these genes contains the metallo-β-lactamase and β-CASP domains, but are otherwise distinct. These genes have been shown to play a variety of roles in cellular metabolism. For example, Artemis is required to cleave DNA hairpins at coding joints during V(D)J recombination, for repair of DSBs by the nonhomologous end joining pathway, and for cell cycle checkpoint regulation in response to DNA damage [11; 12; 13; 14; 15]. SNM1B/Apollo is required for the protection of telomeres during S phase, and in mediating resistance to DNA ICLs [16; 17; 18]. SNM1A has been shown to have the surprising function of being involved in an early mitotic or prophase checkpoint in response to spindle stress, a role that does not appear to involve a DNA damage response [19]. In addition, however, we have shown previously that SNM1A localizes to sites of IR-induced foci (IRIF), and that it strongly colocalizes at these foci with Mre11 and 53BP1 [20]. SNM1A was also shown to co-immunoprecipitate (co-IP) with 53BP1 before and after exposure to IR indicating that these two proteins are physically associated. However, SNM1A-deficient cells are not sensitive to IR, and have only a minor sensitivity to mitomycin C (MMC), but not to other ICL-inducing agents [9; 21]. These findings suggest that SNM1A does not have a direct role in the repair of DSBs, but that it does likely function in some aspect of the DNA damage response (DDR). In this report we confirm that SNM1A localizes to IRIF, and that this localization is dependent upon the checkpoint kinase ATM. In addition, we show that SNM1A is required for the implementation of the G1 phase checkpoint in response to IR treatment.
Materials and methods
Cell culture
Snm1A MEF culture conditions were as previously described [21]. The growth medium for HT-1080 cells, MCF-7 cells, and HEK293 cells was followed according to the ATCC (Global Bioresource Center). ATM-deficient cells (GM5849 and GM9627) were grown in DMEM-F12, 10% FBS, 0.05 mM nonessential amino acids, and 5 mM Na-pyruvate.
siRNA transfection
The siRNA directed against human SNM1A was as previously described [19]. The siRNA target sequence for 53BP1 was AAGCCAGGUUCUAGAGGAUGA, and for H2AX was CAACAAGAAGACGCGAAUC. All siRNAs were transfected into cells with Oligofectamine (Invitrogen). Forty-eight hours after transfection, knockdown of the specific protein was determined by immunoblotting or immunostaining analysis.
G1-phase checkpoint analysis
For Snm1 MEFs, 1 × 106 cells were seeded 24 hours before the experiment. Irradiation was performed in a 137Cs γ-ray irradiator at 3 Gy. After 8 hours untreated and irradiated cells were pulse-labeled with 10 μM BrdU (Roche) for 30 min at 37°C. For HT-1080 cells, 3 × 105 cells were seeded 24 hours before siRNA transfection. Forty-eight hours later, irradiation and BrdU treatment were performed as above. Cells were trypsinized and harvested in cold 10% FBS in PBS, fixed with cold 70% ethanol and kept at 4°C over night. Cells were then incubated with blocking buffer (4% BSA, 0.1% Triton X100 in PBS) for 30 min at room temperature. The cell pellet was suspended with 100 μl PBS plus 2 ml of 2N HCL and incubated for 30 min at room temperature. Cells were stained sequentially with anti-BrdU antibody and fluorescein isothiocyanate conjugated anti-mouse IgG antibody. DNA was stained with propidium iodide and RNA was digested with 50 μg/ml RNase for 30 min at 37°C. Cells were subsequently analyzed by FACS.
Immunofluorescence
Cells were grown on glass slides and exposed to the indicated doses of IR. To generate DSBs by microirradiation, cells were treated with a Micropoint UVA laser (365 nm) (Photonic Instruments) with a power setting of 20% essentially as described [22]. After incubation, cells were fixed with 4% paraformaldehyde for 20 min, and blocked with 4% BSA and 1% Triton X100 in PBS for 1 hr at room temperature. Cells were then immunostained with either γ-H2AX, 53BP1, or SNM1A [20] antibodies. DNA was stained with DAPI.
In vitro kinase assay
HEK293 cells were irradiated with IR (15 Gy) and allowed to recover for 30 min. Cells were harvested with TGN buffer (50 mM Tris pH7.5, 50 mM glycerophosphate, 150 mM NaCl, 10% glycerol, 1% Tween-20, 1 mM NaF, 1 mM NaVO4, 1 mM phenyl-methyl-sulfonyl-fluoride, 2 μg/ml pepstatin A, 5 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 mM DTT). ATM was immunoprecipitated with ATM antibody and protein A beads blocked with 5% BSA. Precipitated ATM was washed with TGN buffer 2 times, RIPA buffer 1 time, and kinase buffer 1 time, and then resuspended in 30 μl kinase buffer. Ten μCi γ-32P-ATP and 1 μg of substrate were added, and the samples were incubated for 30 min at 30°C. Samples were analyzed after SDS PAGE by autoradiography.
