

Abstract
Oxidative stress, an imbalance between oxidants and antioxidants, contributes to the pathogenesis of traumatic brain injury (TBI). Oxidative neurodegeneration is a key mediator of exacerbated morphological responses and deficits in behavioral recoveries. The present study assessed early hippocampal sequential imbalance to possibly enhance antioxidant therapy. Young adult male Sprague-Dawley rats were subjected to a unilateral moderate cortical contusion. At various times post TBI, animals were killed and the hippocampus analyzed for antioxidants (GSH, GSSG, glutathione peroxidase, glutathione reductase, glutathione-S-transferase, glucose-6-phosphate dehydrogenase, superoxide dismutase and catalase), and oxidants (acrolein, 4-hydroxynonenal [4-HNE], protein carbonyl [PC] and 3-nitrotyrosine. Synaptic markers (synapsin-I, post synaptic density-95 [PSD-95], synapse associated protein-97 [SAP-97], growth associated protein-43 were also analyzed. All values were compared to sham operated animals. Significant time-dependent changes in antioxidants were observed as early as 3 h post trauma and paralleled increases in oxidants (4-HNE, acrolein and PC) with peak values obtained at 24–48 h. Time dependent changes in synaptic proteins (synapsin-I, PSD-95 and SAP-97) occurred well after levels of oxidants peaked. These results indicate that depletion of antioxidant systems following trauma could adversely affect synaptic function and plasticity. Early onset of oxidative stress suggests that the initial therapeutic window following TBI appears to be relatively short and it may be necessary to stagger selective types of anti-oxidant therapy to target specific oxidative components.
Keywords: Hippocampus, oxidative stress, synaptic proteins, time course, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is associated with costly health problems, and high mortality and morbidity in previously healthy populations. TBI involves primary and secondary cascades resulting in delayed neuronal dysfunction and death. As part of the secondary cascade, there are large transient increases in excitatory neurotransmitter efflux [1] resulting in excitotoxicity, ionic imbalance, ATP depletion, proteolysis and oxidative stress [2]. The brain is highly sensitive to stress-induced neurodegeneration due to its high content of poly unsaturated fatty acids (PUFA) which is particularly vulnerable to free radical attacks and lipid peroxidation (LPO). Aldehydic products of LPO, 4-hydroxynonenal (4-HNE) and acrolein have recently become of interest in the various neurological ailments [3], because of their involvement in oxidative stress.
A close relationship exists between the degree of oxidative stress and the pathogenesis of TBI [4]. Enhanced production of reactive oxygen/nitrogen species (ROS/RNS) cause oxidative/nitrosative stress [5, 6] leading to damage in lipids, proteins and nucleic acids [7, 8]. Oxidative stress-related cascades resulting from TBI have been implicated in cytoskeletal damage, mitochondrial dysfunction [9] and altered signal transduction [10, 11].
Antioxidants (enzymatic and/or non-enzymatic) can protect the brain against oxidative damage [12] in several ways including: a) removal of ROS/RNS, b) inhibition of ROS/RNS formation, and c) binding metal ions needed for catalysis of ROS generation. Glutathione peroxidase (GPx) and glutathione reductase (GR) are well known intracellular antioxidant enzymes. GPx convert peroxides into nontoxic forms, often with the concomitant oxidation of reduced glutathione (GSH) into the oxidized form (GSSG) and GR recycles GSSG to GSH. Other enzymes and pathways are also involved in the management of cellular defense against oxidative/nitrosative stress. Glutathione-s-transferase (GST) detoxifies deleterious substances and glucose-6-phosphate dehydrogenase (G-6PD) provides electron donors for antioxidant defense system. Enzymes that remove both superoxide and hydrogen peroxide (H2O2) protect the cells from oxidative stress, but when the production of O2−. and H2O2 crosses the normal threshold, the system becomes compromised. Catalase and glutathione peroxidase (GPx), acting in concert with superoxide dismutase (SOD), constitute the major defense enzymes against superoxide radicals [13, 14]. Analysis of these antioxidants, products of LPO and protein damage, may a yield snapshot of oxidative stress.
Oxidative/nitrosative stressed neurons, injured by free radicals, might be present physically but non-functional. As a consequence of oxidative stress both the function and transport of mitochondrial to synaptic regions is impaired, decrease synaptic function [15, 16], which results in neurodegeneration after brain injury [17, 18]. A recent study reported that oxidative stress alters the function of PSD-95 by decreasing a voltage-gated potassium channel that is closely linked to the PSD [19], suggesting that synapse loss and replacement, known to occur following TBI [20], could be modulated by the levels of oxidative stress.
Cortical contusion models of TBI result in cortical neuronal loss and altered function, growth, and plasticity of surviving neurons in non-cortical regions, many of which are involved in memory [21]. A persistent problem associated with TBI is impaired declarative memory, a hippocampal dependant cognitive task. The hippocampus is highly susceptible to TBI due to intrinsic connectivity and/or the presence of high levels of excitatory amino acid (glutamate) receptors [22, 23]. We performed the current study to investigate possible time-dependent alterations in oxidative stress and whether or not they associate with synaptic marker proteins in hippocampus. Oxidative/nitrosative stresses modify proteins via carbonylation, nitration and acrolein and 4-HNE binding. Synaptic proteins also might be affected through these modifications. Our results provide a better understanding of the early oxidative cascades following TBI and have important implications for antioxidant treatment.
Materials and Methods
Chemicals
Mouse monoclonal anti β-actin, goat polyclonal anti PSD-95, synapsin-I and GAP-43 antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse monoclonal anti synapse associated protein-97 (SAP-97) was purchased from Stressgen (Assay Designs, Inc. USA). Alkaline phosphatase conjugated anti-mouse, anti-goat secondary antibodies, and all other chemicals and reagents used were purchased from Sigma (St Louis, MO, USA). Electrophoresis materials and chemicals were purchased from Bio-Rad (Hercules, CA, USA) unless stated otherwise.
Animals and surgical procedures
Adult male Sprague-Dawley rats (250–275g) (Harlan Laboratories, Indianapolis, IN) were used in this study. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Kentucky. Animals were housed 2/cage on a 12 h light/dark cycle and provided free access to pellet diet and water ad libitum. A total of 85 rats were subjected to a unilateral cortical impact utilizing an electronic controlled pneumatic impact device (ECPI) (TBI0310, PSI, Fairfax Station, VA) as previously described [2]. Briefly, each animal was anesthetized with 2% isofluorane and placed in a Kopf stereotaxic frame (Tujunga, CA) with the incisor bar set at −5. Body temperature of each rat were monitored and maintained at 36 °C with a heating pad. Following a midline incision and retraction of the skin, a 6-mm-diameter craniotomy was made approximately midway between bregma and lamda with a Michele hand trephine (Miltex, NY). The skull disk was removed without disturbing the dura. The exposed brain was injured using the ECPI. The impact rod had a 5-mm-diameter beveled tip that was used to compress the cortex to a depth of 2.0 mm at 3.5 m/s. Following injury, Surgicel (Johnson & Johnson, New Brunswick, NJ) was placed over the injury site, and the skull disk replaced and sealed with a thin layer of dental acrylic. The animals were divided into groups (n=6/group; 54 animals) and allowed to survive for (1, 3, 6, 12, 24, 48, 72 and 96 h) post TBI for biochemical investigations. Rests of the animals were used for histological analyses of Fluoro-Jade B (FJB). Sham animals received the craniotomy without TBI, and killed at 12 h.
Tissue processing
For enzymatic and oxidative stress assays, animals were euthanized with CO2, rapidly killed and the brain removed. Hippocampi were isolated from the ipsilateral and the corresponding contralateral side of injury. At the time of dissection, none of the samples were compromised by the presence of surface blood products. None of the hippocampal samples used in these studies had obvious edema or hematomas as a result of the trauma. These samples were immediately frozen in liquid nitrogen, and stored at −80°C until used for analysis. Tissue samples were lysed using an ultrasonic cell disruptor (Microson Farmingdale, NY) on ice in 2.0 ml 0.1M, PBS (pH 7.4) containing 10 mM HEPES, 2.0 mM EDTA, 2.0 mM EGTA, 0.6 mM MgSO4 4.6 mM KCl, pepstatin A, leupeptin, aprotinin, and phenylmethylsulfonyl fluoride (PMSF). Samples were centrifuged at 1000g for 10 min/4°C to remove cell debris and the collected supernatant was centrifuged at 15,000g for 10 min/4°C. Post mitochondrial supernatants (PMS) were used for enzymatic and non-enzymatic analyses. Biochemical assays were completed in 96-well plates (microplate reader) as well as in cuvette (Spectronic™ GENESYS™ 2 and 5 Spectrophotometer). Total protein concentrations were determined using the Pierce BCA method (Sigma, St. Louis, MO).
Estimation of GSH
Determination of GSH was performed as previously described [8]. The reaction mixture containing 0.1 M sodium phosphate buffer (pH 8.0), 5.0 mM EDTA, 10 µl o-phthalaldehyde (1.0 mg/ml), and 10 µl of sample. After incubation for 15 min at room temperature, fluorescence at emission 420 nm was recorded by excitation at 350 nm.
Estimation of GSSG
The estimation of GSSG was performed as previously described [8]. The samples were incubated first with 0.04 M N-ethylmaleimide (NEM) for 30 min to interact with GSH present in sample. The reaction mixture containing 0.1 N NaOH, 5.0 mM EDTA, 10 µl o-phthalaldehyde (1.0 mg/ml), and 10 µl of sample. After incubation for 15 min at room temperature, fluorescence at emission 420 nm was recorded by excitation at 350 nm.
Estimation of GPx activity
GPx (EC 1.11.1.9) activity was measured according to the procedure as previously described [7, 8, 24] with some modifications. The reaction mixture consisted of 0.05 M phosphate buffer (pH 7.0), 1.0 mM EDTA, 1.0 mM sodium azide, 1.4 U of 0.1 ml GR, 1.0 mM GSH, 0.2 mM NADPH, 0.25 mM H2O2, and 0.03 ml of PMS in a final volume of 0.3 ml. The disappearance of NADPH at 340 nm was recorded at room temperature. The enzyme activity was calculated as nmol NADPH oxidized/min/mg protein, using a molar extinction coefficient of 6.22 × 103 M−1 cm−1.
Estimation of GR activity
GR (EC 1.6.4.2) activity was assayed as previously described [8, 24] with some modifications. The assay system consisted of 0.1 M phosphate buffer (pH 7.6), 0.1 mM NADPH, 0.5 mM EDTA, 1.0 mM GSSG, and 0.03 ml PMS in a total volume of 0.3 ml. The enzyme activity was quantitated at room temperature by measuring the disappearance of NADPH at 340 nm and calculated as nmol NADPH oxidized/min/mg protein, using a molar extinction coefficient of 6.22 × 103 M−1 cm−1.
Estimation of GST activity
GST (EC 2.5.1.18) activity was measured by the method as previously described [7, 8, 24] with some modifications. The reaction mixture consisted of 0.1 M phosphate buffer (pH 6.5), 1.0 mM reduced GSH, 1.0 mM CDNB, and 0.03 ml PMS in a final volume of 0.3 ml. The changes in absorbance were recorded at 340 nm, and the enzyme activity calculated as nmol 1-chloro-2,4-dinitrobenzene conjugate formed/min/mg protein, using a molar extinction coefficient of 9.6 × 103 M−1 cm−1.
