

Abstract
Reactive oxygen species (ROS) signal vital physiological processes including cell growth, angiogenesis, contraction, and relaxation of vascular smooth muscle. Because cytochrome P-450 family 4 (CYP4)/20-hydroxyeicosatetraenoic acid (20-HETE) has been reported to enhance angiogenesis, pulmonary vascular tone, and endothelial nitric oxide synthase function, we explored the potential of this system to stimulate bovine pulmonary artery endothelial cell (BPAEC) ROS production. Our data are the first to demonstrate that 20-HETE increases ROS in BPAECs in a time- and concentration-dependent manner as detected by enhanced fluorescence of oxidation products of dihydroethidium (DHE) and dichlorofluorescein diacetate. An analog of 20-HETE elicits no increase in ROS and blocks 20-HETE-evoked increments in DHE fluorescence, supporting its function as an antagonist. Endothelial cells derived from bovine aortas exhibit enhanced ROS production to 20-HETE quantitatively similar to that of BPAECs. 20-HETE-induced ROS production in BPAECs is blunted by pretreatment with polyethylene-glycolated SOD, apocynin, inhibition of Rac1, and a peptide-based inhibitor of NADPH oxidase subunit p47phox association with gp91. These data support 20-HETE-stimulated, NADPH oxidase-derived, and Rac1/2-dependent ROS production in BPAECs. 20-HETE promotes translocation of p47phox and tyrosine phosphorylation of p47phox in a time-dependent manner as well as increased activated Rac1/2, providing at least three mechanisms through which 20-HETE activates NADPH oxidase. These observations suggest that 20-HETE stimulates ROS production in BPAECs at least in part through activation of NADPH oxidase within minutes of application of the lipid.
Keywords: Superoxide, RAC1/2, Hydrogen Peroxide, Tempol, CYP4A, ROS
Introduction
Products of cytochrome P-450 (CYP) ω-hydroxylases (including CYP4 isoforms) mediate key physiological functions including autoregulation of blood flow, tubuloglomerular feedback, Na+ reabsorption in the kidney, and relaxation of pulmonary arterioles (29, 38). Our studies have focused on the role of CYP4 and its arachidonic acid (AA) metabolite, 20-hydroxyeicosatetraenoic acid (20-HETE), in pulmonary vascular function and biology. 20-HETE is the CYP ω-hydroxylation product of AA cleaved from membrane phospholipid sources. Enzymes of the CYP4A, -4B, and -4F families catalyze the ω-hydroxylation of fatty acids, and several isoforms in these families produce 20-HETE when incubated with AA. For example, rat CYP4A1, -4A2, and -4A3 catalyze AA ω- and ω-1-hydroxylations with the highest catalytic efficiency accruing to CYP4A1 (35). Although CYP4A2 and CYP4A3 exhibit an additional arachidonate 11,12-epoxidation activity, CYP4A1 operates solely as an ω-hydroxylase. Most investigators suggest that CYP4 isoforms constitute the major source of 20-HETE synthesis in extrahepatic tissues, including the lung (35, 38). Accordingly, we have investigated the effects of CYP4 product, 20-HETE, in our studies of this system in pulmonary vascular biology.
We have identified a unique role for CYP4/20-HETE in regulating pulmonary artery endothelial cell (PAEC) endothelial nitric oxide synthase (eNOS) (13, 30). Whereas CYP4 expression is widely recognized in vascular smooth muscle cells from systemic circulations, pulmonary endothelium from small arteries as well as vascular smooth muscle cells express CYP4 and convert AA into 20-HETE (45). This subcellular localization suggests distinctive biological opportunities and functions for CYP4 in pulmonary arteries. For example, possibly acting as an eNOS protein partner, CYP4 mediates VEGF-induced relaxation of small pulmonary arteries via enhanced NO release. VEGF and 20-HETE both phosphorylate eNOS and Akt, but neither enhances association of heat shock protein 90 (Hsp90) with eNOS (13, 30). Because 20- HETE mediates VEGF-induced NO release in bovine PAECs (BPAECs), one activity that these lipids might reasonably mediate is angiogenesis. Enhanced survival of PAECs is critical for recovery from lung injury, revascularization of transplanted tissues, growth and development of lungs, and others. Hence, the clinical implications of such actions, if proven, would be high.