Co-immunoprecipitation assay
Co-immunoprecipitation assays were performed with the indicated antibodies as previously described [19].
Results
We have previously shown that SNM1A localizes to sites of DSBs created by exposure to IR [20], however, the function of SNM1A at these sites was not apparent. SNM1A-deficient cells are not sensitive to IR suggesting that it does not play a direct role in the repair of these lesions. Since SNM1A, Artemis and SNM1B/Apollo have all been shown to have roles in various checkpoint pathways in response to cellular stress, we examined whether SNM1A-deficient cells are defective in the G1 cell cycle checkpoint. As shown (Fig. 1A,C), depletion of SNM1A in HT-1080 cells by siRNA transfection resulted in a clear defect in the ability of these cells to arrest in G1 as indicated by BrdU incorporation. To further confirm this phenotype, mouse embryonic fibroblasts (MEFs) were also examined in this assay. A complication of using these cells is that the Snm1A-/- MEFs become rapidly aneuploid with time in culture because of the defect in mitotic function [19]. Nevertheless, as shown (Fig. 1B,C), three different Snm1A-/- MEF clones exhibited a similar defect in the G1 checkpoint in comparison to wild-type MEF clones. Thus, in both human and mouse cell lines a deficiency in expression of SNM1A results in a defective G1 checkpoint response upon exposure to IR.
Fig. 1.
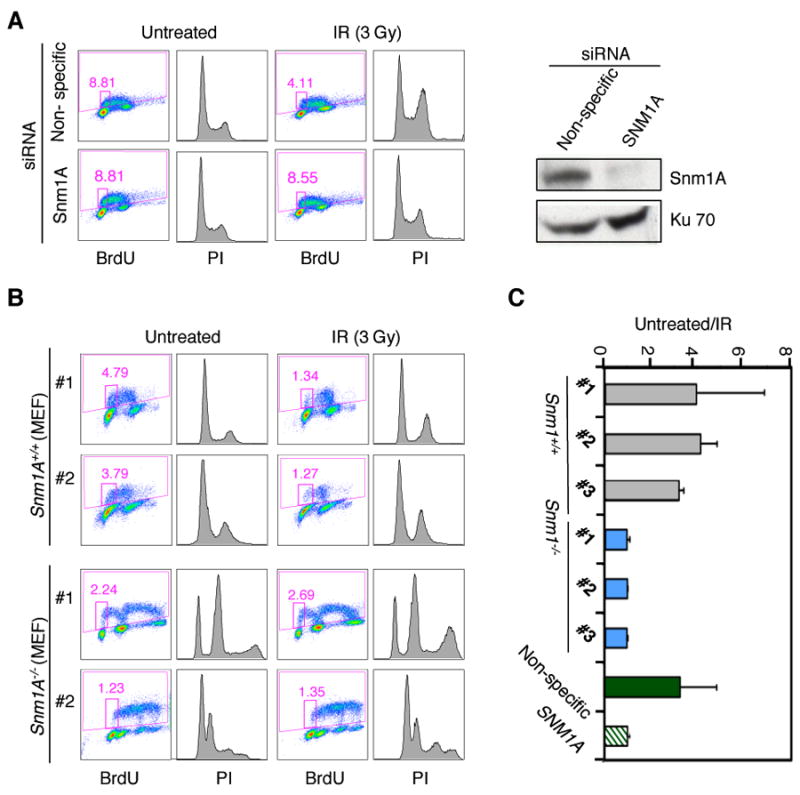
SNM1A is required for the IR-induced G1 phase cell cycle checkpoint. (A) Western blot shows the depletion of SNM1A in HT-1080 cells 48 hours after siRNA transfection (right panel). FACS analysis of siRNA-transfected HT-1080 cells with or without exposure to IR (3 Gy). (B) Spontaneously immortalized Snm1A MEFs were examined as described in A. (C) Graph represents the ratio of untreated BrdU positive cells to IR treated BrdU positive cells shown in A and B. MEF cell lines #1, #2, and #3 represent three independently derived clones from Snm1A+/+ or Snm1A-/- embryos.