Estimation of G-6PD activity
G-6-PD (EC 1.1.1.49) activity was assayed by the method of [25]. Briefly, the assay mixture consisted of 0.05M tris-HCl buffer (pH 7.6), 0.1 mM NADP, 0.8 mM glucose-6-phosphate, 8 mM MgCl2 and 0.03 ml of PMS in total volume of 1.0 ml. Changes in absorbance were recorded at 340 nm and enzyme activity was calculated as nmol NADP reduced/min/mg protein, using a molar extinction coefficient of 6.22 × 103 M−1 cm−1.
Estimation of SOD activity
SOD (EC 1.15.1.1) activity was measured according to the method as previously described [7, 24] with some modifications. Briefly, the assay mixture consisted of 0.05 M glycine buffer (pH 10.4), 6.0 µl (−) epinephrine (20 mg/ml distilled water) and 0.03 ml PMS in a total volume of 0.3 ml. 1.0 mM triethylenetetramine were used as Cu/Zn-SOD inhibitor to measure manganese (Mn)-SOD activity. The absorbance was monitored for 5 min at 480 nm and activity calculated using a molar extinction coefficient of 4.02 × 103 M−1 cm−1 expressed as nmol of epinephine protected from oxidation/min/mg protein.
Estimation of catalase activity
Catalase (CAT, EC 1.11.1.6) activity was assayed by the method as previously described [7, 24]. Briefly, the assay mixture consisted of 0.05 M phosphate buffer (pH 7.0), 0.019 M H2O2, and 0.03 ml PMS in a total volume of 0.3 ml. Changes in absorbance were recorded at 240 nm. CAT activity was calculated in terms of nmol H2O2 consumed/min/mg protein, using a molar extinction coefficient of 43.6 M−1 cm−1.
Estimation of protein carbonyl (an index of protein carbonylation)
Protein carbonyls (PC), markers of protein oxidation, were assessed by following the standard protocol described previously [8]. Samples (5 µl) (normalized to 4 mg/ml), 5 µl of 12% sodium dodecyl sulfate (SDS), and 10 µl of 10 times diluted 2,4-dinitrophenyl hydrazine from 200 mM stock were incubated at room temperature for 20 min. Samples were neutralized with 7.5 µl neutralization solution (2 M Tris in 30% glycerol). The resulting solution was loaded into each well on nitrocellulose membrane under vacuum using a slot-blot apparatus. The membrane was blocked in blocking buffer (3% bovine serum albumin) in PBS/Tween for 1 h and incubated with a 1:100 dilution of anti-DNP polyclonal antibody in PBS/Tween for 1 h. The membrane was washed three times in PBS/Tween and incubated for 1 h with an anti-rabbit IgG alkaline phosphatase secondary antibody diluted in PBS/Tween in a 1:8000 ratio. The membrane was washed three times in PBS/Tween for 5 min and developed in Sigma Fast tablets (BCIP/NBT substrate). Blots were dried, scanned with Adobe PhotoShop, and quantified with Scion Image (PC version of Macintosh-compatible NIH Image). No nonspecific binding of antibody to the membrane was observed.
Estimation of 3-nitrotyrosine (an index of protein nitration)
Samples (5 µl) (normalized to 4 mg/ml), 5 µl of 12% SDS, and 5 µl of modified Laemmli buffer containing 0.125 M Tris base, pH 6.8, 4% (v/v) SDS, and 20% (v/v) glycerol were incubated for 20 min at room temperature. Each sample (250 ng) was loaded into a well on a nitrocellulose membrane in a slot-blot apparatus under vacuum. The membrane was blocked in blocking buffer (3% bovine serum albumin) in PBS/Tween for 1 h and incubated with a 1:2000 dilution of anti-3-nitrotyrosine (3-NT) polyclonal antibody in PBS/Tween for 1 h 30 min. The membrane was washed in PBS/Tween for 5 min three times after incubation. The membrane was incubated for 1 h, after washing, with an anti-rabbit IgG alkaline phosphatase secondary antibody diluted in PBS/Tween in 1:8000 ratio. The membrane was washed three times in PBS/Tween for 5 min and developed in Sigma Fast tablets. Blots were dried, scanned with Adobe PhotoShop, and quantified with Scion Image as above. No nonspecific binding of antibody to the membrane was observed.
Estimation of acrolein and 4-HNE (an index of lipid peroxidation)
Samples (5 µl) (normalized to 4 mg/ml), 5 µl of 12% SDS, and 5 µl of modified Laemmli buffer containing 0.125 M Tris base, pH 6.8, 4% (v/v) SDS, and 20% (v/v) glycerol were incubated for 20 min at room temperature. Samples (250 ng) were loaded into each well on a nitrocellulose membrane in a slot-blot apparatus under vacuum. The membrane was blocked in blocking buffer (3% bovine serum albumin) in PBS/Tween for 1 h and incubated with a 1:5000 dilution of anti-HNE polyclonal antibody in PBS/Tween for 1 h and 30 min. The membrane was washed in PBS/Tween for 5 min three times after incubation. The membrane was incubated for 1 h, after washing, with an anti-rabbit IgG alkaline phosphatase secondary antibody diluted in PBS/Tween in a 1:8000. The membrane was washed three times in PBS/Tween for 5 min and developed in Sigma Fast tablets. Blots were dried, scanned with Adob PhotoShop, and quantified with Scion Image as above. A faint background staining due to the antibody alone was observed, however each sample had a control and, this minor effect was well controlled.
Western-blots for synaptic marker proteins
Synaptic marker proteins were studied by Western-blot method as previously described [8] with some modifications. The above mentioned homogenate samples (0.2 ml) were diluted in 0.2 ml of lysis buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1% Triton X-100, 10% glycerol including above mentioned protease inhibitors) and centrifuged at 13,000 × g for 10 min to remove tissue debris. Supernatants were used for assay of synapsin-I, SAP97, PSD95, and GAP-43. Protein (50µg) was loaded with the appropriate marker (Bio-Rad) on gradient gel (4–20% Tris-HCl), followed by transfer to polyvinylidene fluoride membrane using a semi-dry transfer system (Bio-Rad) in transfer buffer (25 mM Tris, 150 mM glycine, 20% MeOH) 2 h at 15 volt. The membrane was blocked with 5% milk or bovine serum albumin (BSA) in Tris/saline buffer-Tween (TBST). Primary antibodies for synapsin-I, GAP-43, PSD-95, and SAP-97 were added at a concentration of 1:1,000 and incubated overnight at 4°C. The blot was then washed thee times in TBST and incubated for 1 h with alkaline phosphatase conjugated secondary antibodies in a 1:8000 dilution. The membrane was washed three times in TBST for 5 min and developed in Sigma Fast tablets (BCIP/NBT substrate). Blots were dried, scanned with Adobe Photoshop, and quantified with Scion Image (PC version of Macintosh-compatible NIH Image).
Histological investigation
Staining for neuronal death with Fluoro-Jade B (FJB) (Histo-Chem Inc., Jefferson, AR) was performed as previously described [26]. Briefly, the rats were transcardially perfused with 4% paraformaldehyde (PF) and brains postfixed in PF for 24 h at 4°C before cryoprotecting in 20% sucrose/PBS. Cryosections (50 µm) throughout the entire extent of the hippocampal formation, were mounted onto slides and processed for FJB and concurrent 4′,6-diamidino-2-phenylindole (DAPI) (Sigma, St. Louis, MO) histochemistry. Sections were incubated in 1% NaOH in 80% ethanol, hydrated in graded ethanol and distilled water. These sections were then incubated in 0.06% potassium permanganate solution, rinsed in distilled water, and incubated in a 0.0004% solution of FJB and 0.0004% DAPI. The tissue was then rinsed in distilled water (three times for 2 min), air-dried, and placed on a slide warmer until fully dry (5–10 min). The dry slides were cleared in CitriSolv (two times for 5 min) and coverslipped with DPX (Fluka, Sigma, St. Louis).
The total number of FJB-positive neurons was determined blindly with respect to time post injury. A minimum of 12 equally spaced sections were evaluated for each brain as previously described (Anderson et al 2005). Tissue was evaluated under blue (450–490 nm) excitation light, employing a 40X objective. Stereological counting protocols were used with the aid of an image analysis system (Bioquant Nova Prime v. 6.50.10, Nashville, TN) interfaced with an Olympus BX50 microscope and Optronics VI-470 camera (Goleta, CA). The entire reference volume of individual hippocampal subregions (dentate gyrus granule cells, CA3, and CA1 pyramidal cells) was estimated with the Cavalieri method using the anatomical boundaries as previously described [26].
Statistical analysis
Time-dependent differences in enzymatic and non-enzymatic oxidative stress markers, elementary components of synapses, and differences in FJB positive neurons are reported as mean ± SD. Possible differences between group means were evaluated with a one-way ANOVA coupled with a Student Newman-Keuls post-hoc test (StatView 5.0, SAS Institute). For significant differences, alpha was set at 0.05.
Results
Time course of GSH post injury
The level of internal antioxidant tri-peptide molecule GSH was measured in both ipsilateral and contralateral hippocampus following TBI. Analysis of GSH levels demonstrated a significant time-dependant decrease ipsilateral [F (8, 45) = 14.627, p < 0.0001] to the injury (Fig. 1A). A significant decline was observed as early as at 3 h (20.7%) and continued to decrease with maximum depletion observed at 24 h (45.49%), and a moderate increase in later time points. Values observed at 96 h were still significantly below control. GSH levels in the contralateral hippocampus were not significantly changed (p > 0.1) at any time point post injury.
Fig. 1.