In fact, CYP4 has been postulated to promote angiogenesis via NADPH oxidase and reactive oxygen species (ROS)-dependent mechanisms in systemic vascular beds (6, 41). Although deleterious effects of unchecked ROS are well-documented, compelling evidence now exists that ROS play a key role signaling vital physiological processes including cell growth, angiogenesis, contraction and relaxation of vascular smooth muscle, and others (7, 20). Because CYP4/20-HETE promotes angiogenesis, vascular tone, and eNOS function, we explored the potential of this system to enhance pulmonary ROS production. Our assumption is that if 20-HETE-evoked ROS is an important pathway to enhanced survival or proliferation of PAECs, we must first determine the capacity of this lipid product to modulate ROS production and the cellular mechanisms through which this effect is accomplished. We hypothesized that 20-HETE would enhance ROS production in PAECs in a manner that was associated with NADPH activation. Our data demonstrate that 1) 20-HETE increases ROS in isolated BPAECs, and 2) enhanced production is attributable, in part, to NADPH oxidase sources. 20-HETE increases membrane translocation and phosphorylation of p47phox and activation of Rac1/2 in BPAECs.
Materials and Methods
Materials
Apocynin (cat. no. 178385, Calbiochem), DPI (cat. no. D2926, Sigma), Rac assay (cat. no. BK035; Cytoskeleton, Denver, CO), 4-hydroxy-TEMPO (cat. no. 176141, Sigma-Aldrich), dihydroethidium (DHE; D7008, Sigma), dichlorofluorescein diacetate (DCFH-DA; C13293, Molecular Probes), trypsin (cat. no. 15400-054; Gibco, Carlsbad, CA), anti-CYP4 antibody (cat. no. ab622615, Abcam), polyclonal goat anti-p47 antibody (cat. no. 610354, BD Biosciences), and monoclonal anti-p47 antibody (cat. no. sc-17844, Santa Cruz). Mouse monoclonal anti-phosphotyrosine antibody (P-Tyr-100, cat. no. 9411) was obtained from Cell Signaling Technologies (Danvers, MA). Polyethylene-glycolated SOD (PEG-SOD, cat no. S-9549; 685 U/mg solid, 1 unit inhibited rate of reduction of cytochrome c by 50% in a coupled system with xanthine and xanthine oxidase at pH 7.8 at 25°C in a 3-ml reaction volume) and catalase (PEG-cat, cat. no. C-4963; 17,600 U/mg solid, 1 unit decomposed 1 μmol of H2O2/min at pH 7.0 at 25°C, whereas the H2O2 concentration falls from 10.3 to 9.2 mM) were acquired from Sigma. A chemical inhibitor of Rac1, NSC23766, was purchased from EMD Chemicals (cat. no. 553502). An enhanced chemiluminescence (ECL) kit was purchased from Amersham Biosciences, Piscataway, NJ (cat. no. 32106). A protease inhibitor cocktail was obtained from Roche, Mannheim, Germany (cat. no. 836 170). Protein determination kit (cat. no. 500-0006) and Protein Standard I (cat. no. 500-0005) were obtained from Bio-Rad.
A chimeric peptide, which inhibits association of p47phox with gp91 in NADPH oxidase, was synthesized by our protein core according to the sequence defined by Rey et al. (37) to test the contribution of NADPH oxidase to ROS production. The sequence of this peptide is [H]-R-K-K-R-R-Q-R-R-R-C-S-T-R-I-R-R-Q-L-NH2. The sequence of the scrambled (control) peptide is R-R-Q-R-R-R-C-L-R-I-T-R-Q-S-R-NH2. 20-HETE and 20-hydroxyeicosa-6,15-dienoic acid (20-6,15-HEDE, a 20-HETE antagonist) were synthesized in the laboratory of Dr. J. R. Falck (4).