We have previously shown that SNM1A colocalizes with Mre11 and 53BP1 at IRIF, and also co-IPs with 53BP1 before and after induction of DSBs [20]. To determine if 53BP1 is required for the localization of SNM1A at IRIF, we depleted its expression by siRNA. As shown (Fig. 2A and Fig. S1), the absence of 53BP1 did not affect the localization of SNM1A to IRIF. Reciprocally, an examination of Snm1A MEFs indicated that Snm1A was also not required for the localization of 53BP1 to IRIF (Fig. 2B). H2AX is a variant histone that is phosphorylated (γ-H2AX) at sites of DSBs, and is responsible for the recruitment of proteins involved in checkpoint and DNA repair functions [23]. However, depletion of H2AX did not affect the localization of SNM1A to IRIF indicating that SNM1A does not function downstream of γ-H2AX (Fig. 2C and Fig. S1).
Fig. 2.
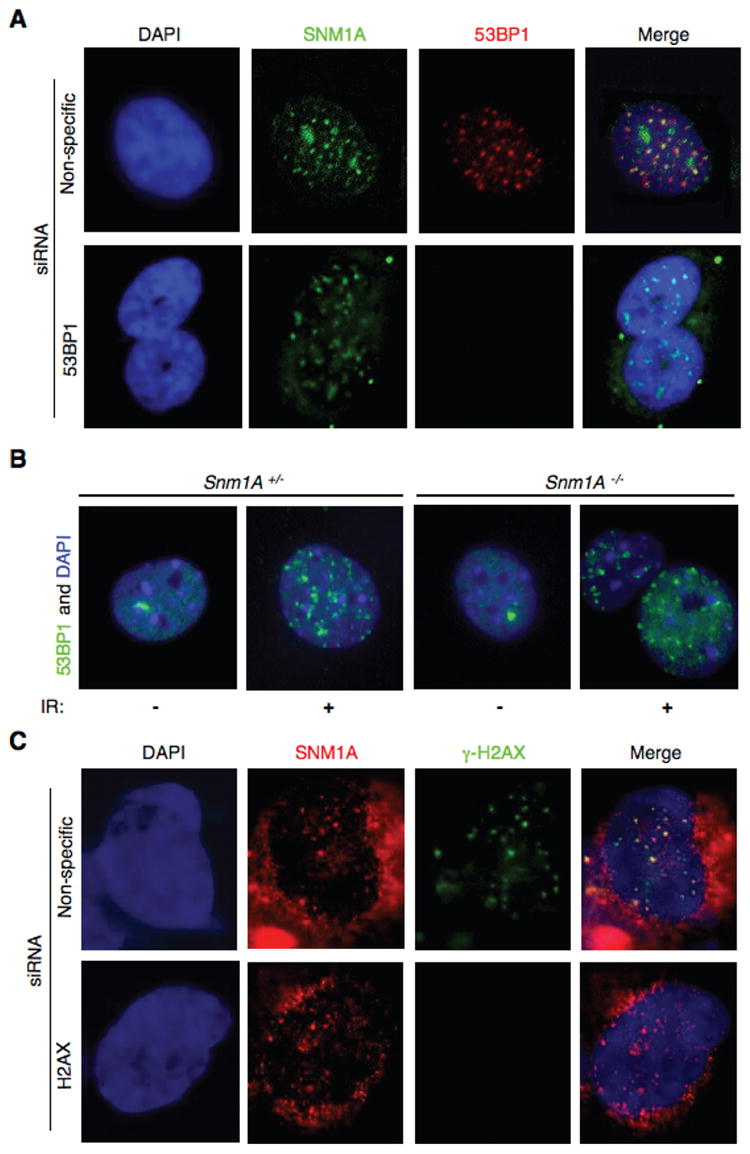
IR-induced SNM1A foci do not depend on γ-H2AX or 53BP1. (A) HT-1080 cells were depleted of 53BP1 by siRNA, and exposed to IR (10 Gy). Four hrs later cells were fixed and immunostained with the indicated antibodies. (B) IR-induced 53BP1 foci formation was not affected in in Snm1A-/- MEF cells. Cells were processed as in A. (C) HT-1080 cells were depleted of H2AX by siRNA, and processed as described in A.
In response to IR the upstream kinase ATM, a member of the phosphatidylinositol-3-OH kinase-like (PIK) family, phosphorylates a number of checkpoint and DNA repair proteins involved in mediating the DDR [24; 25]. To determine if ATM affects the localization of SNM1A to IRIF, we examined this response in two ATM-deficient cell lines. Interestingly, as shown (Fig. 3A), the absence of ATM activity resulted in a failure of SNM1A to localize to IRIF. The single large foci found in the ATM-deficient cells likely represents an SNM1A body, which are typically the localization of SNM1A observed in approximately half of untreated cells [20]. After IR treatment these bodies appear to dissolve and SNM1A relocates to sites of DSBs. To further confirm that ATM is required for the localization of SNM1A to sites of DSBs, we used microirradiation to form DSBs in control and AT cells. Again, SNM1A failed to relocalize to sites of DSBs (Fig. 3B). These findings indicate that ATM is required for the localization of SNM1A to sites of DSBs.