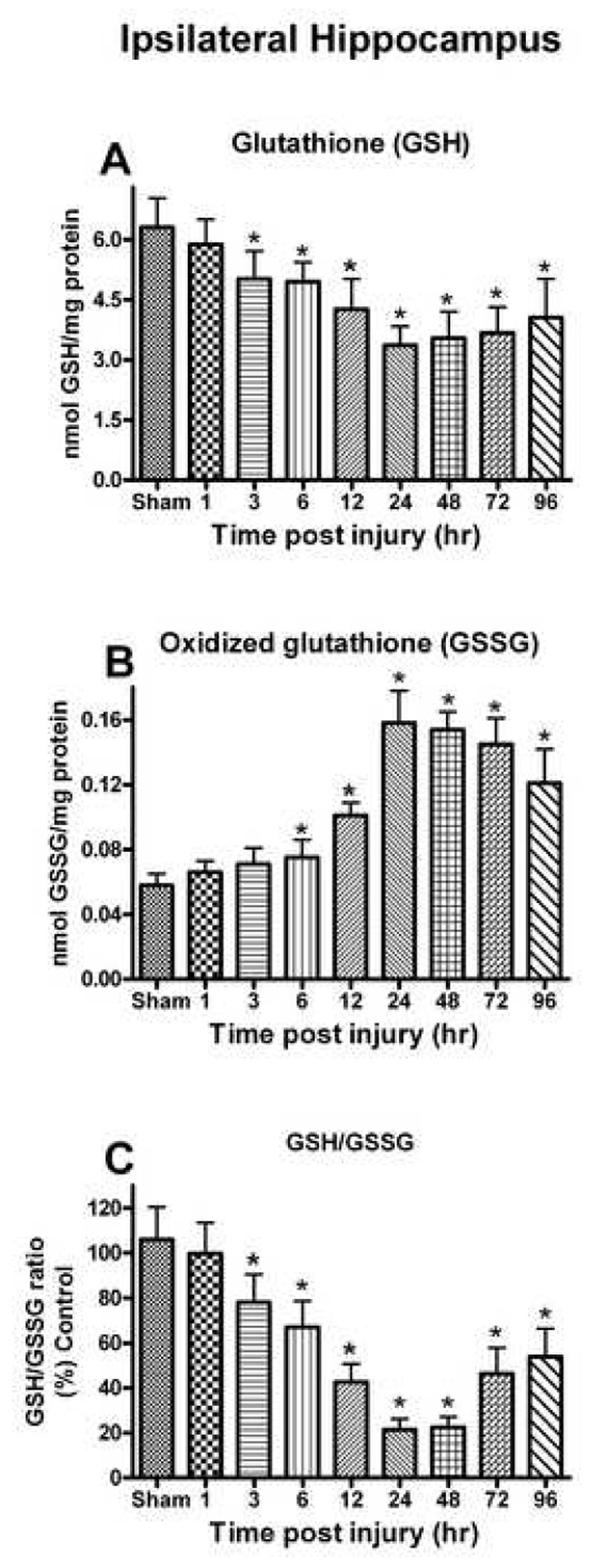
Changes in the GSH system (GSH, GSSG and GSH/GSSG) after a unilateral cortical contusion in male SD rats. (A) As early as 3h post trauma, levels of GSH were significantly decreased and depletion of GSH levels persisted for up to 96h. (B) The levels of GSSG were significantly increased at 6h and remained significantly elevated for up to 96h. (C) At the 3h post injury, levels of GSH/GSSG were significantly altered and this depletion in ratio remained significant at 96h. Each bar represents the group mean ± SD of six animals/group. *p < 0.05 versus sham.
Time course of GSSG post injury
Analysis of GSSG levels in the ipsilateral hippocampus demonstrated a significant time post-injury increase [F (8, 45) = 52.316, p < 0.0001] (Fig. 1B). GSSG levels increased significantly at 6 h (74.13%) and continued to increase with the maximum at 24–48 h (172.41-165.52%). The levels of GSSG declined at later time points, but remained significantly above control levels at 96 h. In the corresponding contralateral hippocampus, GSSG did not change significantly post injury (p > 0.1).
Time course on the GSH/GSSG ratio post injury
The GSH/GSSG ratio significantly declined in a time-dependent fashion in the ipsilateral hippocampus [F (8, 45) = 47.120, p < 0.0001] (Fig. 1C). The ratio was significantly depleted as early as 3 h (26.31%) and continued with maximum values decreased at 24–48 h (78.61-77.54%). In the corresponding contralateral hippocampus, the GSH/GSSG ratio did not significantly change (p > 0.1).
Time course of antioxidant enzymes activity post injury
To further evaluate the response of the antioxidant process following TBI, antioxidant enzyme activity was evaluated in ipsilateral and contralateral hippocampus. Only the ipsilateral hippocampus demonstrated significant changes in antioxidant enzymes compared to the sham group. GPx detoxifies peroxides, including lipid peroxide, and uses NADPH as an electron contributor. The overall analysis of GPx activity revealed a significant time-dependent decline [F(8,45) = 3.607, p < 0.0026] with the lowest levels observed at 24–48 h (30.7-29.7%) post injury. By 96 h post injury, the levels had returned to control values (Fig. 2A).
Fig. 2.
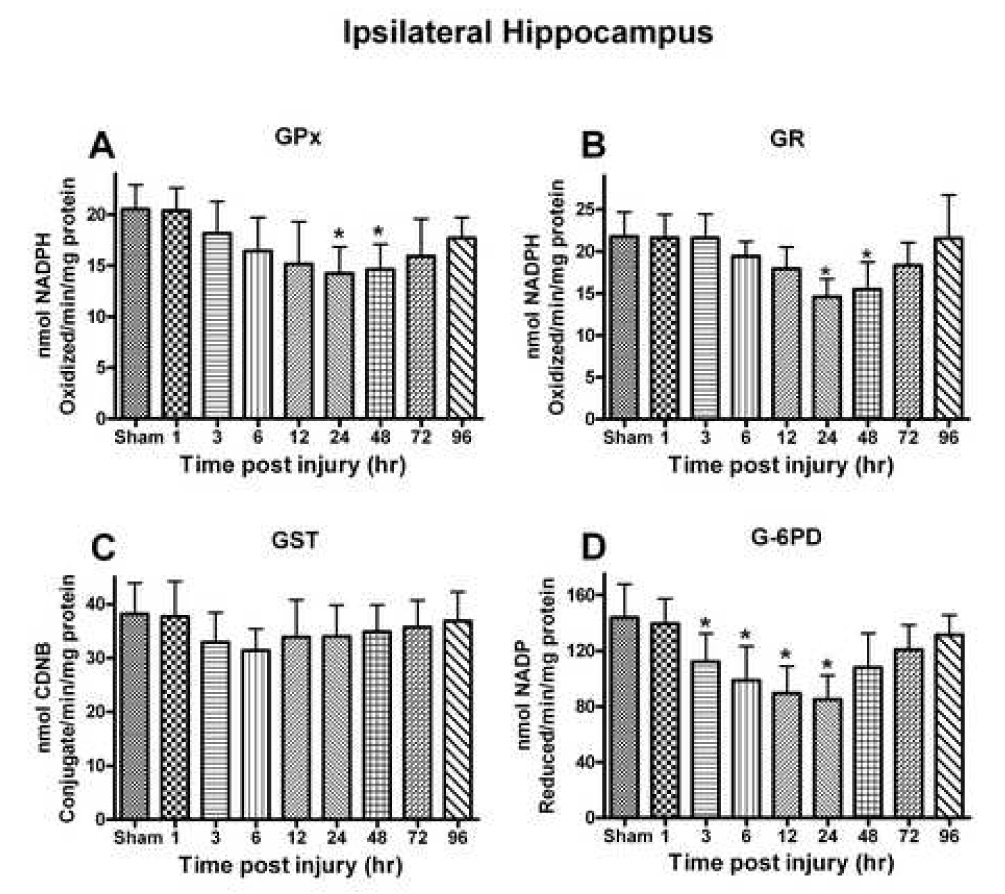
Several different antioxidant enzymes were quantitatively assessed following a moderate unilateral cortical contusion. (A) Glutathione peroxidase (GPx) activity demonstrated a time-dependent change with significant reductions observed at 24–48h post injury. (B) Glutathione reductase (GR) activity demonstrated a time-dependent decline that mirrored that observed for GPx. (C) Glutathione-S-transferase (GST) activity had non-significant depletion very early following the trauma and remained relatively unchanged. (D) Glucose-6-phosphate dehydrogenase (G-6PD) activity altered significantly at 3h with maximum depletion by 24h, eventually returning to control levels at 96h post injury. Each bar represents the group mean ± SD of six animals/group. *p < 0.05 versus sham.
GR reduces GSSG into GSH to continue the antioxidant cycle during detoxification of oxidants. The analysis demonstrated a significant time-dependent change in GR levels [F (8, 45) = 5.528, p < 0.0001] (Fig. 2B). A significant decline was observed as early as 6 h post injury in the ipsilateral hippocampus with maximum depletion (33.03%) observed at 24 h post injury. The levels of GR approached control values by 96 h post injury.
GST is used for detoxification of external or internal toxicants. The analysis failed to reveal a significant change in GST activity [F (8, 45) = 1.051, p < 0.4133] (Fig. 2C). There was a modest decline (17.82%) in activity (p < 0.05) at 6 h post injury that subsequently approached control levels.
G-6-PD is used to provide electrons for reduction of oxidized nicotinamide adenine dinucleotides (NAD+/NADP+). Their activity showed a significant time-dependent change [F (8, 45) = 6.551, p < 0.0001] in the hippocampus following injury (Fig. 2D). A significant decline (p < 0.01) could be observed as early as 6 h (31.34%) post-injury with the maximum depletion observed at 24 h (40.85%) (p < 0.001). The activity of G-6-PD approached that of control by 96h post injury.
SOD is an important enzyme that reduces the superoxide burden in injured tissue. Analysis of the Cu/Zn-SOD in ipsilateral hippocampus demonstrated a significant time-dependent change in activity [F (8, 45) = 4.912, p < 0.0002] (Fig. 3A). The Cu/Zn-SOD levels were significantly reduced at 12 h post-injury (p< 0.001) and remained low until 72 h post-injury. The maximum depletion was observed at 24 h (41.81%) (p < 0.001). The activity of Mn-SOD was also affected [F (8, 45) = 7.501, p < 0.0001] (Fig. 3B). The activity of Mn-SOD was significantly reduced at 12 h post-injury (p< 0.001) and remained low until 96 h post-injury with maximum depletion at 24 h (50.60%). The activity SOD was not decreased in contralateral hippocampus at any time post-injury.
Fig. 3.
