

Abstract
Abnormal centrosome numbers are detected in virtually all cancers. The molecular mechanisms that underlie centrosome amplification, however, are poorly characterized. Based on the model that each maternal centriole serves as a template for the formation of one and only one daughter centriole per cell division cycle, the prevailing view is that centriole overduplication arises from successive rounds of centriole reproduction. Here, we provide evidence that a single maternal centriole can concurrently generate multiple daughter centrioles. This mechanism was initially identified in cells treated with the peptide vinyl sulfone proteasome inhibitor Z-L3VS. We subsequently found that the formation of more than one daughter at maternal centrioles required cyclin E/cyclin-dependent kinase 2 (CDK2) as well as Polo-like kinase 4 (PLK4) and that overexpression of these proteins mimics this phenotype in the absence of a proteasome inhibitor. To corroborate that a concurrent formation of multiple daughter centrioles is potentially relevant for centriole overduplication in human cancer, we show that the human papillomavirus type 16 (HPV-16) E7 oncoprotein stimulates aberrant daughter centriole numbers in part through the formation of more than one daughter centriole at single maternal templates. These results help to explain how oncogenic stimuli can rapidly induce abnormal centriole numbers within a single cell division cycle and provide insights into the regulation of centriole duplication.
Introduction
Centrosomes are the major microtubule organizing centers in most animal and human cells (Bornens, 2002; Nigg, 2004). The single centrosome duplicates precisely once prior to mitosis through mechanisms that remain to be understood. In a normal cell cycle, each of the two centrioles that make up a G1 phase centrosome is thought to function as a template for the formation of precisely one newly synthesized daughter centriole (Sluder, 2004). The restriction to a single round of centriole duplication per cell division cycle (Tsou & Stearns, 2006a) contributes to the prevention of aberrant centrosome numbers, multipolar mitoses and chromosomal instability (Brinkley, 2001; Nigg, 2002; Salisbury et al., 1999).
In contrast to normal cells, tumor cells frequently contain abnormal centrosome numbers (Lingle et al., 1998; Pihan et al., 1998). Although various oncogenic stimuli have been found to provoke abnormal centrosome and centriole numbers in vitro, relatively little is known about the precise mechanisms of their origin. In principal, aberrant centrosome numbers can arise through cell division failure or a genuine disruption of the centriole duplication cycle itself (Duensing, 2005; Nigg, 2002). Only the second category should be considered as centriole overduplication and it has been proposed that overduplication can be distinguished from centriole accumulation through the presence of excessive numbers of immature daughter centrioles (Guarguaglini et al., 2005).
The premise that each maternal centriole serves as a template for the formation of one and only one daughter centriole during each cell division cycle does not readily explain the rapid induction of aberrant centriole numbers detected under certain experimental conditions. The HPV-16 E7 oncoprotein has been found to rapidly stimulate supernumerary centrosomes in primary human cells and tumor cell lines within 48 h following transient transfection (Duensing et al., 2000). A 24 h treatment interval with either a CDK inhibitor (Duensing et al., 2004) or an inhibitor of RNA polymerase II (Duensing et al., 2006a) was sufficient to suppress HPV-16 E7-induced aberrant centriole numbers. These results suggest that significant centriole overduplication can occur within a 24 h time period that corresponds to approximately one cell division cycle in the cells used for these experiments. HPV-16 E7 binds and degrades the retinoblastoma tumor suppressor protein (pRB), inactivates cyclin-dependent kinase inhibitors such as p21Cip1 and causes a deregulated expression of cyclin E (Martin et al., 1998; McIntyre et al., 1996) thereby creating an aberrant S-phase like state that supports viral replication (Munger et al., 2001). How these activities can trigger centriole overduplication within a single cell cycle in currently unknown.
In the present report, we provide evidence for centriole overduplication through the concurrent formation of multiple daughter centrioles at single maternal centrioles in human cells. This mechanism was first identified using the proteasome inhibitor Z-L3VS (carboxybenzyl-leucyl-leucyl-leucine vinyl sulfone) but further analyses showed a requirement for cyclin E/CDK2 and PLK4. Combined overexpression of these proteins in fact mimicked the Z-L3VS-induced phenotype in both tumor cells and in non-transformed cells, even in the absence of a proteasome inhibitor. Finally, we discovered that HPV-16 E7-induced centriole overduplication also involves the formation of more than one daughter centriole per maternal centriole. These findings help to explain the rapid induction of abnormal centriole numbers by this viral oncogene and provide evidence for a role of proteolysis in the regulation of centriole duplication.
Results
Induction of abnormal centriole duplication by the proteasome inhibitor Z-L3VS
Components of the ubiquitin-proteasome machinery localize to centrosomes and ubiquitin-mediated proteolysis has been implicated in the regulation of centriole duplication (Fabunmi et al., 2000; Freed et al., 1999; Gstaiger et al., 1999; Nakayama et al., 2000; Wigley et al., 1999; Wojcik et al., 2000). Thus, we performed a morphological screen for aberrant centriole duplication events using a panel of proteasome inhibitors in U-2 OS cell populations stably expressing centrin-GFP (U-2 OS/centrin-GFP) to visualize individual centrioles (please see Supplemental Material for Material and Methods). Centrin-GFP signals in DMSO-treated cells typically consisted of large centrin-GFP dots (indicating the maternal centriole) with a single smaller centrin-GFP dot (indicating the daughter centriole) in close proximity to the maternal centriole (Fig. 1A). Treatment of cells for 48 h with the proteasome inhibitor Z-L3VS (Bogyo et al., 1997) was found to induce multiple centrin-GFP dots in a significant proportion of cells (Fig. 1A,B). Centrin-GFP dots were frequently arranged in a “flower”-like pattern with a large centrin-GFP dot in the center surrounded by multiple smaller centrin-GFP dots (Fig. 1A). We scored cells with more than one small centrin-GFP dot adjacent to a large centrin-GFP dot as aberrant daughter centriole formation at a maternal template in all subsequent experiments.