Fig. 3.
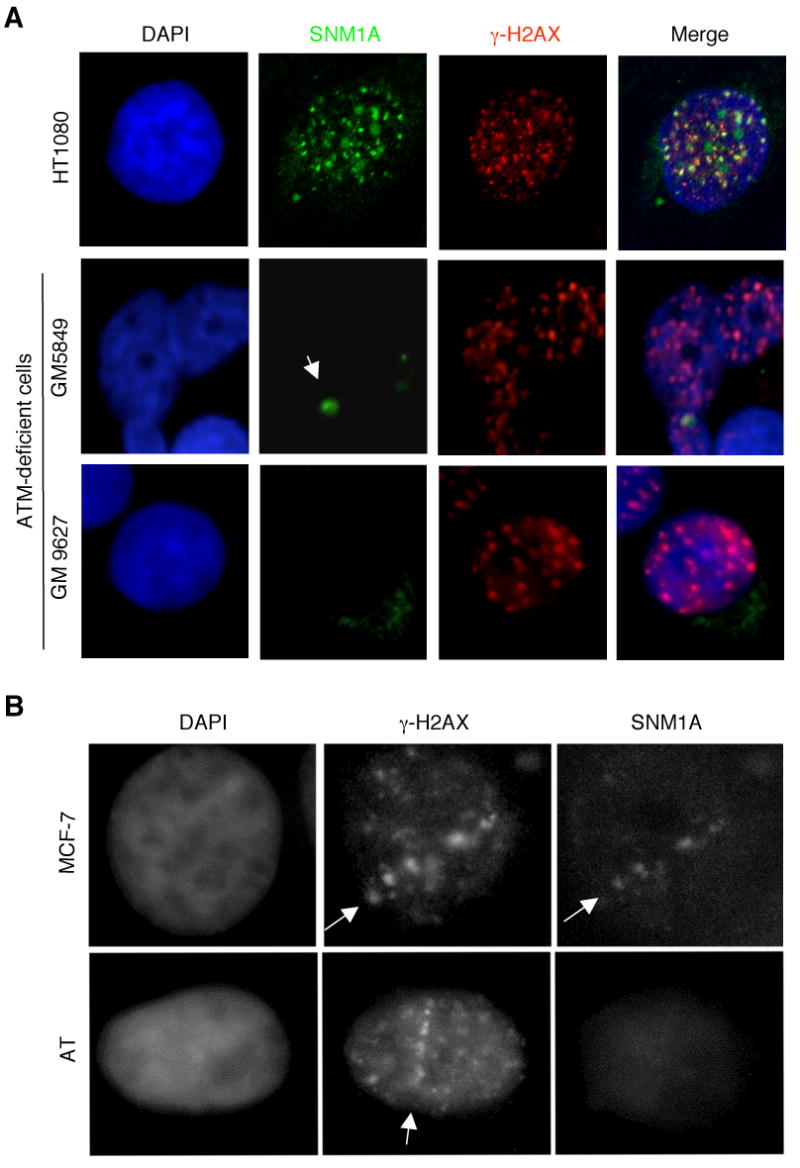
IR-induced SNM1A foci formation is dependent upon ATM. (A) ATM-deficient cells were irradiated with IR (10 Gy). Four hrs later cells were fixed and immunostained with the indicated antibodies. The arrow indicates an SNM1A body. (B) Induction of DSBs by UVA laser microirradiation indicates defective localization of SNM1A in AT cells. MCF-7 or AT cells were microirradiated at 365 nm and 4 hrs later cells were fixed and immunostained with the indicated antibodies. Arrows indicate the line of laser microirradiation.