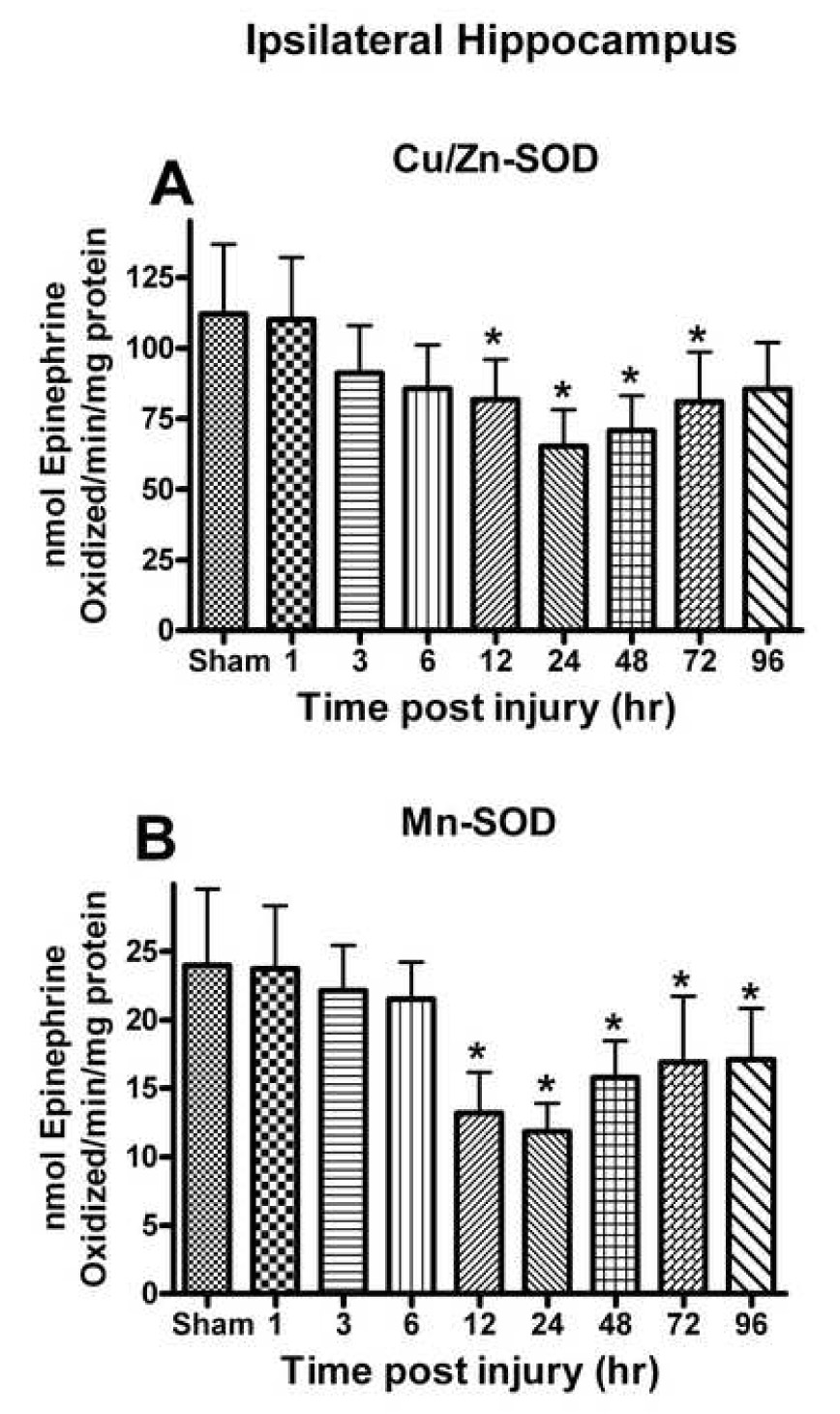
(A) Cu/Zn-SOD and (B) Mn-SOD activity in the ipsilateral hippocampus was affected post TBI. Cu/Zn-SOD activity declined at 12h following the trauma and approached sham levels by 96h post injury. Mn-SOD activity demonstrated a time-dependent decline that was greater than that observed for Cu/Zn-SOD and failed to return to control levels. Each bar represents the group mean ± SD of six animals/group. *p < 0.05 versus sham.
CAT is an important enzyme that breaks down H2O2 into H2O and O2. Analysis revealed a significant [F (8, 45) = 5.729, p < 0.0001] time-dependent change in CAT activity with the earliest significant loss observed at 12 h post injury (p < 0.05) (Fig. 4). The maximum significant depletion in CAT activity was observed at 24 h (28.17% compared to sham). At 96 h post injury, levels were no longer significantly different from controls. The contralateral side failed to show any significant change in CAT activity.
Fig. 4.
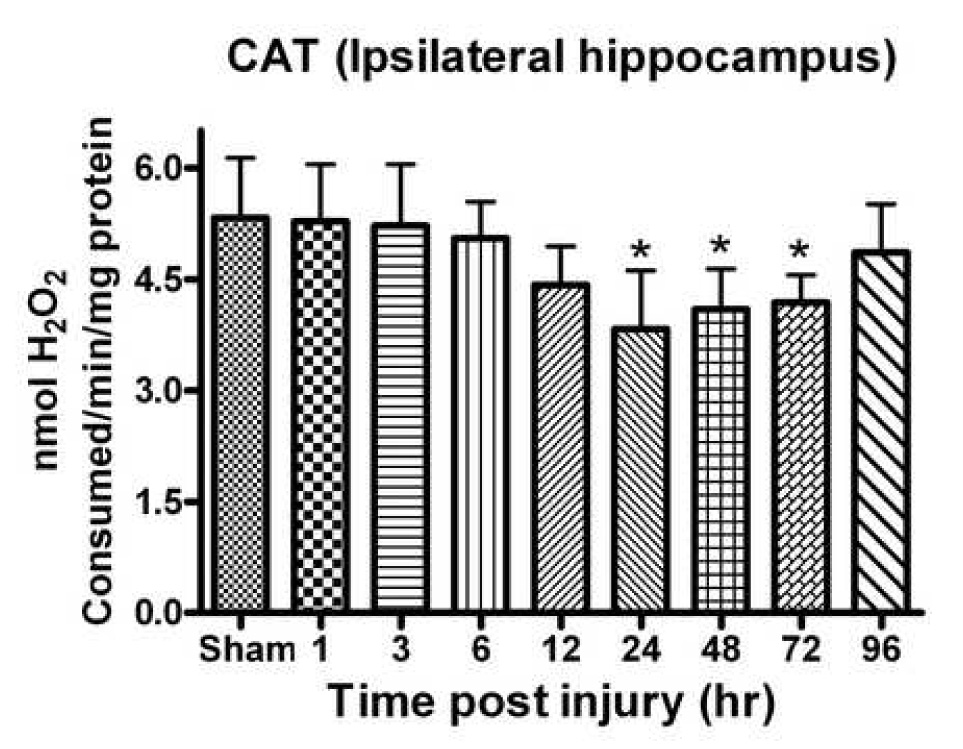
CAT activity in the hippocampus significantly declined following a unilateral cortical contusion. Significant depletion was observed by 24h post TBI with a return to control levels by 96h. Enzyme activity was measured with H2O2. Each bar represents group mean ± SD of six animals/group. *p < 0.05 versus sham.
Time course of protein oxidation, nitration and lipid peroxidation post injury
Levels of modified proteins with lipid peroxidation products (4-HNE and acrolein) were evaluated in the ipsilateral and contralateral hippocampus following cortical contusion. The analysis revealed a significant effect of time post-injury effect for 4-HNE ipsilateral to the impact site [F (8, 45) = 9.703, p < 0.0001] (Fig. 5A). The levels increased significantly (p < 0.05) at 3 h (26.59%) and continued to increase with peak levels observed at 48–72 h (73.43-75.02%). Levels remained elevated at 96 h. Similar to 4-HNE, the levels of acrolein demonstrated a significant time-dependent change ipsilateral to the injury [F (8,45) = 5.176, p < 0.0001]. Peak values were observed at 24–48 h (46.8-43.5%) (Fig. 5B). At 96 h the levels were still significantly elevated. The contralateral side failed to demonstrate a significant alteration in acrolein.
Fig. 5.
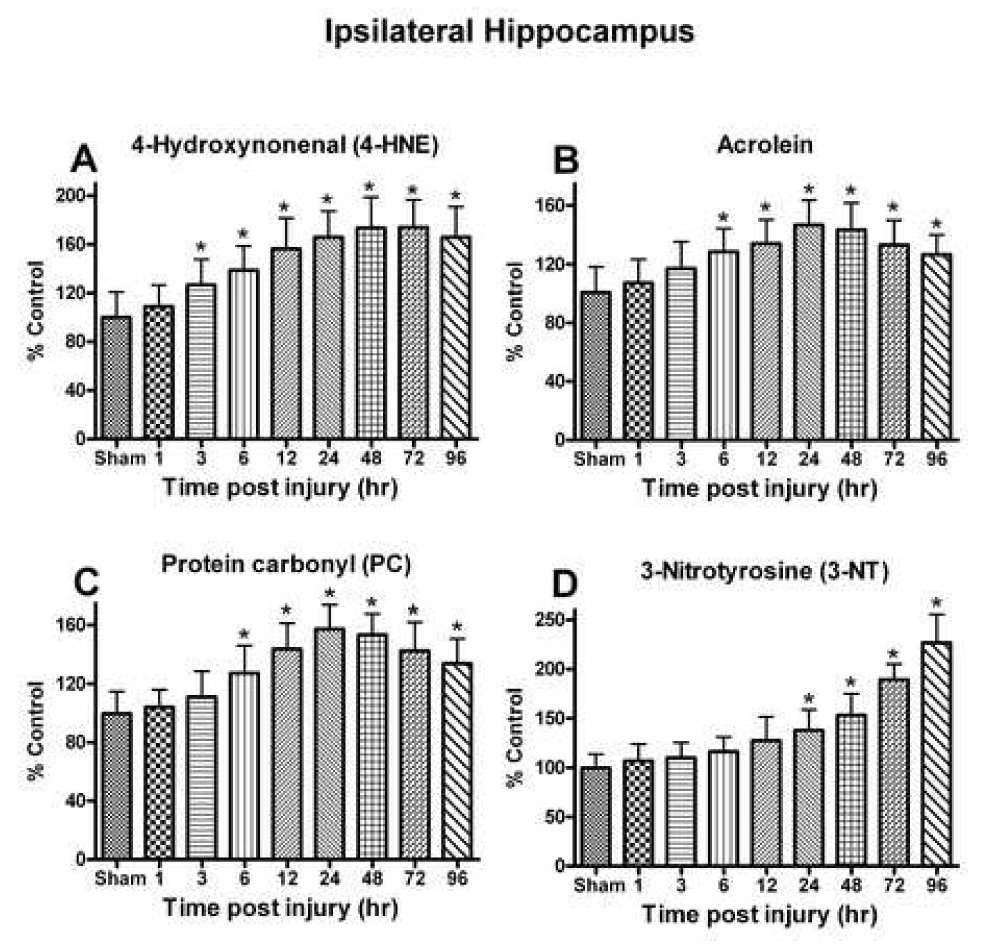
The effect of a moderate unilateral cortical contusion on lipid peroxidation and protein modification at different time points post TBI. (A) 4-HNE levels demonstrated a time-dependent increase in the ipsilateral hippocampus following trauma and remained elevated at 96h post trauma. (B) Acrolein levels were increased significantly as early as 6h following trauma and never returned to control levels by 96h. (C) PC levels demonstrated a time-dependent change that mirrored that observed for acrolein with significant increase observed as early as 6h post trauma. (D) Changes in 3-NT levels were delayed in response to the trauma with a significant increase first observed at 24h that failed to returne to control levels by 96h post injury. Each bar represents the group mean ± SD of six animals/group. *p < 0.05 versus sham.
The levels of protein oxidation and nitration products (PC and 3-NT) were evaluated in the ipsilateral and contralateral hippocampus following TBI. The analysis of PC demonstrated a significant effect of time post-injury ipsilateral to the impact site [F (8, 45) = 9.898, p < 0.0001] (Fig. 5C). Levels increased significantly (p < 0.05) at 6 h (27.29%) and continued to increase with peak levels observed at 24–48 (57.33-53.41%). At 72 h and 96 h post injury, PC values remained significantly (p < 0.01) elevated. The level of 3-NT was significantly elevated in a time-dependant fashion [F (8, 45) = 29.859, p < 0.0001] (Fig. 5D). Analysis showed that the 3-NT levels were highest at 96 h post-injury. The contralateral hippocampus failed to reveal any significant change.
Time course of synaptic proteins post injury
Synaptic proteins (synapsin-I, PSD-95, SAP97 and GAP-43) were evaluated in the ipsilateral and contralateral hippocampus. Western-blot analysis shows time dependent changes in the ipsilateral hippocampus (Fig. 6), while the contralateral side failed to show any significant change in synaptic proteins.
Fig. 6.