Figure 1. Aberrant centriole configuration following treatment with the proteasome inhibitor Z-L3VS.

(A) Fluorescence microscopic analysis of U-2 OS cells stably expressing centrin-GFP to visualize centrioles (arrows; inserts) after either treatment with 0.1% DMSO (top panels) or 1 μM Z-L3VS (bottom panels) for 48 h. Nuclei stained with DAPI. Scale bar indicates 10 μm.
(B) Quantification of U-2 OS/centrin-GFP cells with more than one daughter centriole per maternal centriole after treatment with either 0.1% DMSO or 1 μM Z-L3VS for 48 h. Each bar represents mean and standard error of at least three independent experiments with a minimum of 100 cells counted per experiment.
Quantification of cells with more than one small centrin-GFP dot adjacent to a large centrin-GFP dot revealed a 55.7-fold increase in populations treated with 1 μM Z-L3VS for 48 h (39%; p≤0.0001) compared to DMSO-treated controls (0.7%; Fig. 1B). This increase was dose-dependent since cell populations treated with lower concentrations of Z-L3VS contained a reduced fraction of cells with aberrant daughter centriole formation (data not shown). An increase of cells with excessive daughter centriole formation, albeit to a lesser extent, was also detected in cells treated with the proteasome inhibitors MG262 or MG132 but not in cells treated with epoxomicin or lactacystin. The Z-L3VS-induced aberrant formation of daughter centrioles was significantly less frequently detected in HeLa or T98G cells and was absent in IMR-90 normal human fibroblasts (data not shown).
To further determine why U-2 OS cells but not HeLa or T98G tumor cell lines showed excessive daughter centriole synthesis, we performed immunoblot experiments using whole cell lysates from DMSO-treated cells or cells treated with 1 μM Z-L3VS for 48 h (Suppl. Fig. 1). We analyzed the expression of cyclin E, cyclin A, CDK2 and polo-like kinase 4 (PLK4) since these proteins have previously been implicated in centrosome amplification (Bettencourt-Dias et al., 2005; Habedanck et al., 2005; Hinchcliffe et al., 1999; Lacey et al., 1999; Matsumoto et al., 1999; Meraldi et al., 1999). U-2 OS cells treated with Z-L3VS showed a marked increase of cyclin E protein levels in comparison to DMSO-treated controls (Suppl. Fig. 1A). A minor increase of cyclin E expression was detected in Z-L3VS-treated HeLa cells but not in T98G cells when compared to DMSO-treated controls. Changes in cyclin A, CDK2 and PLK4 levels were considered insignificant based on protein loading. Furthermore, flow cytometric analyses revealed an accumulation of Z-L3VS-treated U-2 OS cells in G2 phase (Suppl. Fig. 1B), which is in line with a previous report showing an accumulation of this cell type in G2 upon cyclin E overexpression (Bartkova et al., 2005). Such changes in the cell cycle profile were not detected in Z-L3VS-treated HeLa or T98G cells (Suppl. Fig. 1B). To corroborate the accumulation in G2, an immunofluorescence analysis of Z-L3VS-treated U-2 OS cells for cyclin B was performed (Suppl. Fig. 1C). Cyclin B accumulates in the cytoplasm in S and G2 phase (Pines & Hunter, 1991). We found an increase of U-2 OS cells expressing cytoplasmic cyclin B from 16.6% in DMSO-treated controls to 30.4% in Z-L3VS-treated cells.
Several lines of evidence suggest, however, that an accumulation in G2 phase is not required for excessive daughter centriole formation at maternal templates and that this process begins before cells reach G2. First, there was no strict correlation between cyclin B expression and the formation of more than one daughter centriole per maternal template since 22.3% of cells with this phenotype had undetectable levels of cyclin B. Such cells frequently showed an incomplete phenotype as illustrated in (Suppl. Fig 1C; top panels) suggesting that aberrant centriole synthesis starts before cells accumulate in G2. Moreover, when U-2 OS cells treated with Z-L3VS were analyzed for centriole maturation using CEP170 as a marker (Suppl. Fig. 2), we found that supernumerary daughter centrioles can form prior to or while the second maternal centriole reaches maturity. This indicates that a prolonged arrest in G2 phase is not required for excessive daughter centriole formation and that this process is likely to start before cells reach G2 phase.
Since our results suggest that cyclin E accumulation may be a distinctive feature of cells that form excessive numbers at maternal centrioles following Z-L3VS treatment, we analyzed next whether ectopic expression of cyclin E in HeLa and T98G cells induced this phenotype (Suppl. Fig. 3). A significant increase of cells with more than one daughter per maternal centriole was detected in Z-L3VS-treated T98G cells transfected with cyclin E (16.1%) in comparison to empty vector-transfected cells (6.2%). A similar increase was also found in HeLa cells (17.6% in cyclin E-transfected cells versus 6.2% in empty vector controls). We hasten to add that these results are lower than what we detected in U-2 OS cells treated with Z-L3VS suggesting that cyclin E may not be solely responsible and that changes in the expression level of additional proteins are involved.