Growth and culture of PAEC
BPAEC from small pulmonary arteries (<5 mm diameter) and bovine aortic endothelial cells (BAECs) were isolated (45) and cultured in RPMI media (cat. no. 11875-093, Gibco) containing 10% fetal bovine serum (cat. no. 16000-044, Gibco) and 1% penicillin-streptomycin (cat. no. 15140-122, Gibco) in 100-mm2 dishes. Tissues from which these cells were primarily isolated were obtained from a local abattoir with all protocols reviewed and approved by the Medical College of Wisconsin. When the cells were 80% confluent, they were washed with sterile PBS (cat. no. 14190-144, Gibco). The media was changed to serum-free RPMI before experiments, unless otherwise mentioned, to remove the effects of lipids and growth factors in the serum, and 0.1% bovine serum albumin was added instead as a lipid carrier. Treatments included 10 nM to 10 μM 20-HETE, 10 nM to 1 μM antagonist 20-6,15-HEDE, 1 μM apocynin, 20 μM NSC23766, PEG-SOD (100 or 250 U) or PEG catalase (500 U), and 50 μM peptide-based inhibitor of NADPH oxidase activity.
Detection of ROS by fluorescence microscopy
Primarily isolated BPAECs from passages 2 to 7 were used. Inhibitors of ROS were applied 30 min before loading with fluorescent dyes, DHE (final concentration 10 μM), or 2′,7′-dichlorodihydrofluorescein-DA (final concentration 5 μM). DHE is widely used as a superoxide probe since two-electron oxidation by superoxide of membrane-permeable DHE results in the impermeable fluorescent product ethidium (22). Likewise, the oxidation of the nonfluorescent 2′,7′-DCFH-DA to the intensely fluorescent 2′,7′-DCF is commonly used to detect the generation of reactive oxygen intermediates in neutrophils and macrophages, particularly hydrogen peroxide (12). However, DCF itself can also act as a photosensitizer for dichlorodihydrofluorescein-DA oxidation; in a cell-free system, dichlorodihydrofluorescein has been reported to be oxidized to DCF by peroxynitrite anion (ONOO−; Ref. 8). Therefore, we tested 20-HETE-induced fluorescence with both indicators as well as membrane-permeant superoxide dismutase and catalase to determine the contribution of superoxide and hydrogen peroxide, respectively, to the signals.
Images were acquired with a Nikon Eclipse TE200 microscope equipped with fluorescence attachment (Lambda DG-4, Sutter Instrument), captured using a Hamamatsu digital camera C4742-95, and processed using MetaMorph version 6.2 software as previously described (18, 19). Background fluorescence was estimated by capturing an image in an area free of cells and subtracted from the fluorescence intensity of cells on the same slide.
Western blot analysis
Western blot analyses were performed according to our previously published protocols (34, 45). Protein determination was estimated using a protein assay kit (Bio-Rad; see MATERIALS AND METHODS). Specific antibodies for Rac1/2, p47phox, and phosphotyrosine were matched with secondary antibodies and visualized with ECL Plus detection reagent (cat. no. RPN 2133, Amersham Biosciences). The blots were scanned using an Alpha Image 220 Analysis System (Alpha Innotech), and the relative densities of the bands from the same blot were compared. Each experiment was repeated at least four times using separate batches of cells to ensure reproducibility.
Cell fractionation
BPAECs were lifted with trypsin, washed with PBS, and lysed with 25 mM Tris·HCl, 1 mM EDTA, 1 mM EGTA, and cocktail of protease inhibitors, pH 7.4, sonicated for 5–10 s on ice in VirSonic 60 and centrifuged at 18,200 g for 10 min. The pellets contain the membrane fractions, which are solubilized with RIPA buffer (50 mM Tris·HCl, 150 mM NaCl, 0.25% deoxycholic acid, 1% Nonidet P-40, 1 mM EDTA, and cocktail of protease inhibitors, pH 7.4).
Rac assays
Rac activation assay Biochem Kit was purchased from Cytoskeleton. Two milligrams of protein from BPAEC lysates were used for each pull down. Cells were studied between 80–90% confluence in passages 4–5. In this assay, an epitope protein binds to the PAK-PBD domain (Rac effector protein, p21 activated kinase 1-p21 binding domain) in activated Rac associated with GTP. Positive controls were recombinant Rac1-His tagged protein. Proteins were separated by SDS-PAGE and identified in Western blots probed with a primary polyclonal antibody to Rac provided in the kit and used in a 1:250 dilution. Total Rac1 in cell lysates was estimated by density of Western blots obtained using the primary antibody in this kit.