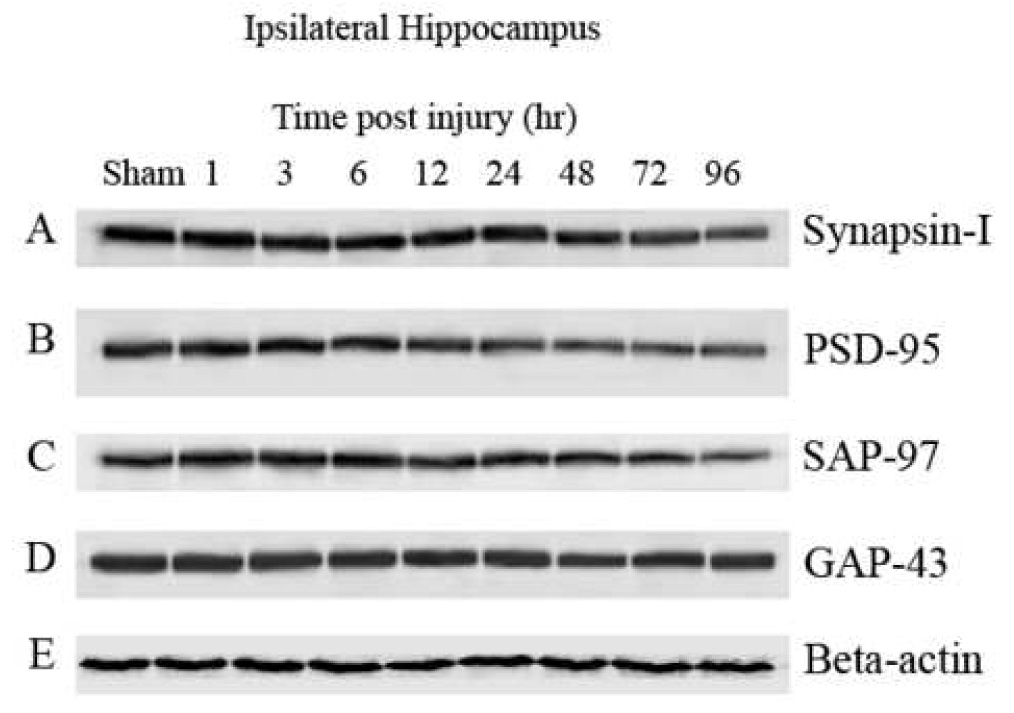
Synaptic proteins in the ipsilateral hippocampus were significantly affected following a unilateral cortical contusion. Hippocampal sample from six animals/group were processed for immunobloting followed by Western-blot. Immunoblotes were developed with 5- Bromo-4-chloro-3-indolyl phosphate/Nitro blue tetrazolium (alkaline phosphatase substrate; SIGMA FAST™ BCIP/NBT) and densities of bands were analyzed using Scion Image. Synapsin-I, PSD-95, and synapse associated protein -97 (SAP-97) demonstrated a time-dependent decrease in signal. Growth associated protein-43 (GAP-43) and Beta-actin failed to demonstrate any time-dependent decline in levels in the ipsilateral hippocampus.
Synapsin-I, a major pre synaptic protein showed a time dependent change [F (8, 36) = 4.329, p < 0.0010] in ipsilateral hippocampus post injury (Fig. 7A). Synapsin-I significantly declined at 48 h (21.28%) with the lowest levels observed at 96 h post injury (30.02%). PSD-95 is a core scaffolding component of post synaptic elements, and has been implicated in various roles in synaptic function and plasticity. PSD-95 showed a significant time-dependent decline [F (8, 36) = 6.378, p > 0.0001] in ipsilateral to the injury (Fig. 7B). Like synapsin-I, the earliest changes observed in PSD-95 levels were at 48 h and continued to 96 h post injury (p < 0.01). SAP-97 is a membrane associated phosphoprotein, which participates in a variety of functions with AMPA-type glutamate receptors. SAP-97 demonstrated a protracted decline [F (8, 36) = 4.329, p < 0.0010] with the lowest levels observed at 96 h post injury (22.94%) (Fig. 7C). GAP-43 is a phosphoprotein which is associated with neuronal growth and synaptic regeneration. GAP-43 failed to demonstrate significant change (p > 0.05) at any time up to 96 h (Fig. 7D).
Fig. 7.
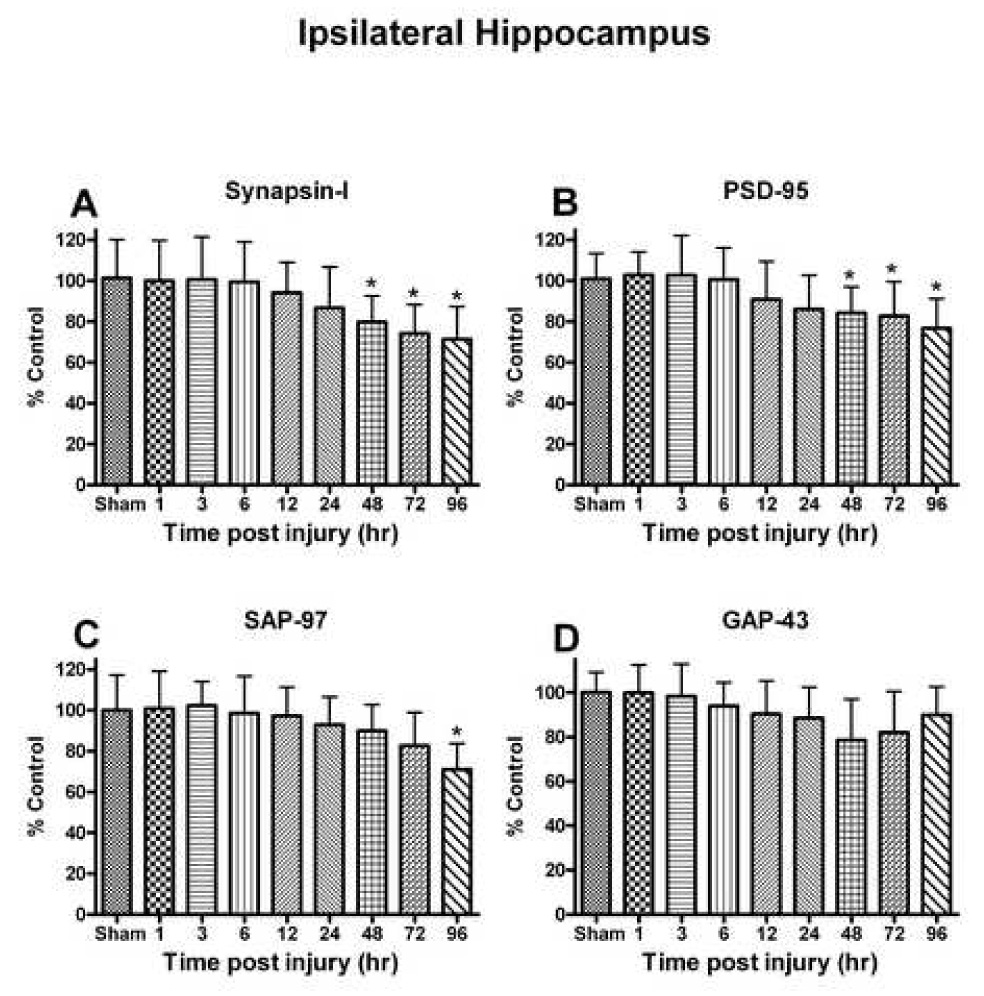
(A) Pre-synaptic marker protein synapsin-I levels in the ipsilateral hippocampus declined following a unilateral cortical contusion and failed to return to sham injury levels by 96h. (B) Post-synaptic density protein -95 (PSD-95) levels mirrored the changes observed with synapsin-I and remained significantly decreased by 96h. (C) SAP-97 levels demonstrated a significant decline following the trauma at 96h. (D) Growth associated protein-43 (GAP-43) failed to demonstrate a significant change following TBI up to 96hr. Each bar represents the group mean ± SD of six animals/group. *p < 0.05 versus sham.
Time course of hippocampal neuronal death
In all tissue stained for FJB, the degenerating neurons that stained positive were easily discernable from non-degenerating neurons. The total number of FJB-positive staining cells was evaluated in three different subregions of the hippocampus both ipsilateral and contralateral to the cortical injury. FJB-positive cells were not observed in any of the cases in the contralateral hippocampus. The analysis revealed a significant time-dependent change in the dentate gyrus granule cells [F (6,24) = 5.986, p < 0.0006] (Fig. 8). Significant numbers of FJB-positive granule cells could be observed as early as 1 h post injury, with peak values observed at 24 h (Fig. 9). Significant numbers were still observable at 7 days post injury. In the CA3 subregion, the analysis revealed a significant time -dependent change [F (6,24) = 6.492, p < 0.0004]. Post hoc analysis revealed significant accumulation at 1 h post injury and peak values were observed at 24 h. Similar to the dentate gyrus, significant numbers of FJB-positive neurons were observed at 7 days. Finally, the analysis of the CA1 subregion also revealed a significant time-dependent accumulation of FJB-positive cells [F (6,24) = 3.610, p < 0.01] with earliest significant accumulation occurring at 6 h post injury.
Fig. 8.
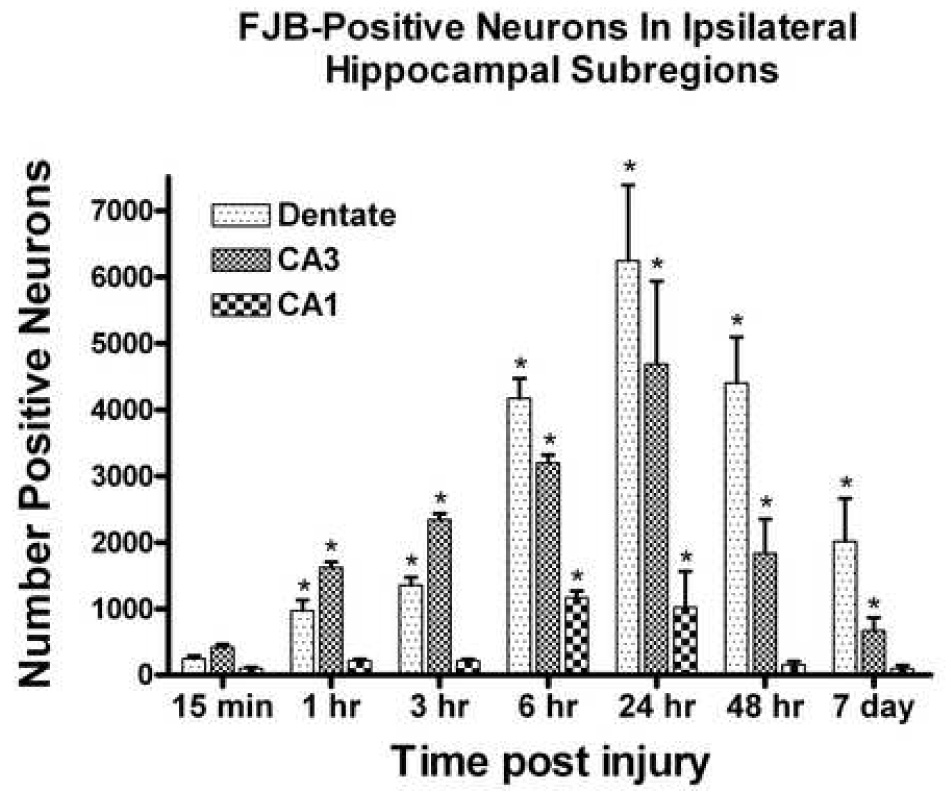
The total number of Fluoro-Jade B positive neurons in the different regions of the ipsilateral hippocampus following a unilateral cortical contusion. As early as 1h following TBI, significant number of FJB-positive neurons were observed in the dentate gyrus granule cell layer and CA3. The CA1 pyramidal cells showed the least number of positive cells with significant increase observed at 6h post contusion. Peak values were observed at 24h with significant number of FJB-positive cells still present at 7 day post trauma. Each bar represents the group mean ± SD. *p < 0.05 versus contralateral hippocampus as sham. 24h, 48h and 7 day data included from earlier published our lab findings (Anderson et al., 2005).