Ultimately, we sought to determine whether the aberrant centrin-GFP dots were in fact intact daughter centrioles, we examined Z-L3VS treated U-2 OS cells by electron microscopy. Over one hundred cells were screened by electron microscopy. A total of twenty-four centrioles were analyzed in serial sections of which three had multiple daughter centrioles. Figure 2 demonstrates multiple daughter centrioles radially arranged around a central maternal centriole in comparison to a normal mother-daughter centriole pair.
Figure 2. Concurrent formation of multiple daughter centrioles at a single maternal centriole following Z-L3VS.

Electron microscopic analysis of U-2 OS/centrin-GFP cells either DMSO-treated with 0.1% DMSO (A) or treated with 1 μM Z-L3VS for 48 h (B). Scale bars indicate 500 nm.
Taken together, our results suggest that Z-L3VS leads to an accumulation of proteins in U-2 OS cells that trigger an excessive daughter centriole formation at single maternal templates.
Aberrant centriole duplication requires cyclin E/CDK2 and PLK4
To determine the role of cyclin E, CDK2 and PLK4 in Z-L3VS-induced abnormal daughter centriole formation in greater detail, small-interfering RNA (siRNA) experiments were performed (Fig. 3A). As expected, U-2 OS/centrin-GFP cells transfected with control siRNA duplexes and treated with 1 μM Z-L3VS for 48 h showed excessive daughter centrioles at maternal centrioles in 46.6% of cells in comparison to 1.3% in DMSO-treated controls (p≤0.0001; Fig. 3B,C). The proportion of cells showing this configuration was significantly reduced in cells transfected with siRNA duplexes targeting cyclin E1 (21.9%; p≤0.0001), or, to a lesser extent, cyclin E2 (36.3%; p≤0.05). Depletion of CDK2 caused a significant decrease of cells with aberrant centriole formation (29.9%; p≤0.005), whereas knock-down of cyclin A2 had no effect (Fig. 3C).
Figure 3. Z-L3VS-induced formation of excessive daughter centrioles at maternal centrioles requires cyclin E, CDK2 and PLK4.
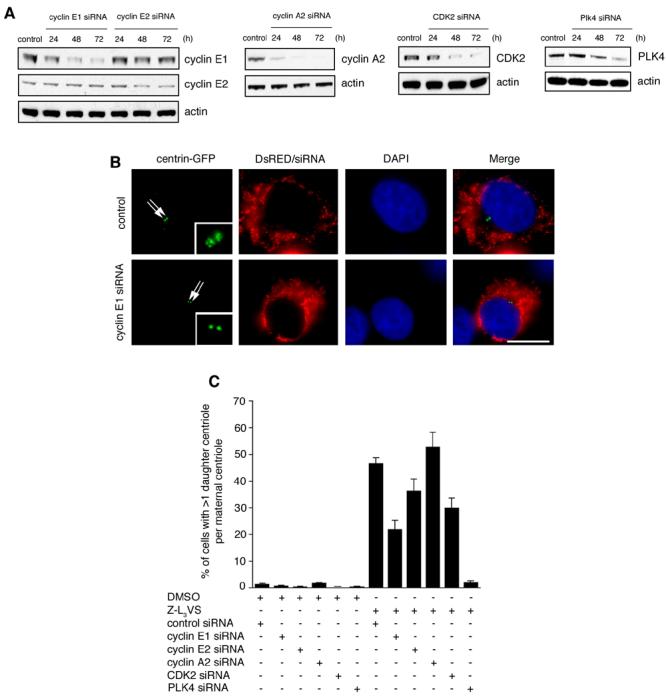
(A) Immunoblot analyses of U-2 OS/centrin-GFP cells transfected with either control siRNA duplexes (control) or siRNAs targeting cyclin E1, cyclin E2, cyclin A2, CDK2 or PLK4 for the indicated time intervals. Immunoblots for actin are shown to demonstrate loading of equal amounts of protein.
(B) Fluorescence microscopic analysis of U-2 OS/centrin-GFP cells transfected with control siRNA duplexes (top panels) or siRNAs targeting cyclin E1 (bottom panels) following treatment with 1 μM Z-L3VS for 48 h. Cells were co-transfected with DsRED fluorescent protein as transfection marker. Nuclei stained with DAPI. Scale bar indicates 10 μm.
(C) Quantification of U-2 OS/centrin-GFP cells transfected with the indicated siRNAs followed by either control treatment with 0.1% DMSO or 1 μM Z-L3VS for 48 h. Each bar represents mean and standard error of at least three independent experiments with a minimum of 100 cells counted per experiment.
Importantly, PLK4 has recently not only been implicated in centriole overduplication (Bettencourt-Dias et al., 2005; Habedanck et al., 2005) but has furthermore been shown to trigger a deposition of centriole precursor material in a rosette-like arrangement around maternal centrioles (Habedanck et al., 2005). Since the aberrant centriole configuration observed here in Z-L3VS-treated cells was highly reminiscent of the rosette-like pattern of centriolar precursor material in cells overexpressing PLK4, we tested whether depletion of PLK4 interferes with the induction of this phenotype. We found that siRNA duplexes targeting PLK4 abolished the abnormal centriole formation induced by Z-L3VS (2%; p≤0.0001; Fig. 3C).