Identification of phosphotyrosine in immunoprecipitated p47phox from BPAECs
BPAECs were grown to ∼80% confluence, washed, starved for 8 h, and then treated with vehicle or 1 μM 20-HETE for times as indicated for each experiment. After treatments, cells were lysed, and 500-μg proteins were subjected to immunoprecipitation with polyclonal goat anti-p47phox (cat. no. 610354, BD Biosciences) for 16 h. Immunoprecipitates were analyzed by Western blot with mouse monoclonal anti-phosphotyrosine antibody (1:000 dilution, P-Tyr-100, cat. no. 9411; Cell Signaling Technologies) as well as p47phox using a second monoclonal anti-p47 antibody (sc-17844). The relative densities of phospho-p47phox bands were compared in scanned and quantitated images of the blots using the Alpha Image 220 Analysis System detailed above.
Statistical analysis
For all DHE or DCF fluorescence studies, a minimum of 60 cells for each test condition and two separate isolations of cells were used. For all Western blots, a minimum of four studies from separate cell isolates were performed. Pooled data from each experiment were used to calculate the means ± SE for control (vehicle treated) or experimental (treated with 20-HETE, inhibitors, or inhibitors + 20-HETE) samples. The data were tested for significance by a Student's t-test (for 2-paired or unpaired samples) or Mann-Whitney rank sum test for three or more groups using the Jandel SigmaStat software. Experiments with P < 0.05 were considered significant.
Results
20-HETE increases superoxide in primary cultures of BPAECs in a concentration- and time-dependent manner
BPAECs (passage ≤7) were loaded with DHE (10 μM) and imaged with fluorescence microscopy (Fig. 1, A and B). In these experiments, 20-HETE (from 10 nM to 10 μM) or vehicle was added to the bath after baseline values were obtained, and then images were captured after 5 min. Vehicle (ethanol) alone did not change DHE fluorescence. 20-HETE at all concentrations >10 nM significantly increased fluorescence (Fig. 1A). The fluorescence response peaked at 1 μM 20-HETE and decreased at higher concentrations.
Fig. 1. Concentration-related response to 20-hydroxyeicosatetraenoic acid (20-HETE).
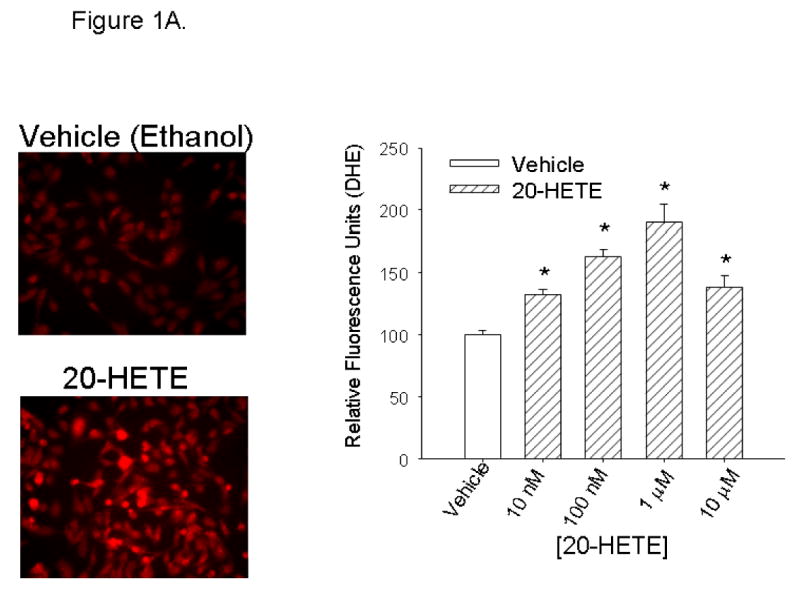
A: After baseline images were obtained, bovine pulmonary artery endothelial cells (BPAECs) were treated with ethanol vehicle or 20-HETE (concentrations appear on the x-axis) and then reimaged at 5 min. Not less than 60 cells for each experimental condition from at least 2 separate BPAEC isolations were imaged and analyzed for these experiments. MetaMorph analysis was used for semiquantitation of fluorescence intensity. Representative images of cells treated with ethanol vehicle or 20-HETE appear on the left with averaged data on the right. Coarse diagonal hatched bars represent data from groups of cells that were treated with 20-HETE; open bars represent data from vehicle-treated cells. Error bars represent SE of the mean in this and all figures. *Denote values that are different from vehicle control baseline (P < 0.05). The value labeled as “vehicle” in the graph, and to which fluorescence of other treatment groups were normalized, represents the normalized mean fluorescence of ethanol-treated cells 5 min after application of this vehicle. The vehicle (ethanol) resulted