Fig. 9.
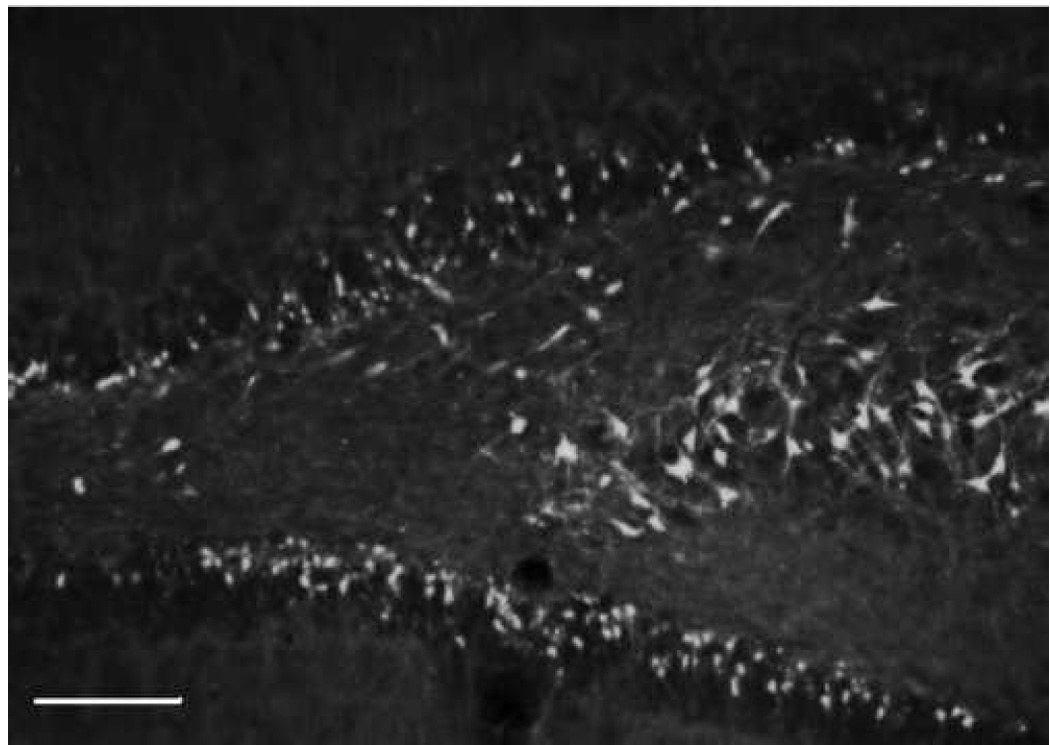
Ipsilateral coronal section through the hippocampal dentate gyrus and CA3 region at 24h following a unilateral cortical cortical contusion. Sections were stained with flour-Jade B (FJB) an anionic fluorescein derivative that specifically identifies degenerating neurons. Stereological techniques were used to determine the total number of FJB-positive neurons in different subregions of the hippocampal formation. Calibration bar = 500um
Discussion
This study documents early time-dependent changes in cellular oxidative markers (GSSG, protein carbonyls, 3-NT, 4-HNE and acrolein), the anti-oxidant system (GSH, GST, GPx, GR, G-6PD, Cu/Zn or Mn-SOD and CAT), and synaptic proteins (synapsin-I, PSD-95, and SAP-97) in the adult rat hippocampus following experimental TBI. Oxidative stress is a key participant, along with metabolic compromise and exitotoxicity, in apoptotic and necrotic neurodegenerative processes [27]. Oxidative stress reflects a marked imbalance between ROS and their removal by antioxidant systems. This imbalance may originate from an overproduction of ROS and/or from a reduction in antioxidant defenses [8, 24, 28, 29]. The brain is particularly vulnerable to oxidative injury because of its high rate of oxygen consumption, intense production of reactive radicals, and high levels of transition metals, such as iron that catalyze the production of reactive radicals. Moreover, neuronal membranes are rich in PUFA that are a source of LPO reaction [30]. Establishing the time course of oxidative stress provides information on a possible therapeutic window for protection against oxidative and/or nitrosative insults in the hippocampus. The present results suggest that the increased ROS is a rapidly occurring event with a protracted time course. The levels of oxidative markers were significantly elevated in ipsilateral hippocampus; concomitantly, antioxidants were depleted along with synaptic marker proteins.
Cellular defense, such as GSH system and antioxidant enzymes, protects tissue from the damaging effect of free radicals. GSH is a ubiquitous tri-peptide and a major intracellular antioxidant molecule, which constitutes an important mechanism against oxidative stress. It plays a crucial role in detoxification of peroxides and electrophilic toxins as a substrate for GPx and GST, in conjunction with NADPH and forms GSSG in this process [31, 32]. Reduced GSH levels 3 h post injury in the ipsilateral hippocampus remained significantly lower over the course of 96 h. A decrease in GSH levels on GSSG production is linked to apoptotic neuronal death. The GSH/GSSG ratio is an indicator of oxidative and/or nitrosative burden in the system [8], which was decreased significantly within 3 h post-injury, and remained low over the study period of 96 h. The level of protein damages through lipid peroxidation (4-hydroxynonenal and acrolein-bound proteins) remains significantly higher throughout this same time course, suggesting that the antioxidant defense were not able to offset oxidative stress. The low levels of GSH may be directly related to the increased ROS/RNS, lipid peroxides, and highly reactive hydroxyl radicals [7, 33]. A low GSH/GSSG ratio could contribute to promoting free radial load and oxidative stress [34]. These circumstances would lead to prolonged neuronal dysfunction [8, 35]. During oxidative stress, several lipid peroxidation products are formed that are involved in neuronal death, and become an immediate substrate for GSH [36] and aggravates further oxidative stress.
To eliminate the peroxides, GSH works in conjunction with GPx and produces GSSG, which is reconverted to GSH by GR at the consumption of NADPH. GPx provides protection against oxidative stress in TBI; reduced GPx activity could directly affect the clearance of ROS and lipid peroxides as indicated by elevated levels of 4-HNE and acrolein in the present study. GST, another detoxification enzyme [24, 36], can alleviate damage from lipid peroxidation by-products (4-HNE and acrolein) by catalyzing their conjugation with glutathione. The depletion of GST could result in increased protein modification/dysfunction and lead to further oxidative stress and decline of GSH [8]. Balancing the GSH/GSSG level requires an important cofactor, NADPH, which is produced from reactions catalyzed by G-6PD and 6-phosphogluconate dehydrogenase. Thus, GST, GR and G-6PD are secondary antioxidant enzymes that play an important role in detoxifying ROS/RNS by maintaining a ready supply of intermediates such as GSH and NADPH [25]. Decreased activity of GR would directly affect GSH as would a reduction in the levels of GST, resulting in overall low levels of antioxidant defense, thus predicting the overpowering influences of ROS/RNS in brain injuries [7, 8, 24].
The most abundant ROS formed in the course of cellular metabolism is the superoxide radical (O2−•), which has been implicated in brain edema and hippocampal neuronal death [37]. The first enzymatic reaction in the reduction pathway is dismutation of two molecules of O2−• when they are converted into H2O2 and O2. The enzyme Cu/Zn-SOD and/or Mn-SOD protect neurons from high O2−• environment after TBI [28, 38]. Moreover, GPx and CAT participate in the elimination of H2O2, which is one of the most toxic molecules in the brain. Because GPx and CAT both control H2O2, they represent the major defense against ROS [14]. Decreased activity of Mn-SOD is a likely consequence of peroxynitrite after TBI, and could lead to further oxidative stress and progressively enhance peroxynitrite production as part of the secondary damage cascade [10]. Decreased activity of Cu/Zn-SOD in ipsilateral hippocampus of young animals post injury compromises defense capacities against oxidative stress [39].
In this study, we demonstrated a time course in FJB staining among the hippocampal subregions to highlight dead or dying neurons after TBI [26]. Results show large numbers of FJB-positive neurons in all three major subregions of the ipsilateral hippocampus. FJB-positive neurons can be readily observed at 1 h and continue to die until 96 h, matching the observed decline in synaptic proteins. All subregions of the hippocampus show maximum neuronal death at 24 h post injury, which is a mirror image of our oxidative stress findings. Oxidative stress as a mediator of secondary sequelae after TBI might be an important factor contributing to subsequent pathology and neurobehavioral shortfalls [40]. The pathophysiological mechanisms of secondary injury are complex and include inflammation, production of ROS/RNS, increased intracellular calcium and lipid peroxidation [41, 42]. The breakdown of the blood-brain-barrier and a neuroinflammatory response after brain injury also contribute to neuronal death [43, 44]. Oxidative stress and peroxidation of PUFA damage neuronal membranes, leading to the generation of 4-HNE and acrolein, which in turn can damage proteins. Levels of 4-HNE and acrolein were significantly elevated by 6 h post trauma and peaked 24–72 h in ipsilateral hippocampus, indicating that LPO products significantly increased very early following injury, and remained elevated for several days in young animals [11, 45]. Elevated oxidative stress resulting from PC, 3-NT, 4-HNE, or acrolein can lead to delayed neuronal death, and may be responsible for prolonged accumulation of FJB-positive neurons observed in the ipsilateral hippocampus in the present study.
Altered energy metabolism and oxidative stress negatively affects synaptic plasticity and cognitive function though oxidation of synaptic proteins [18, 20]. Oxidative stress contributes to synaptic dysfunction and disconnection, with impaired mitochondrial transport to the synapses contributing to both neuronal death and neuritic degenerative cascades [15, 16]. Exitotoxicity contributes to energy failure resulting in further oxidative stress by enhanced metabolism of excitatory amino acids, leading to secondary cascades of injuries following TBI [46]. These cascades implicate mitochondrial bioenergetic dysfunction, disruption of Ca2+ homeostasis, and over production of ROS, adversely affecting synaptic functions and plasticity and neuronal loss [47]. CNS neurons injured by reactive radicals may be physically present but non-functional due to impaired energy metabolism, neuronal sickness, or impaired mitochondrial transport to the synaptic regions [15].