Taken together, these results indicate that both cyclin E/CDK2 and PLK4 are required for the formation of multiple daughter centrioles at maternal templates.
Overexpression of cyclin E/CDK2 and PLK4 stimulates the concurrent formation of multiple daughter centrioles at single maternal centrioles
We asked whether ectopic expression of cyclin E/CDK2 and/or PLK4 could recapitulate a centriole phenotype with more than one daughter per maternal centriole in the absence of a proteasome inhibitor. Transient transfection of U-2 OS/centrin-GFP cells with cyclin E/CDK2 did not lead to a significant increase in the percentage of cells with excessive daughter centriole formation at maternal templates (Fig. 4A), although in line with previous experiments (Duensing et al., 2004), a 2.2-fold increase in cells with more than four centrioles arranged in a random fashion was detected (data not shown). Overexpression of PLK4, however, caused a significant 17.9-fold increase of abnormal centriole formation at maternal centrioles from 1% to 17.9% (p≤0.0005) in agreement with previous results (Habedanck et al., 2005). This increase was further enhanced when cyclin E/CDK2 and PLK4 were expressed simultaneously as evidenced by the 27-fold increase of excessive daughter centriole formation (27%; p≤0.0005). These results suggest that cyclin E/CDK2 activity can enhance the PLK4-mediated formation of multiple centrioles at maternal centrioles.
Figure 4. Overexpression of cyclin E/CDK2 and PLK4 stimulates the concurrent formation of multiple daughter centrioles at single maternal centrioles.
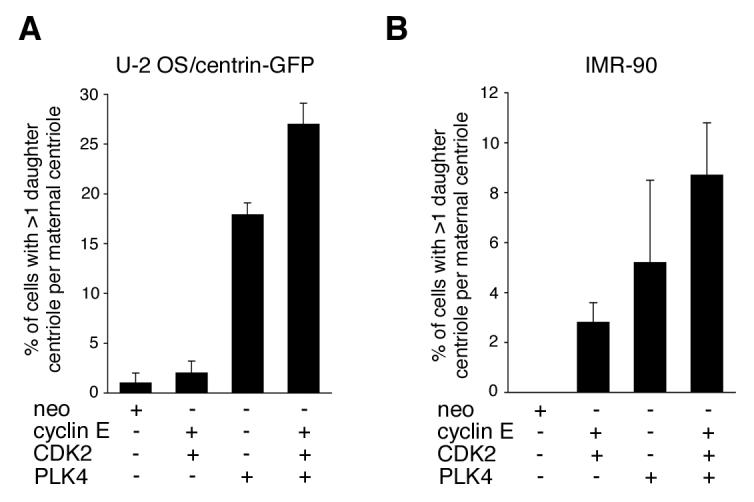
(A) Quantification of U-2 OS/centrin-GFP cells with more than one daughter centriole per maternal centriole at 48 h after transfection with empty vector (neo), cyclin E, CDK2 and PLK4 either alone or in combination. Each bar represents mean and standard error of at least three independent experiments with a minimum of 100 cells counted per experiment.
(B) Quantification of IMR-90 normal human fibroblasts with more than one daughter centriole per maternal centriole at 48 h after transfection with empty vector (neo), cyclin E, CDK2 and PLK4 either alone or in combination. Centrioles were visualized by immunofluorescence microscopy for centrin. Each bar represents mean and standard error of at least three independent experiments with a minimum of 50 cells counted per experiment.
IMR-90 normal human fibroblasts did not show aberrant daughter centriole formation following treatment with Z-L3VS. Nonetheless, ectopic expression of PLK4 either alone or in combination with cyclin E/CDK2 caused a strikingly similar phenotype after immunofluorescence staining for centrin with centrioles arranged in a “flower”-like pattern. Overexpression of cyclin E/CDK2 resulted in an increase of cells with abnormal daughter centriole formation from 0% in controls to 2.8% (p≤0.05; Fig. 4B). Cells transfected with PLK4 alone showed abnormal centriole formation in 5.2% of cells, whereas co-expression of cyclin E/CDK2 and PLK4 led to 8.7% of IMR-90 cells displaying more than one daughter centriole per maternal template (p≤0.01).
Taken together, these results show that overexpression of PLK4 can stimulate the formation of multiple daughter centrioles at each maternal centriole (Habedanck et al., 2005) and, in addition, they show that cyclin E/CDK2 enhances this process. The cooperative effects between cyclin E/CDK2 complexes and PLK4 appear to be particularly important for aberrant daughter centriole formation in non-transformed cells.