in no significant change in dihydroethidium (DHE) fluorescence. In contrast, 20-HETE increases fluorescence of BPAECs over vehicle control for all concentrations greater than or equal to 10 nM in a manner that peaks at a concentration of 1 μM. B: time-dependent response to 20-HETE. To determine the optimum time to capture DHE fluorescence in BPAECs treated with 20-HETE, we performed additional experiments to investigate the peak time-dependent response to this lipid. More than 60 cells from 2 or more isolates per experimental condition were imaged and analyzed for these experiments. After loading with DHE and washing unincorporated DHE from the cells, baseline images were acquired. Then, vehicle or 1 μM 20-HETE was added to media, and images were acquired at the time indicated on the x-axis (in minutes). Open bars represent data from cells treated with ethanol (vehicle). Diagonal crossed bars show data from cells treated with 20-HETE. Data acquired at time 0 were obtained within 5–30 s after the addition of test substance. For values at 20, 30, and 60 min, values of cells treated with 20-HETE are normalized to baseline values of cells treated with vehicle. *P < 0.05 relative to paired controls for the same time period. Baseline data were not different between the groups, and the addition of either vehicle or 20-HETE had no immediate effect on DHE fluorescence (t = 0). Fluorescence values obtained 5 min after the addition of test substance were substantially higher in cells treated with 20-HETE than vehicle. DHE fluorescence in 20-HETE treated cells peaked in ∼5 min and is maintained above baseline values for at least 60 min thereafter. DHE fluorescence of BPAECs treated with ethanol alone did not have fluorescence values different from baseline at any time through 60 min. ns, not significant.
Next, we tested time-dependent responses to a fixed concentration of 20-HETE, which gave maximal increments in DHE fluorescence (Fig. 1B). In these experiments, cells were preincubated with DHE for 20 min and washed, and then 1 μM 20-HETE was added. Images were acquired at the times indicated on the x-axis of Fig. 1B after addition of 20-HETE. Vehicle (ethanol alone) had no effect on DHE fluorescence. Similarly, values obtained within the first 15 s after the addition of 20-HETE were not different from those of vehicle alone (t = 0; see Fig. 1B). The peak intensity in DHE fluorescence was elicited within 5 min of application of the lipid (Fig. 1B). Thereafter, increased DHE fluorescence was noted for at least 60 min in cells treated with 20-HETE relative to that of cells treated with vehicle. For further experiments detailed below, we evaluated the effect of 1 μM 20-HETE on DHE fluorescence 5 min after application to the bath unless otherwise noted in the text.
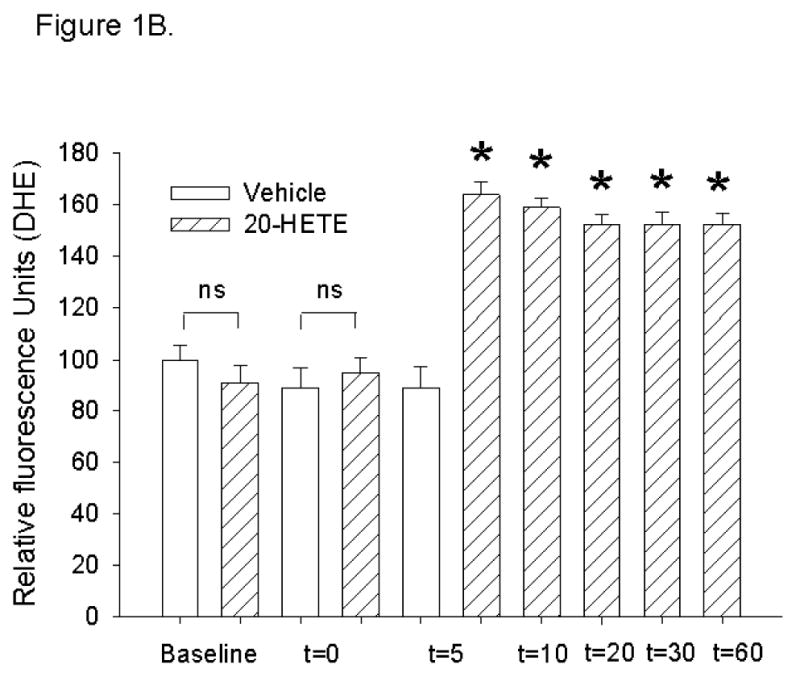
To examine the dependence of increase in DHE fluorescence in BPAECs treated with 20-HETE on superoxide, we tested the capacity of PEG-SOD, a cell-permeable superoxide dismutase, to blunt DHE increases in cells treated with 20-HETE (24). In cells pretreated with 100 or 250 U PEG-SOD, the addition of 20-HETE to the bathing solution evoked no increase in the DHE signal (Fig. 2). These data support a contribution of superoxide to 20-HETE-evoked DHE fluorescence signals.