Artemis is a member of the SNM1 gene family and has been shown to be phosphorylated in response to DNA damage by the PIK kinases ATM, DNA-PK, and ATR both in vitro and in vivo [12; 13; 14; 15; 26; 27; 28; 29]. PIK kinases have a strong preference for phosphorylation at (S/T)Q motifs [30; 31], and Artemis contains a cluster of these sites (referred to as a (S/T)Q cluster domain or SCD) in its nonconserved region. Likewise, SNM1A also has a cluster of these sites in its nonconserved domain (Fig. 4A). To determine if SNM1A is a potential substrate of ATM, we expressed a recombinant form of the nonconserved region in E. coli and purified it by means of a fused maltose binding protein (MBP) tag. For a negative control we used MBP-SNM1B. As shown (Fig. 4B), ATM was able to phosphorylate MBP-SNM1A-NT in vitro, but not MBP-SNM1B. These results indicate that SNM1A may be a direct phosphorylation substrate of ATM, which is consistent with a requirement for this kinase in the localization of SNM1A to sites of DSBs.
Fig. 4.
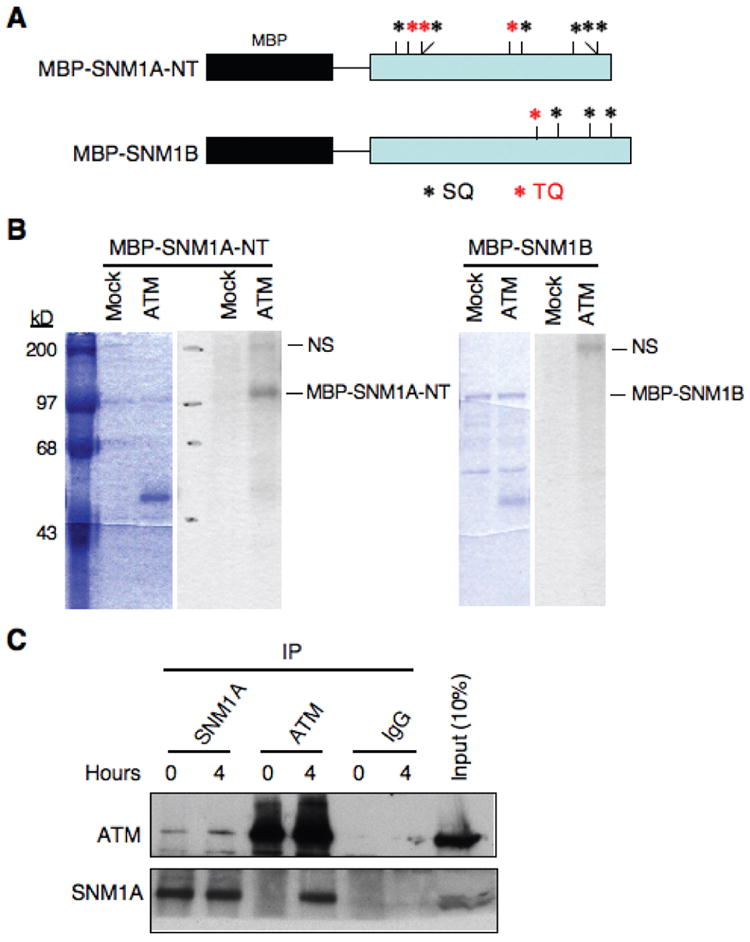
SNM1A is a phosphorylation substrate of ATM in vitro and interacts with ATM in vivo. (A) The SQ and TQ sites present in the N-terminus (NT) region of SNM1A and in full-length SNM1B are shown. (B) An IP kinase assay shows that recombinant MBP-SNM1A-NT (SNM1A residues 1-393), but not SNM1B is an ATM substrate. Left side of each panel shows Coomassie blue stained gels, and right sides show autoradiograms. “Mock” indicates an immunoprecipitation performed with a nonspecific IgG. “NS” indicates a nonspecific substrate of ATM. (C) Immunoblot showing reciprocal co-IP of ATM and SNM1A with or without IR treatment (10 Gy).
The results described above suggest that SNM1A may be a substrate of ATM in vivo. If so, then it is expected that SNM1A and ATM interact. Reciprocal co-immunoprecipitations (co-IP) of these proteins showed that this prediction was correct (Fig. 4C). Furthermore, the interaction was greatly enhanced after exposure of cells to IR particularly as shown by the co-IP with an ATM antibody. These results suggest that ATM recruits SNM1A to sites of DSBs and likely phosphorylates it at these sites. We have established that SNM1A is required for implementation of the G1 checkpoint and that it interacts with ATM. Thus, we examined markers that are activated downstream of ATM to effectuate the G1 checkpoint to determine if SNM1A plays a role in their activation. P53 is a direct phosphorylation target of ATM in response to DNA damage, and p21 is a transcriptional target of p53 [23]. Depletion of SNM1A reduced ATM-mediated phosphorylation of p53 and the downstream activation of p21 (Fig. S2). Taken together these findings suggest that SNM1A is required for the full implementation of the G1 checkpoint.