Supernumerary daughter centrioles contribute to mitotic spindle pole formation
To determine whether supernumerary daughter centrioles are able to mature and to become functional, we assessed mitotic spindle pole abnormalities in U-2 OS/centrin-GFP cells treated with 1 μM Z-L3VS in comparison to cells treated with 0.1% DMSO (Fig. 5A). Cells were treated for 24 h, washed thoroughly and released into normal media for an additional 24 h. A significant decrease in normal bipolar mitotic spindle formation was detected from 80.5% in DMSO-treated controls to 20% in Z-L3VS-treated populations (p≤0.005; Fig. 5B). At the same time, a significant increase of multipolar mitoses from 5.6% in controls to 30.7% in Z-L3VS-treated cells was detected (p≤0.05). Even more significant was the increase of pseudobipolar mitoses with multiple centrioles at each spindle pole of an apparently bipolar spindle (Brinkley, 2001). Such cell division abnormalities increased from 13.1% in controls to 48.7% in Z-L3VS-treated cells (p≤0.005). These findings suggest that supernumerary centrioles that are induced by Z-L3VS can become functional and contribute to mitotic spindle pole formation.
Figure 5. Aberrantly synthesized daughter centrioles can function as mitotic spindle poles.

(A) Fluorescence microscopic analysis of mitotic U-2 OS/centrin-GFP cells. A normal bipolar metaphase is shown in comparison to a pseudobipolar metaphase cell and a multipolar (tripolar) anaphase cell. More than two images are merged for middle and right panels to show all centrioles. Chromosomes stained with DAPI.
(B) Quantification of mitotic abnormalities in U-2 OS/centrin-GFP populations after treatment with 0.1% DMSO or 1 μM Z-L3VS for 24 h and incubation in normal media for an additional 24 h. Only dividing cells were evaluated. Each bar represents mean and standard error of three independent experiments with at least 100 mitotic cells counted per experiment.
HPV-16 E7-induced centriole overduplication involves concurrent formation of more than one daughter at maternal centrioles
The HPV-16 E7 oncoprotein has been shown to rapidly stimulate centriole overduplication in a CDK2-dependent manner (Duensing et al., 2006b; Duensing et al., 2004). We therefore determined whether HPV-16 E7-induced centriole overduplication involves the formation of more than one daughter centriole per maternal centriole.
U-2 OS/centrin-GFP cells transiently transfected with HPV-16 E7 were not previously reported to form flower- or rosette-like daughter centrioles at maternal centrioles, except that immature daughters were occasionally seen to surround mature centrioles (Guarguaglini et al., 2005). We performed a detailed analysis of HPV-16 E7-expressing U-2 OS/centrin-GFP cells in comparison to controls and determined the formation of more than one daughter centriole at single maternal centrioles (Fig. 6). At 24 h after transfection, a significant 2.9-fold increase of cells with abnormal daughter centriole formation at maternal centrioles (mostly two) was detected from 1.1% in empty vector controls to 3.2% in HPV-16 E7-expressing cells (p≤0.05; Fig. 6A,B). At 48 h after transfection, the difference in cells with abnormal daughter centriole formation at maternal centrioles was 2.2-fold increased in HPV-16 E7-expressing cells (1.7%) when compared to controls (0.3%; p≤0.05). At the same time point, cells with a random arrangement of supernumerary centrioles was 2.2-fold increased from 3.6% in controls to 8% in HPV-16 E7-expressing cells (p≤0.0001).
Figure 6. HPV-16 E7 rapidly stimulates the concurrent formation of more than one daughter centriole at maternal templates.
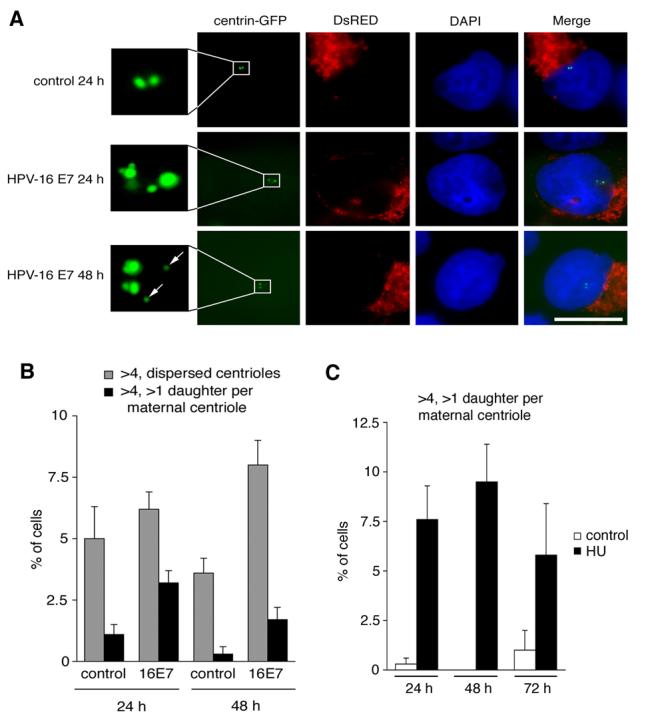
(A) Fluorescence microscopic analysis of U-2 OS/centrin-GFP cells transiently transfected with either empty vector (control) or HPV-16 E7 after 24 h or 48 h. Note the formation of two daughters at a single maternal centriole after 24 h in a HPV-16 E7-transfected cell (middle panels, insert). After 48 h, this pattern was less frequent and the majority of cells with abnormal centriole numbers showed a more dispersed arrangement (bottom panels, insert, arrows). Cells were co-transfected with DsRED fluorescent protein as transfection marker. Nuclei stained with DAPI. Scale bar indicates 10 μm.