Fig. 2. The increase in DHE in cells treated with 20-HETE is blocked by pretreatment with polyethylene-glycolated SOD (PEG-SOD).
To examine whether the increase in DHE fluorescence in BPAECs treated with 20-HETE represents increased superoxide, we tested the potential of PEG-SOD, a cell-permeable superoxide dismutase, to blunt the signal evoked in cells treated with 20-HETE. More than 60 cells for each test condition were analyzed. Cells pretreated with 100 or 250 U PEG-SOD (cat. no. S-9549; 685 U/mg solid, 1 unit inhibited rate of reduction of cytochrome c by 50% in a coupled system with xanthine and xanthine oxidase at pH 7.8 at 25°C in a 3-ml reaction volume) and 20-HETE vehicle (cross hatched fill) had no change in DHE fluorescence after the addition of 20-HETE (*P < 0.05). These data support a contribution of superoxide to 20-HETE-evoked DHE signals in our cells.
20-HETE increases H2O2 in BPAECs
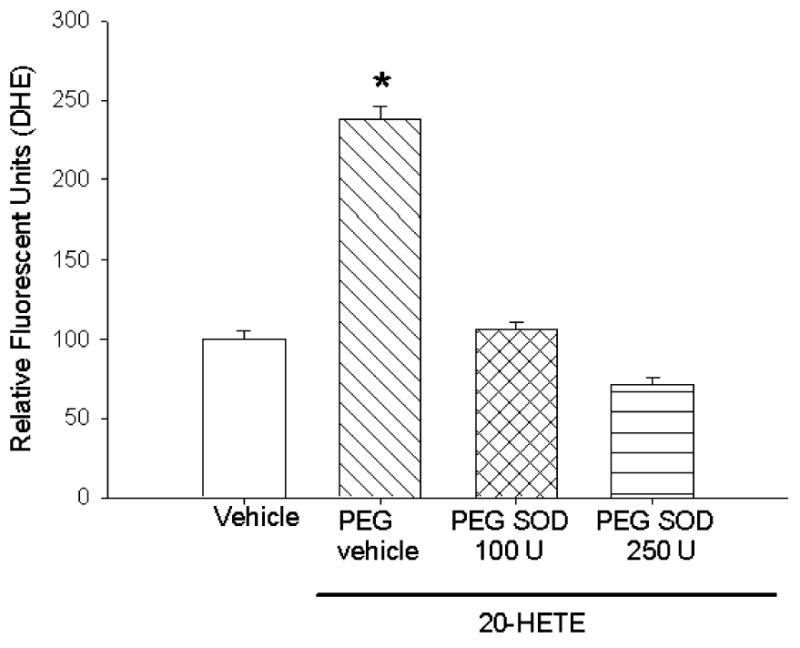
Because superoxide is rapidly dismuted in vivo to hydrogen peroxide, we tested the capacity of 20-HETE to increase H2O2 in BPAECs. 2′,7′-Dichlorodihydrofluorescein-DA is a fluoroprobe that, after deesterification by intracellular esterases, undergoes oxidation to a fluorescent compound, DCF. DCF fluorescence in BPAECs preincubated with 5 μM probe was increased by the addition of 20-HETE, but not vehicle, to the bath (Fig. 3). To help exclude potential nonspecific fluorescence of DCF, we tested the potential of PEG catalase (21) to blunt the 20-HETE-induced DCF signal in these cells. We observed effective inhibition of 20-HETE-induced increase in fluorescence in cells treated with 500 U PEG catalase.