Discussion
Our previous studies on the mammalian members of the SNM1 gene family have revealed cell cycle checkpoint functions for SNM1A, SNM1B/Apollo, and Artemis. These functions are wide ranging and involve responses not only to DNA damage but also to other forms of cellular stress such as spindle poisons. Artemis functions in the G2/M checkpoint in response to IR to regulate the activation of Cdk1-cyclin B and thereby recovery from the induced cell cycle arrest [12; 15]. We have shown that SNM1B/Apollo functions in an S phase checkpoint specifically in response to DNA interstrand cross-linking agents [32]. In this case SNM1B/Apollo is required for the formation of DSBs that occur during S phase when a replication fork encounters an interstrand cross-link. The formation of the DSB activates ATM and thus prolongs the S phase arrest allowing for removal of the cross-link. Perhaps most surprising are our findings showing that SNM1A is involved in an early mitotic checkpoint that arrests cells in prophase in response to spindle poisons [19]. This pathway also requires a gene termed CHFR which has been shown to be highly mutated in many different forms of cancer indicating the importance of this pathway in tumorigenesis (Scolnick and Halazonetis, 2000; type [33; 34; 35; 36; 37; 38]. Both CHFR and SNM1A have been shown to act as tumor suppressors in the mouse [21; 39].
In this report we have added to the growing body of evidence that SNM1 family members act as cell cycle regulators in response to cellular stress. In our previous studies we had shown that SNM1A is recruited to IRIF and that it interacts with 53BP1 [20]. We have shown here that SNM1A is required for implementation of the G1 checkpoint. Surprisingly, the recruitment of SNM1A to IRIF does not require 53BP1 or vice versa despite the fact that these proteins interact. This finding is consistent with the result that γ-H2AX is also not required for the recruitment of SNM1A since the recruitment of 53BP1 requires γ-H2AX [23]. Rather the recruitment of SNM1A to IRIF appears to be mediated by ATM perhaps directly since SNM1A and ATM interact, and this interaction is enhanced upon IR treatment. Our results also suggest that SNM1A is a likely substrate of ATM. The exact role that SNM1A plays at IRIF is unclear, however, our evidence indicates that it does affect the activation of downstream effectors of the G1 checkpoint such as p53 that is a direct phosphorylation target of ATM.
Supplementary Material
Acknowledgments
This work was supported by NCI grants CA052461 and CA097175. DNA sequencing resources were supported by the Cancer Center Support (Core) Grant CA16672.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ma Y, Schwarz K, Lieber MR. The Artemis:DNA-PKcs endonuclease cleaves DNA loops, flaps, and gaps. DNA Repair (Amst) 2005;4:845–51. doi: 10.1016/j.dnarep.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 2.Pannicke U, Ma Y, Hopfner KP, Niewolik D, Lieber MR, Schwarz K. Functional and biochemical dissection of the structure-specific nuclease ARTEMIS. Embo J. 2004;23:1987–97. doi: 10.1038/sj.emboj.7600206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, Hejna J, Moses RE. The yeast Snm1 protein is a DNA 5′-exonuclease. DNA Repair (Amst) 2005;4:163–70. doi: 10.1016/j.dnarep.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Hazrati A, Ramis-Castelltort M, Sarkar S, Barber LJ, Schofield CJ, Hartley JA, McHugh PJ. Human SNM1A suppresses the DNA repair defects of yeast pso2 mutants. DNA Repair (Amst) 2008;7:230–8. doi: 10.1016/j.dnarep.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 5.Ruhland A, Kircher M, Wilborn F, Brendel M. A yeast mutant specifically sensitive to bifunctional alkylation. Mutat Res. 1981;91:457–62. doi: 10.1016/0165-7992(81)90052-x. [DOI] [PubMed] [Google Scholar]