In our study, oxidative stress most likely contributes to the loss of synapsin-I and PSD-95, which are known to affect neuronal survival and function. Synapses are formed and stabilized by concerted interactions of pre-, intra- and post-synaptic components [48]. Synapsins and synaptotagmin (preynaptic proteins), as well as PSD-95, represent the synaptic complex [49, 50]. The synapsin-I present in synaptic vesicles, plays a wide range of neuronal functions including neurotransmitter release, synaptogenesis, cytoskeletal structural dynamics, energy metabolism, ion homeostasis, and protein folding [51, 52]. PSD-95 interacts with N-methyl-D-aspartate receptors [53] and are implicated in various important roles in the regulation of ion-channel function, neuronal differentiation, synaptogenesis, synaptic plasticity, and the processes of learning and memory [54, 55]. PSD-95 could target membrane addition to polarized synaptic compartments, formation of cellular junctions as well as synaptic protein anchoring and trafficking [56]. The synaptic proteins and their modifications play a dynamic role in learning and memory [18, 57]. The loss of PSD-95 could adversely affect synaptic connectivity and the neuronal network [58]. The depletion of SAP-97 in our study, most likely is a measure of lost synaptic connectivity in hippocampus. It would adversely affect the functions of hippocampus following brain injuries and may underlie behavioral deficits. It is a synaptic membrane-associated protein, localized to post synaptic densities [59], and thought to play a role in trafficking of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors [60, 61]. Recently SAP-97 depletion has been linked with cerebral ischemia [62].
GAP-43 is a growth associated protein that has been linked with synaptic regeneration in response to injury associated behavioral deficits [63, 64]. Synaptic and axonal regeneration after brain injury is essential for predicting the neural compensation that could be achieved after various types of injury [65]. In our study, GAP-43 failed to show any significant change post injury.
In summary, our data show alterations in the antioxidant capacity in the hippocampus following TBI. Examination of ipsilateral hippocampus showed that 4-HNE, acrolein, 3-NT, and protein carbonyl levels increase, whereas antioxidant enzyme activity decreases post TBI. Decreased antioxidant enzyme activity and increased oxidative stress lead to neuronal death. Loss of synaptic proteins in hippocampus might be through modifications with 4-HNE, acrolein, 3-NT, and carbonylation, which could contribute to behavioral abnormalities after TBI.
Acknowledgments
This research was supported by the NIH AG21981 and the Kentucky Spinal Cord Head Injury Research Trust #5-A.
Abbreviations used
- CAT
catalase
- FJB
Fluoro-Jade B
- G-6PD
glucose-6-phosphate dehydrogenase
- GPx
glutathione peroxidase
- GR
glutathione reductase
- GST
glutathione-S-transferase
- GAP-43
growth associated protein-43
- 4-HNE
4-hydroxynonenal
- LPO
lipid peroxidation
- NEM
N-ethyleimide
- 3-NT
3-nitrotyrosine
- PF
paraformaldehyde
- PBS
phosphate-buffered saline
- PMSF
phenylmethylsulfonyl fluoride
- PUFA
poly unsaturated fatty acid
- PMS
post-mitochondrial supernatant
- PSD-95
post synaptic density-95
- PC
protein carbonyl
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- Cu/Zn-Mn SOD
copper/zinc-manganese superoxide dismutase
- SAP-97
synapse associated protein-97
- TBI
TBI, traumatic brain injury
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Biegon A, Fry PA, Paden CM, Alexandrovich A, Tsenter J, Shohami E. Dynamic changes in N-methyl-D-aspartate receptors after closed head injury in mice: Implications for treatment of neurological and cognitive deficits. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5117–5122. doi: 10.1073/pnas.0305741101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sullivan PG, Keller JN, Mattson MP, Scheff SW. Traumatic brain injury alters synaptic homeostasis: implications for impaired mitochondrial and transport function. J. Neurotrauma. 1998;15:789–798. doi: 10.1089/neu.1998.15.789. [DOI] [PubMed] [Google Scholar]
- 3.Engle MR, Singh SP, Czernik PJ, Gaddy D, Montague DC, Ceci JD, Yang Y, Awasthi S, Awasthi YC, Zimniak P. Physiological role of mGSTA4-4, a glutathione S-transferase metabolizing 4-hydroxynonenal: generation and analysis of mGsta4 null mouse. Toxicol. Appl. Pharmacol. 2004;194:296–308. doi: 10.1016/j.taap.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Shao C, Roberts KN, Markesbery WR, Scheff SW, Lovell MA. Oxidative stress in head trauma in aging. Free Radic. Biol. Med. 2006;41:77–85. doi: 10.1016/j.freeradbiomed.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Ozdemir D, Uysal N, Gonenc S, Acikgoz O, Sonmez A, Topcu A, Ozdemir N, Duman M, Semin I, Ozkan H. Effect of melatonin on brain oxidative damage induced by traumatic brain injury in immature rats. Physiol. Res. 2005;54:631–637. [PubMed] [Google Scholar]
- 6.Solaroglu I, Okutan O, Kaptanoglu E, Beskonakli E, Kilinc K. Increased xanthine oxidase activity after traumatic brain injury in rats. J. Clin. Neurosci. 2005;12:273–275. doi: 10.1016/j.jocn.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Ansari MA, Ahmad AS, Ahmad M, Salim S, Yousuf S, Ishrat T, Islam F. Selenium protects cerebral ischemia in rat brain mitochondria. Biol. Trace Elem. Res. 2004;101:73–86. doi: 10.1385/BTER:101:1:73. [DOI] [PubMed] [Google Scholar]
- 8.Ansari MA, Joshi G, Huang Q, Opii WO, Abdul HM, Sultana R, Butterfield DA. In vivo administration of D609 leads to protection of subsequently isolated gerbil brain mitochondria subjected to in vitro oxidative stress induced by amyloid beta-peptide and other oxidative stressors: relevance to Alzheimer's disease and other oxidative stress-related neurodegenerative disorders. Free Radic. Biol. Med. 2006;41:1694–1703. doi: 10.1016/j.freeradbiomed.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh IN, Sullivan PG, Hall ED. Peroxynitrite-mediated oxidative damage to brain mitochondria: Protective effects of peroxynitrite scavengers. J. Neurosci. Res. 2007;85:2216–2223. doi: 10.1002/jnr.21360. [DOI] [PubMed] [Google Scholar]
- 10.Bayir H, Kagan VE, Clark RS, Janesko-Feldman K, Rafikov R, Huang Z, Zhang X, Vagni V, Billiar TR, Kochanek PM. Neuronal NOS-mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J. Neurochem. 2007;101:168–181. doi: 10.1111/j.1471-4159.2006.04353.x. [DOI] [PubMed] [Google Scholar]
- 11.Hall ED, Detloff MR, Johnson K, Kupina NC. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J. Neurotrauma. 2004;21:9–20. doi: 10.1089/089771504772695904. [DOI] [PubMed] [Google Scholar]
- 12.Ozturk E, Demirbilek S, Kadir But A, Saricicek V, Gulec M, Akyol O, Ozcan Ersoy M. Antioxidant properties of propofol and erythropoietin after closed head injury in rats. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2005;29:922–927. doi: 10.1016/j.pnpbp.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 13.DeKosky ST, Taffe KM, Abrahamson EE, Dixon CE, Kochanek PM, Ikonomovic MD. Time course analysis of hippocampal nerve growth factor and antioxidant enzyme activity following lateral controlled cortical impact brain injury in the rat. J. Neurotrauma. 2004;21:491–500. doi: 10.1089/089771504774129838. [DOI] [PubMed] [Google Scholar]
- 14.Dringen R, Pawlowski PG, Hirrlinger J. Peroxide detoxification by brain cells. J. Neurosci. Res. 2005;79:157–165. doi: 10.1002/jnr.20280. [DOI] [PubMed] [Google Scholar]
- 15.de la Monte SM, Neely TR, Cannon J, Wands JR. Oxidative stress and hypoxialike injury cause Alzheimer-type molecular abnormalities in central nervous system neurons. Cell Mol. Life Sci. 2000;57:1471–1481. doi: 10.1007/PL00000630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matzilevich DA, Rall JM, Moore AN, Grill RJ, Dash PK. High-density microarray analysis of hippocampal gene expression following experimental brain injury. J. Neurosci. Res. 2002;67:646–663. doi: 10.1002/jnr.10157. [DOI] [PubMed] [Google Scholar]
- 17.Kotulska K, LePecheur M, Marcol W, Lewin-Kowalik J, Larysz-Brysz M, Paly E, Matuszek I, London J. Overexpression of copper/zinc-superoxide dismutase in transgenic mice markedly impairs regeneration and increases development of neuropathic pain after sciatic nerve injury. J. Neurosci. Res. 2006;84:1091–1097. doi: 10.1002/jnr.21000. [DOI] [PubMed] [Google Scholar]
- 18.Wu A, Ying Z, Gomez-Pinilla F. Dietary curcumin counteracts the outcome of traumatic brain injury on oxidative stress, synaptic plasticity, and cognition. Exp. Neurol. 2006;197:309–317. doi: 10.1016/j.expneurol.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 19.Cayabyab FS, Khanna R, Jones OT, Schlichter LC. Suppression of the rat microglia Kv1.3 current by src-family tyrosine kinases and oxygen/glucose deprivation. Eur. J. Neurosci. 2000;12:1949–1960. doi: 10.1046/j.1460-9568.2000.00083.x. [DOI] [PubMed] [Google Scholar]
- 20.Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J. Neurotrauma. 2005;22:719–732. doi: 10.1089/neu.2005.22.719. [DOI] [PubMed] [Google Scholar]
- 21.Fineman I, Giza CC, Nahed BV, Lee SM, Hovda DA. Inhibition of neocortical plasticity during development by a moderate concussive brain injury. J. Neurotrauma. 2000;17:739–749. doi: 10.1089/neu.2000.17.739. [DOI] [PubMed] [Google Scholar]
- 22.Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- 23.Redell JB, Moore AN, Dash PK. Expression of the prodynorphin gene after experimental brain injury and its role in behavioral dysfunction. Exp. Biol. Med. (Maywood, N.J. 2003;228:261–269. doi: 10.1177/153537020322800304. [DOI] [PubMed] [Google Scholar]
- 24.Ahmad AS, Ansari MA, Ahmad M, Saleem S, Yousuf S, Hoda MN, Islam F. Neuroprotection by crocetin in a hemi-parkinsonian rat model. Pharmacol. Biochem. Behav. 2005;81:805–813. doi: 10.1016/j.pbb.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 25.Shah ZA, Vohora SB. Antioxidant/restorative effects of calcined gold preparations used in Indian systems of medicine against global and focal models of ischaemia. Pharmacol. Toxicol. 2002;90:254–259. doi: 10.1034/j.1600-0773.2002.900505.x. [DOI] [PubMed] [Google Scholar]