Fig. 3. 20-HETE increases dichlorofluorescein (DCFH) fluorescence.
Similar to experiments with DHE, BPAECs were preincubated with 5 μM DCF for 20 min and then imaged for semiquantitation of fluorescent values using MetaMorph 5 min after application of 1 μM 20-HETE (diagonal crossed bars) or vehicle. More than 60 cells for each experimental condition were imaged and analyzed for these experiments. Representative images appear on the left with averaged values shown in bar graphs on the right. Application of ethanol vehicle alone had no effect on DCF fluorescence (open bars), whereas 20-HETE (diagonal cross hatched bars) increased these values 5 min after the addition of the lipid to the bath. Pretreatment with 500 U PEG catalase (PEG-cat; cat. no. C-4963, 17,600 U/mg solid, 1 unit decomposed 1 μmol of H2O2/min at pH 7.0 at 25°C, whereas the H2O2 concentration falls from 10.3 to 9.2 mM) effectively blocked 20-HETE-induced DCF fluorescence. *P < 0.05 relative to vehicle control.
20-HETE-evoked ROS production is not mimicked by structurally similar lipids
The specificity of 20-HETE in producing ROS in BPAECs was tested by use of a structurally similar lipid, 20-6,15-HEDE, which, compared with 20-HETE, is missing two double bonds (between carbons 8-9 and 11-12) and has the NH2 and COOH terminus CO2 H and OH groups switched (Fig. 4). This compound does not cause constriction of renal or cerebral arteries as does 20-HETE but is reported to function as an antagonist to 20-HETE in renal arteries (4). As opposed to 20-HETE, this compound did not evoke increases in ROS production over that of vehicle control (Fig. 4) with 10 nM 20-6,15-HEDE decreasing ROS production over that of vehicle alone. Moreover, pretreatment with this analog significantly blunted 20-HETE-evoked increases in ROS, the most effective concentration in this regard being 100 nM. These data support the position that 20-HETE-induced change in ROS production in BPAECs does not represent a bulk or nonspecific lipid effect and that this analog acts as an antagonist in BPAECs as well as renal arteries.
Fig. 4. 20-HETE response is specific.
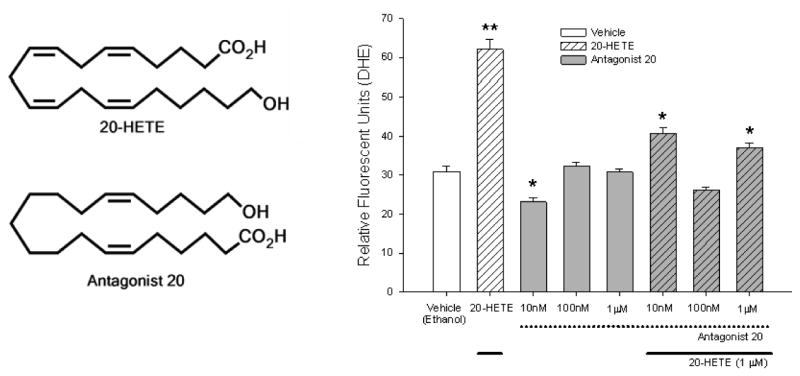
The specificity of 20-HETE in producing reactive oxygen species (ROS) in BPAECs was tested by use of a structurally similar lipid, antagonist 20-hydroxyeicosa-6,15-dienoic acid (20-6,15-HEDE; Antagonist 20 in the figure). The chemical structures of 20-HETE and antagonist 20-6,15-HEDE are shown on the left. More than 20 cells for each experimental group and with no less than 60 cells per group were imaged and analyzed for these experiments. For each 20-HETE or vehicle, measurements were obtained 5 min after adding test substance. 