- 6.Henriques JA, Moustacchi E. Isolation and characterization of pso mutants sensitive to photo-addition of psoralen derivatives in Saccharomyces cerevisiae. Genetics. 1980;95:273–88. doi: 10.1093/genetics/95.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barber LJ, Ward TA, Hartley JA, McHugh PJ. DNA interstrand cross-link repair in the Saccharomyces cerevisiae cell cycle: overlapping roles for PSO2 (SNM1) with MutS factors and EXO1 during S phase. Mol Cell Biol. 2005;25:2297–309. doi: 10.1128/MCB.25.6.2297-2309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tavtigian SV, Simard J, Teng DH, Abtin V, Baumgard M, Beck A, Camp NJ, Carillo AR, Chen Y, Dayananth P, Desrochers M, Dumont M, Farnham JM, Frank D, Frye C, Ghaffari S, Gupte JS, Hu R, Iliev D, Janecki T, Kort EN, Laity KE, Leavitt A, Leblanc G, McArthur-Morrison J, Pederson A, Penn B, Peterson KT, Reid JE, Richards S, Schroeder M, Smith R, Snyder SC, Swedlund B, Swensen J, Thomas A, Tranchant M, Woodland AM, Labrie F, Skolnick MH, Neuhausen S, Rommens J, Cannon-Albright LA. A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet. 2001;27:172–80. doi: 10.1038/84808. [DOI] [PubMed] [Google Scholar]
- 9.Dronkert ML, de Wit J, Boeve M, Vasconcelos ML, van Steeg H, Tan TL, Hoeijmakers JH, Kanaar R. Disruption of mouse SNM1 causes increased sensitivity to the DNA interstrand cross-linking agent mitomycin C. Mol Cell Biol. 2000;20:4553–61. doi: 10.1128/mcb.20.13.4553-4561.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jenny A, Minvielle-Sebastia L, Preker PJ, Keller W. Sequence similarity between the 73-kilodalton protein of mammalian CPSF and a subunit of yeast polyadenylation factor I. Science. 1996;274:1514–7. doi: 10.1126/science.274.5292.1514. [DOI] [PubMed] [Google Scholar]
- 11.Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, Fischer A, de Villartay JP. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–86. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 12.Geng L, Zhang X, Zheng S, Legerski RJ. Artemis links ATM to G2/M checkpoint recovery via regulation of Cdk1-cyclin B. Mol Cell Biol. 2007;27:2625–35. doi: 10.1128/MCB.02072-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–94. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 14.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, Parker AR, Jackson SP, Gennery A, Jeggo PA, Lobrich M. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–24. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, Succi J, Feng Z, Prithivirajsingh S, Story MD, Legerski RJ. Artemis is a phosphorylation target of ATM and ATR and is involved in the G2/M DNA damage checkpoint response. Mol Cell Biol. 2004;24:9207–20. doi: 10.1128/MCB.24.20.9207-9220.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenain C, Bauwens S, Amiard S, Brunori M, Giraud-Panis MJ, Gilson E. The Apollo 5′ exonuclease functions together with TRF2 to protect telomeres from DNA repair. Curr Biol. 2006;16:1303–10. doi: 10.1016/j.cub.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 17.Szilard RK, Durocher D. Telomere protection: an act of God. Curr Biol. 2006;16:R544–6. doi: 10.1016/j.cub.2006.06.037. [DOI] [PubMed] [Google Scholar]
- 18.van Overbeek M, de Lange T. Apollo, an Artemis-related nuclease, interacts with TRF2 and protects human telomeres in S phase. Curr Biol. 2006;16:1295–302. doi: 10.1016/j.cub.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 19.Akhter S, Richie CT, Deng JM, Brey E, Zhang X, Patrick C, Jr, Behringer RR, Legerski RJ. Deficiency in SNM1 abolishes an early mitotic checkpoint induced by spindle stress. Mol Cell Biol. 2004;24:10448–55. doi: 10.1128/MCB.24.23.10448-10455.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richie CT, Peterson C, Lu T, Hittelman WN, Carpenter PB, Legerski RJ. hSnm1 colocalizes and physically associates with 53BP1 before and after DNA damage. Mol Cell Biol. 2002;22:8635–47. doi: 10.1128/MCB.22.24.8635-8647.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahkter S, Richie CT, Zhang N, Behringer RR, Zhu C, Legerski RJ. Snm1-deficient mice exhibit accelerated tumorigenesis and susceptibility to infection. Mol Cell Biol. 2005;25:10071–8. doi: 10.1128/MCB.25.22.10071-10078.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, Mari PO, van Gent DC, Chen BP, Chen DJ. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol. 2007;177:219–29. doi: 10.1083/jcb.200608077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 24.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–96. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 25.Lukas J, Bohr VA, Halazonetis TD. Cellular responses to DNA damage: current state of the field and review of the 52nd Benzon Symposium. DNA Repair (Amst) 2006;5:591–601. doi: 10.1016/j.dnarep.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Pluth JM, Cooper PK, Cowan MJ, Chen DJ, Yannone SM. Artemis deficiency confers a DNA double-strand break repair defect and Artemis phosphorylation status is altered by DNA damage and cell cycle progression. DNA Repair (Amst) 2005;4:556–70. doi: 10.1016/j.dnarep.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 27.Poinsignon C, de Chasseval R, Soubeyrand S, Moshous D, Fischer A, Hache RJ, de Villartay JP. Phosphorylation of Artemis following irradiation-induced DNA damage. Eur J Immunol. 2004;34:3146–55. doi: 10.1002/eji.200425455. [DOI] [PubMed] [Google Scholar]
- 28.Chen L, Morio T, Minegishi Y, Nakada S, Nagasawa M, Komatsu K, Chessa L, Villa A, Lecis D, Delia D, Mizutani S. Ataxia-telangiectasia-mutated dependent phosphorylation of Artemis in response to DNA damage. Cancer Sci. 2005;96:134–41. doi: 10.1111/j.1349-7006.2005.00019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nojima K, Hochegger H, Saberi A, Fukushima T, Kikuchi K, Yoshimura M, Orelli BJ, Bishop DK, Hirano S, Ohzeki M, Ishiai M, Yamamoto K, Takata M, Arakawa H, Buerstedde JM, Yamazoe M, Kawamoto T, Araki K, Takahashi JA, Hashimoto N, Takeda S, Sonoda E. Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res. 2005;65:11704–11. doi: 10.1158/0008-5472.CAN-05-1214. [DOI] [PubMed] [Google Scholar]
- 30.Traven A, Heierhorst J. SQ/TQ cluster domains: concentrated ATM/ATR kinase phosphorylation site regions in DNA-damage-response proteins. Bioessays. 2005;27:397–407. doi: 10.1002/bies.20204. [DOI] [PubMed] [Google Scholar]
- 31.Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274:37538–43. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- 32.Bae JB, Mukhopadhyay SS, Liu L, Zhang N, Tan J, Akhter S, Liu X, Shen X, Li L, Legerski RJ. Snm1B/Apollo mediates replication fork collapse and S Phase checkpoint activation in response to DNA interstrand cross-links. Oncogene. 2008 doi: 10.1038/onc.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheung HW, Ching YP, Nicholls JM, Ling MT, Wong YC, Hui N, Cheung A, Tsao SW, Wang Q, Yeun PW, Lo KW, Jin DY, Wang X. Epigenetic inactivation of CHFR in nasopharyngeal carcinoma through promoter methylation. Mol Carcinog. 2005;43:237–45. doi: 10.1002/mc.20106. [DOI] [PubMed] [Google Scholar]
- 34.Corn PG, Summers MK, Fogt F, Virmani AK, Gazdar AF, Halazonetis TD, El-Deiry WS. Frequent hypermethylation of the 5′ CpG island of the mitotic stress checkpoint gene Chfr in colorectal and non-small cell lung cancer. Carcinogenesis. 2003;24:47–51. doi: 10.1093/carcin/24.1.47. [DOI] [PubMed] [Google Scholar]
- 35.McPherson JP, Lemmers B, Chahwan R, Pamidi A, Migon E, Matysiak-Zablocki E, Moynahan ME, Essers J, Hanada K, Poonepalli A, Sanchez-Sweatman O, Khokha R, Kanaar R, Jasin M, Hande MP, Hakem R. Involvement of mammalian Mus81 in genome integrity and tumor suppression. Science. 2004;304:1822–6. doi: 10.1126/science.1094557. [DOI] [PubMed] [Google Scholar]
- 36.Mariatos G, Bothos J, Zacharatos P, Summers MK, Scolnick DM, Kittas C, Halazonetis TD, Gorgoulis VG. Inactivating mutations targeting the chfr mitotic checkpoint gene in human lung cancer. Cancer Res. 2003;63:7185–9. [PubMed] [Google Scholar]
- 37.Mizuno K, Osada H, Konishi H, Tatematsu Y, Yatabe Y, Mitsudomi T, Fujii Y, Takahashi T. Aberrant hypermethylation of the CHFR prophase checkpoint gene in human lung cancers. Oncogene. 2002;21:2328–33. doi: 10.1038/sj.onc.1205402. [DOI] [PubMed] [Google Scholar]
- 38.Morioka Y, Hibi K, Sakai M, Koike M, Fujiwara M, Kodera Y, Ito K, Nakao A. Aberrant methylation of the CHFR gene is frequently detected in non-invasive colorectal cancer. Anticancer Res. 2006;26:4267–70. [PubMed] [Google Scholar]
- 39.Yu X, Minter-Dykhouse K, Malureanu L, Zhao WM, Zhang D, Merkle CJ, Ward IM, Saya H, Fang G, van Deursen J, Chen J. Chfr is required for tumor suppression and Aurora A regulation. Nat Genet. 2005;37:401–6. doi: 10.1038/ng1538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.