- 26.Anderson KJ, Miller KM, Fugaccia I, Scheff SW. Regional distribution of fluorojade B staining in the hippocampus following traumatic brain injury. Exp. Neurol. 2005;193:125–130. doi: 10.1016/j.expneurol.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 27.Alexi T, Borlongan CV, Faull RL, Williams CE, Clark RG, Gluckman PD, Hughes PE. Neuroprotective strategies for basal ganglia degeneration: Parkinson' and Huntington's diseases. Prog. Neurobiol. 2000;60:409–470. doi: 10.1016/s0301-0082(99)00032-5. [DOI] [PubMed] [Google Scholar]
- 28.Freeman BA, Crapo JD. Biology of disease: free radicals and tissue injury. Lab. Invest. 1982;47:412–426. [PubMed] [Google Scholar]
- 29.Halliwell B, Gutteridge JM. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reiter RJ. Oxidative damage in the central nervous system: protection by melatonin. Prog. Neurobiol. 1998;56:359–384. doi: 10.1016/s0301-0082(98)00052-5. [DOI] [PubMed] [Google Scholar]
- 31.Choe H, Hansen JM, Harris C. Spatial and temporal ontogenies of glutathione peroxidase and glutathione disulfide reductase during development of the prenatal rat. J. Biochem. Mol. Toxicol. 2001;15:197–206. doi: 10.1002/jbt.17. [DOI] [PubMed] [Google Scholar]
- 32.Sies H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999;27:916–921. doi: 10.1016/s0891-5849(99)00177-x. [DOI] [PubMed] [Google Scholar]
- 33.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U. S. A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bains JS, Shaw CA. Neurodegenerative disorders in humans: the role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Brain Res. Rev. 1997;25:335–358. doi: 10.1016/s0165-0173(97)00045-3. [DOI] [PubMed] [Google Scholar]
- 35.Ates O, Cayli S, Gurses I, Yucel N, Iraz M, Altinoz E, Kocak A, Yologlu S. Effect of pinealectomy and melatonin replacement on morphological and biochemical recovery after traumatic brain injury. Int. J. Dev. Neurosci. 2006;24:357–363. doi: 10.1016/j.ijdevneu.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 36.Xie C, Lovell MA, Markesbery WR. Glutathione transferase protects neuronal cultures against four hydroxynonenal toxicity. Free Radic. Biol. Med. 1998;25:979–988. doi: 10.1016/s0891-5849(98)00186-5. [DOI] [PubMed] [Google Scholar]
- 37.Yunoki M, Kawauchi M, Ukita N, Sugiura T, Ohmoto T. Effects of lecithinized superoxide dismutase on neuronal cell loss in CA3 hippocampus after traumatic brain injury in rats. Surg. Neurol. 2003;59:156–160. doi: 10.1016/s0090-3019(02)01040-6. discussion 160-151. [DOI] [PubMed] [Google Scholar]
- 38.Yunoki M, Kawauchi M, Ukita N, Noguchi Y, Nishio S, Ono Y, Asari S, Ohmoto T, Asanuma M, Ogawa N. Effects of lecithinized SOD on sequential change in SOD activity after cerebral contusion in rats. Acta Neurochir. Suppl. 1998;71:142–145. doi: 10.1007/978-3-7091-6475-4_42. [DOI] [PubMed] [Google Scholar]
- 39.Azbill RD, Mu X, Bruce-Keller AJ, Mattson MP, Springer JE. Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res. 1997;765:283–290. doi: 10.1016/s0006-8993(97)00573-8. [DOI] [PubMed] [Google Scholar]
- 40.Kline AE, Massucci JL, Ma X, Zafonte RD, Dixon CE. Bromocriptine reduces lipid peroxidation and enhances spatial learning and hippocampal neuron survival in a rodent model of focal brain trauma. J. Neurotrauma. 2004;21:1712–1722. doi: 10.1089/neu.2004.21.1712. [DOI] [PubMed] [Google Scholar]
- 41.Acarin L, Peluffo H, Barbeito L, Castellano B, Gonzalez B. Astroglial nitration after postnatal excitotoxic damage: correlation with nitric oxide sources, cytoskeletal, apoptotic and antioxidant proteins. J. Neurotrauma. 2005;22:189–200. doi: 10.1089/neu.2005.22.189. [DOI] [PubMed] [Google Scholar]
- 42.Gahm C, Holmin S, Wiklund PN, Brundin L, Mathiesen T. Neuroprotection by selective inhibition of inducible nitric oxide synthase after experimental brain contusion. J. Neurotrauma. 2006;23:1343–1354. doi: 10.1089/neu.2006.23.1343. [DOI] [PubMed] [Google Scholar]
- 43.Martin LJ, Price AC, McClendon KB, Al-Abdulla NA, Subramaniam JR, Wong PC, Liu Z. Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J. Neurochem. 2003;85:234–247. doi: 10.1046/j.1471-4159.2003.01659.x. [DOI] [PubMed] [Google Scholar]
- 44.Wagner KR, Dean C, Beiler S, Bryan DW, Packard BA, Smulian AG, Linke MJ, de Courten-Myers GM. Plasma infusions into porcine cerebral white matter induce early edema, oxidative stress, pro-inflammatory cytokine gene expression and DNA fragmentation: implications for white matter injury with increased blood-brain-barrier permeability. Curr. Neurovasc. Res. 2005;2:149–155. doi: 10.2174/1567202053586785. [DOI] [PubMed] [Google Scholar]
- 45.Tyurin VA, Tyurina YY, Borisenko GG, Sokolova TV, Ritov VB, Quinn PJ, Rose M, Kochanek P, Graham SH, Kagan VE. Oxidative stress following traumatic brain injury in rats: quantitation of biomarkers and detection of free radical intermediates. J. Neurochem. 2000;75:2178–2189. doi: 10.1046/j.1471-4159.2000.0752178.x. [DOI] [PubMed] [Google Scholar]
- 46.Yi JH, Hazell AS. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 2006;48:394–403. doi: 10.1016/j.neuint.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 47.Pandya JD, Pauly JR, Nukala VN, Sebastian AH, Day KM, Korde AS, Maragos WF, Hall ED, Sullivan PG. Post-Injury Administration of Mitochondrial Uncouplers Increases Tissue Sparing and Improves Behavioral Outcome following Traumatic Brain Injury in Rodents. J. Neurotrauma. 2007;24:798–811. doi: 10.1089/neu.2006.3673. [DOI] [PubMed] [Google Scholar]
- 48.Egles C, Claudepierre T, Manglapus MK, Champliaud MF, Brunken WJ, Hunter DD. Laminins containing the beta2 chain modulate the precise organization of CNS synapses. Mol. Cell. Neurosci. 2007;34:288–298. doi: 10.1016/j.mcn.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 49.Krapivinsky G, Mochida S, Krapivinsky L, Cibulsky SM, Clapham DE. The TRPM7 ion channel functions in cholinergic synaptic vesicles and affects transmitter release. Neuron. 2006;52:485–496. doi: 10.1016/j.neuron.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 50.Lechuga-Sancho AM, Arroba AI, Frago LM, Garcia-Caceres C, de Celix AD, Argente J, Chowen JA. Reduction in the number of astrocytes and their projections is associated with increased synaptic protein density in the hypothalamus of poorly controlled diabetic rats. Endocrinology. 2006;147:5314–5324. doi: 10.1210/en.2006-0766. [DOI] [PubMed] [Google Scholar]
- 51.Fassio A, Merlo D, Mapelli J, Menegon A, Corradi A, Mete M, Zappettini S, Bonanno G, Valtorta F, D'Angelo E, Benfenati F. The synapsin domain E accelerates the exoendocytotic cycle of synaptic vesicles in cerebellar Purkinje cells. J. Cell Sci. 2006;119:4257–4268. doi: 10.1242/jcs.03194. [DOI] [PubMed] [Google Scholar]
- 52.Futai K, Kim MJ, Hashikawa T, Scheiffele P, Sheng M, Hayashi Y. Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95-neuroligin. Nat. Neurosci. 2007;10:186–195. doi: 10.1038/nn1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gascon S, Sobrado M, Roda JM, Rodriguez-Pena A, Diaz-Guerra M. Excitotoxicity and focal cerebral ischemia induce truncation of the NR2A and NR2B subunits of the NMDA receptor and cleavage of the scaffolding protein PSD-95. Mol. Psychiatry. 2007 doi: 10.1038/sj.mp.4002017. [DOI] [PubMed] [Google Scholar]
- 54.Chen P, Gu Z, Liu W, Yan Z. Glycogen synthase kinase 3 regulates N-methyl-D-aspartate receptor channel trafficking and function in cortical neurons. Mol. Pharmacol. 2007;72:40–51. doi: 10.1124/mol.107.034942. [DOI] [PubMed] [Google Scholar]
- 55.Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proc. Natl. Acad. Sci. U. S. A. 2007;104:4176–4181. doi: 10.1073/pnas.0609307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsuriel S, Geva R, Zamorano P, Dresbach T, Boeckers T, Gundelfinger ED, Garner CC, Ziv NE. Local sharing as a predominant determinant of synaptic matrix molecular dynamics. PLoS Biol. 2006;4:e271. doi: 10.1371/journal.pbio.0040271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gorczyca D, Ashley J, Speese S, Gherbesi N, Thomas U, Gundelfinger E, Gramates LS, Budnik V. Postsynaptic membrane addition depends on the Discs-Large-interacting t-SNARE Gtaxin. J. Neurosci. 2007;27:1033–1044. doi: 10.1523/JNEUROSCI.3160-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lang SB, Stein V, Bonhoeffer T, Lohmann C. Endogenous brain-derived neurotrophic factor triggers fast calcium transients at synapses in developing dendrites. J. Neurosci. 2007;27:1097–1105. doi: 10.1523/JNEUROSCI.3590-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Valtschanoff JG, Burette A, Davare MA, Leonard AS, Hell JW, Weinberg RJ. SAP97 concentrates at the postsynaptic density in cerebral cortex. Eur. J. Neurosci. 2000;12:3605–3614. doi: 10.1046/j.1460-9568.2000.00256.x. [DOI] [PubMed] [Google Scholar]
- 60.Sans N, Racca C, Petralia RS, Wang YX, McCallum J, Wenthold RJ. Synapse-associated protein 97 selectively associates with a subset of AMPA receptors early in their biosynthetic pathway. J. Neurosci. 2001;21:7506–7516. doi: 10.1523/JNEUROSCI.21-19-07506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen L, Liang F, Walensky LD, Huganir RL. Regulation of AMPA receptor GluR1 subunit surface expression by a 4. 1N-linked actin cytoskeletal association. J. Neurosci. 2000;20:7932–7940. doi: 10.1523/JNEUROSCI.20-21-07932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res. Rev. 2007;54:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 63.Benowitz LI, Routtenberg A. GAP-43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997;20:84–91. doi: 10.1016/s0166-2236(96)10072-2. [DOI] [PubMed] [Google Scholar]
- 64.Hughes-Davis EJ, Cogen JP, Jakowec MW, Cheng HW, Grenningloh G, Meshul CK, McNeill TH. Differential regulation of the growth-associated proteins GAP-43 and superior cervical ganglion 10 in response to lesions of the cortex and substantia nigra in the adult rat. Neuroscience. 2005;135:1231–1239. doi: 10.1016/j.neuroscience.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 65.Buffo A, Carulli D, Rossi F, Strata P. Extrinsic regulation of injury/growth-related gene expression in the inferior olive of the adult rat. Eur. J. Neurosci. 2003;18:2146–2158. doi: 10.1046/j.1460-9568.2003.02940.x. [DOI] [PubMed] [Google Scholar]