(B) Quantification of U-2 OS/centrin-GFP cells with more than four centrioles in a random arrangement (gray bars) in comparison to cells with more than four centrioles and a concurrent formation of more than one daughter per maternal centriole (black bars). Cells were transfected with empty vector (control) or HPV-16 E7 for 24 h or 48 h. Each bar represents mean and standard error of at least three independent experiments with a minimum of 100 cells counted per experiment.
(C) Quantification of U-2 OS/centrin-GFP cells with more than four centrioles and concurrent formation of more than one daughter centriole at a maternal centriole after treatment with 1 mM HU for the indicated time intervals. dH2O-treated cells are shown as controls. Each bar represents mean and standard error of at least three independent experiments with a minimum of 100 cells counted per experiment.
These results suggest that abnormal daughter centriole synthesis at single maternal centrioles is involved in centriole overduplication induced by HPV-16 E7, in particular at early time points after transfection. Since HPV-16 E7 is not known to inhibit centriole separation (disengagement), centrioles may then be released from maternal centrioles giving rise to the more dispersed centriole arrangement typically found in HPV-16 E7-expressing cells (Fig. 6A; bottom panels).
Besides HPV-16 E7, treatment of cells with the ribonucleotide reductase hydroxyurea (HU) has been shown to stimulate a bona fide centriole overduplication (Guarguaglini et al., 2005). We therefore asked whether aberrant daughter centriole formation at single maternal centrioles is also involved in HU-induced centriole overduplication. U-2 OS/centrin-GFP cells with more than one daughter per maternal centriole were detected after 24 h (7.6%), 48 h (9.5%) and 72 h (5.8%) treatment with HU (Fig. 6C). Such cells were increased when compared to mock-treated controls (0.3%, 0% and 1%, respectively; differences at 24 h and 48 h were statistically significant, p≤0.05).
Taken together, these findings suggest that excessive daughter centriole formation at maternal templates contributes to centriole overduplication induced by both, HU or HPV-16 E7.
Discussion
Here, we present evidence for a mechanism of centriole overduplication in human cells that involves the concurrent formation of more than one daughter centriole at single maternal centrioles. We first identified this mechanism in cell populations treated with the peptide vinyl sulfone proteasome inhibitor Z-L3VS (Bogyo et al., 1997). Other proteasome inhibitors tested, for example lactacystin, did not produce this phenotype. Z-L3VS covalently inhibits the trypsin-like and chymotrypsin-like activity in cells by modifying the active site threonine of the catalytic subunit of β subunits of the proteasome (Bogyo et al., 1997). Unlike lactacystin, Z-L3VS also inhibits the peptidylglutamyl-peptide hydrolyzing (PGPH) activity of the 20S proteasome. Although speculative, such biochemical differences between the individual inhibitors may account for the observed properties of Z-L3VS.
U-2 OS cells treated with Z-L3VS contained markedly higher protein levels of cyclin E and accumulated in G2 phase of the cell division cycle when compared to other tumor cell lines. Overexpression of cyclin E and simultaneous treatment with Z-L3VS could induce the formation of more than one daughter at maternal centrioles in cells that did not respond to Z-L3VS, albeit not to the level detected in Z-L3VS-trated U-2 OS cells. These results suggest that changes in the expression of additional proteins may be important for the induction of aberrant daughter centriole formation. Importantly, overexpression of cyclin E/CDK2 together with PLK4 mimicked the centriole overduplication phenotype in the absence of Z-L3VS and even in cell types in which this inhibitor has no effect. It is therefore possible that subtle changes in PLK4 protein levels and/or changes in its subcellular localization contribute to the observed phenotype.
It has recently been reported that centrioles undergo disengagement at the end of mitosis or early in G1 phase in a process that involves separase. This mechanism restricts centriole duplication to once per cell cycle (Tsou & Stearns, 2006b). Our finding that the formation of “centriole flowers” occurred predominantly in cells that contained two large centrin-GFP dots suggests that maternal centrioles were disengaged and therefore in a duplication-competent state. However, we provide evidence that maternal centrioles are not limited to nucleate only one daughter centriole. Moreover, a simultaneous growth of two or more progeny is not only found in Z-L3VS-treated cells but is in fact involved in centriole overduplication triggered by an oncogenic stimulus, the HPV-16 E7 oncoprotein.
The surprisingly rapid induction of aberrant centriole numbers by the HPV-16 E7 oncoprotein has been difficult to reconcile with the idea that centriole overduplication arises from successive rounds of daughter centriole reproduction. By showing that HPV-16 E7 can trigger the concurrent formation of more than one daughter centriole at maternal centrioles, we provide an explanation of how multiple daughter centrioles can be generated within a single S phase. The latter notion represents an important difference to a previous study showing the formation of two daughter centrioles adjacent to a single maternal centriole in Drosophila wing disc cells undergoing repeated rounds of S phase (Vidwans et al., 2003). Additional support for the notion that mammalian cells have the potential to generate multiple centrioles adjacent to pre-existing centrioles stems from early studies on basal body formation in ciliated epithelia (Hagiwara et al., 2004). In such cells, most basal bodies form through acentriolar pathways, but flower-like structures similar to those described here have also been observed. (Anderson & Brenner, 1971; Brinkley et al., 1981; Dirksen, 1971).
In conclusion, results shown here provide evidence for a concurrent formation of more than one daughter centriole at single maternal centrioles. These aberrantly synthesized daughter centrioles are functional and can contribute to mitotic spindle pole formation. Our results imply that the normal limitation to one and only one daughter per mother centriole and cell division cycle is not due to inherent structural constraints or limited initiation sites but rather reflects regulation by cyclin E/CDK2 complexes and PLK4. Our results help to explain the rapid centriole overduplication induced by the HPV-16 E7 oncoprotein and, most likely, also other oncogenic insults. Further experiments to elucidate how cyclin E/CDK2 and PLK4 cooperate will provide important insights into the regulation of the centriole duplication cycle.
Supplementary Material
(A) Immunoblot analysis of U-2 OS/centrin-GFP cells treated with 0.1% DMSO or 1 μM Z-L3VS (48 h) for cyclin E, cyclin A, CDK2 and PLK4. Immunoblot for actin is shown to demonstrate protein loading.
(B) Flow cytometric analysis of U-2 OS/centrin-GFP, HeLa or T98G cells treated with 0.1% DMSO or 1 μM Z-L3VS for 48 h.
(C) Immunofluorescence analysis of U-2 OS/centrin-GFP cells for cyclin B expression following 48 h treatment with 1 μM Z-L3VS. Note the absence of cyclin B in cells that contain more than one daughter centriole per maternal centriole (top panels). Arrows indicate centrioles shown in the inserts. Nuclei stained with DAPI. Scale bar indicates 10 μm.
Immunofluorescence analysis of U-2 OS/centrin-GFP cells for CEP170, a marker for mature centrioles (Guarguaglini et al., 2005). Arrows indicate centrioles shown in inserts. Note that supernumerary daughters can form at single maternal centrioles that have not fully matured (arrow in merged bottom panel). Nuclei stained with DAPI. Scale bar indicates 10 μm.
Quantification of Z-L3VS-treated HeLa or T98G cells with concurrent formation of more than one daughter centriole per maternal centriole after transfection with empty vector (control) or cyclin E. Cells were treated with 1 μM Z-L3VS at 24 h after transfection for an additional 48 h. Each bar represents mean and standard error of triple quantification of at least 50 cells of a representative experiment.
Acknowledgements
We would like to thank Michel Bornens (Institut Curie), William R. Brinkley (Baylor College of Medicine), Philip W. Hinds (Tufts University), Karl Münger (Channing Laboratory, Brigham and Women's Hospital), Jeffrey L. Salisbury (Mayo Clinic) and Jens Westendorf (Max Planck Institute for Biochemistry) for sharing important reagents and/or helpful suggestions. We are extremely grateful to Joseph Suhan (Carnegie Mellon University) for invaluable help and support with electron microscopy. We thank Huib Ovaa (Harvard Medical School) for helpful discussions. This work was supported by NIH/NCI grant R01 CA112598, the Susan G. Komen Breast Cancer Foundation and a Research Scholar Grant from the American Cancer Society (to S.D.).
References
- Anderson RG, Brenner RM. J Cell Biol. 1971;50:10–34. doi: 10.1083/jcb.50.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bettencourt-Dias M, Rodrigues-Martins A, Carpenter L, Riparbelli M, Lehmann L, Gatt MK, et al. Curr Biol. 2005;15:2199–207. doi: 10.1016/j.cub.2005.11.042. [DOI] [PubMed] [Google Scholar]
- Bogyo M, McMaster JS, Gaczynska M, Tortorella D, Goldberg AL, Ploegh H. Proc Natl Acad Sci U S A. 1997;94:6629–6634. doi: 10.1073/pnas.94.13.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornens M. Curr Opin Cell Biol. 2002;14:25–34. doi: 10.1016/s0955-0674(01)00290-3. [DOI] [PubMed] [Google Scholar]
- Brinkley BR. Trends Cell Biol. 2001;11:8–21. doi: 10.1016/s0962-8924(00)01872-9. [DOI] [PubMed] [Google Scholar]
- Brinkley BR, Cox SM, Pepper DA, Wible L, Brenner SL, Pardue RL. J Cell Biol. 1981;90:554–62. doi: 10.1083/jcb.90.3.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen ER. J Cell Biol. 1971;51:286–302. doi: 10.1083/jcb.51.1.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing A, Liu Y, Spardy N, Bartoli K, Tseng M, Kwon JA, Teng X, Duensing S. Oncogene. 2006a;26:215–223. doi: 10.1038/sj.onc.1209782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing A, Liu Y, Tseng M, Malumbres M, Barbacid M, Duensing S. Oncogene. 2006b;25:2943–2949. doi: 10.1038/sj.onc.1209310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing S. Cell Biol Int. 2005;29:352–359. doi: 10.1016/j.cellbi.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Duensing S, Duensing A, Lee DC, Edwards KM, Piboonniyom S, Manuel E, et al. Oncogene. 2004;23:8206–8215. doi: 10.1038/sj.onc.1208012. [DOI] [PubMed] [Google Scholar]
- Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S, Gonzalez S, et al. Proc Natl Acad Sci U S A. 2000;97:10002–10007. doi: 10.1073/pnas.170093297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Fabunmi RP, Wigley WC, Thomas PJ, DeMartino GN. J Biol Chem. 2000;275:409–13. doi: 10.1074/jbc.275.1.409. [DOI] [PubMed] [Google Scholar]
- Freed E, Lacey KR, Huie P, Lyapina SA, Deshaies RJ, Stearns T, et al. Genes Dev. 1999;13:2242–2257. doi: 10.1101/gad.13.17.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gstaiger M, Marti A, Krek W. Exp Cell Res. 1999;247:554–562. doi: 10.1006/excr.1999.4386. [DOI] [PubMed] [Google Scholar]
- Guarguaglini G, Duncan PI, Stierhof YD, Holmstrom T, Duensing S, Nigg EA. Mol Biol Cell. 2005;16:1095–1107. doi: 10.1091/mbc.E04-10-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. Nat Cell Biol. 2005;7:1140–1146. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- Hagiwara H, Ohwada N, Takata K. Int Rev Cytol. 2004;234:101–141. doi: 10.1016/S0074-7696(04)34003-9. [DOI] [PubMed] [Google Scholar]
- Hinchcliffe EH, Li C, Thompson EA, Maller JL, Sluder G. Science. 1999;283:851–854. doi: 10.1126/science.283.5403.851. [DOI] [PubMed] [Google Scholar]
- Lacey KR, Jackson PK, Stearns T. Proc Natl Acad Sci U S A. 1999;96:2817–2822. doi: 10.1073/pnas.96.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL. Proc Natl Acad Sci U S A. 1998;95:2950–2955. doi: 10.1073/pnas.95.6.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LG, Demers GW, Galloway DA. J Virol. 1998;72:975–985. doi: 10.1128/jvi.72.2.975-985.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y, Hayashi K, Nishida E. Curr Biol. 1999;9:429–432. doi: 10.1016/s0960-9822(99)80191-2. [DOI] [PubMed] [Google Scholar]
- McIntyre MC, Ruesch MN, Laimins LA. Virology. 1996;215:73–82. doi: 10.1006/viro.1996.0008. [DOI] [PubMed] [Google Scholar]
- Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA. Nat Cell Biol. 1999;1:88–93. doi: 10.1038/10054. [DOI] [PubMed] [Google Scholar]
- Munger K, Basile JR, Duensing S, Eichten A, Gonzalez SL, Grace M, et al. Oncogene. 2001;20:7888–7898. doi: 10.1038/sj.onc.1204860. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, et al. EMBO J. 2000;19:2069–2081. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA. Nature Rev. Cancer. 2002;2:1–11. doi: 10.1038/nrc924. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Centrosomes in Development and Disease. Wiley-VCH; Weinheim, Germany: 2004. [Google Scholar]
- Pihan GA, Purohit A, Wallace J, Knecht H, Woda B, Quesenberry P, Doxsey SJ. Cancer Res. 1998;58:3974–3985. [PubMed] [Google Scholar]
- Pines J, Hunter T. J Cell Biol. 1991;115:1–17. doi: 10.1083/jcb.115.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury JL, Whitehead CM, Lingle WL, Barrett SL. Biol Cell. 1999;91:451–460. [PubMed] [Google Scholar]
- Sluder G. In: Centrosomes in Development and Disease. Nigg EA, editor. Wiley-VCH; Weinheim, Germany: 2004. pp. 167–189. [Google Scholar]
- Tsou MF, Stearns T. Curr Opin Cell Biol. 2006a;18:74–78. doi: 10.1016/j.ceb.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Tsou MF, Stearns T. Nature. 2006b;442:947–951. doi: 10.1038/nature04985. [DOI] [PubMed] [Google Scholar]
- Vidwans SJ, Wong ML, O'Farrell PH. J. Cell Sci. 2003;116:137–143. doi: 10.1242/jcs.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigley WC, Fabunmi RP, Lee MG, Marino CR, Muallem S, DeMartino GN, et al. J Cell Biol. 1999;145:481–490. doi: 10.1083/jcb.145.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik EJ, Glover DM, Hays TS. Curr Biol. 2000;10:1131–1134. doi: 10.1016/s0960-9822(00)00703-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Immunoblot analysis of U-2 OS/centrin-GFP cells treated with 0.1% DMSO or 1 μM Z-L3VS (48 h) for cyclin E, cyclin A, CDK2 and PLK4. Immunoblot for actin is shown to demonstrate protein loading.
(B) Flow cytometric analysis of U-2 OS/centrin-GFP, HeLa or T98G cells treated with 0.1% DMSO or 1 μM Z-L3VS for 48 h.
(C) Immunofluorescence analysis of U-2 OS/centrin-GFP cells for cyclin B expression following 48 h treatment with 1 μM Z-L3VS. Note the absence of cyclin B in cells that contain more than one daughter centriole per maternal centriole (top panels). Arrows indicate centrioles shown in the inserts. Nuclei stained with DAPI. Scale bar indicates 10 μm.
Immunofluorescence analysis of U-2 OS/centrin-GFP cells for CEP170, a marker for mature centrioles (Guarguaglini et al., 2005). Arrows indicate centrioles shown in inserts. Note that supernumerary daughters can form at single maternal centrioles that have not fully matured (arrow in merged bottom panel). Nuclei stained with DAPI. Scale bar indicates 10 μm.
Quantification of Z-L3VS-treated HeLa or T98G cells with concurrent formation of more than one daughter centriole per maternal centriole after transfection with empty vector (control) or cyclin E. Cells were treated with 1 μM Z-L3VS at 24 h after transfection for an additional 48 h. Each bar represents mean and standard error of triple quantification of at least 50 cells of a representative experiment.