20-HETE was applied at a final concentration of 1 μM for all test cells receiving this treatment in this set of experiments. The concentrations on the x-axis refer to final concentrations of Antagonist 20, which were applied 15 min before addition of 20-HETE (20 min total before imaging). Dotted lines below the x-axis and gray fills in the bars denote groups of cells pretreated with Antagonist 20. Solid lines below the x-axis and diagonal cross hatches in the bars identify cells treated with 20-HETE. *P < 0.05 relative to vehicle alone; **P < 0.05 relative to all other treatments. 20-HETE nearly doubled DHE fluorescence over that of ethanol vehicle alone. A concentration response to Antagonist 20 was performed next, which demonstrated ROS production below that of vehicle control for cells treated with 10 nM and values indistinguishable from those of vehicle control for final concentrations of 100 nM and 1 μM. Next, the capacity of Antagonist 20 to block 20-HETE-induced increases in DHE fluorescence was tested. Although all concentrations, including the lowest concentration we tested, 10 nM, blocked 20-HETE-stimulated DHE fluorescence relative to cells untreated with Antagonist 20, the most effective concentration of Antagonist 20 in blocking 20-HETE-evoked fluorescence was 100 nM. These data support the specificity of the ROS response to 20-HETE in these cells as well as the function of 20-6,15-HEDE as an antagonist with respect to stimulated DHE fluorescence in BPAECs.
Vascular bed-independent effects of 20-HETE on ROS production
To test the effect of 20-HETE on ROS production in endothelial cells from a different vascular bed, we examined the capacity of this lipid to change DHE fluorescence in BAECs, which do not express CYP4A (45). BAECs treated with 1 μM 20-HETE exhibited an ∼2-fold increase in DHE fluorescence above baseline (Fig. 5), quantitatively similar to the response of BPAECs albeit with a peak at 10 rather than 5 min. These data demonstrate that endothelial cells from systemic as well as pulmonary vascular beds respond to 20-HETE with increased ROS production. They also suggest that endogenously generated 20-HETE is not essential for the effect of exogenous 20-HETE to increase acutely intracellular superoxide.
Fig. 5. 20-HETE increases DHE fluorescence in bovine aortic endothelial cells (BAECs).
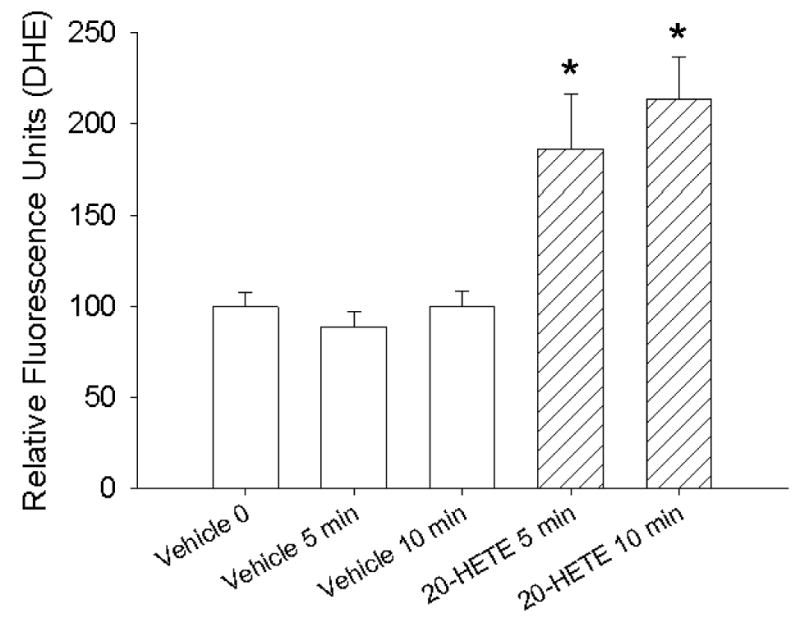
We examined the response of BAECs to 20-HETE. These investigations were completed, as were those in Fig. 1 for BPAECs, with loading of the cells for 20 min with DHE, removing unincorporated DHE from the media, and then adding vehicle or 20-HETE with image captured 5 or 10 min later. Fluorescence values for all groups are normalized to that of cells loaded with DHE immediately after the addition of vehicle (ethanol, open bars) at time 0. n = 3 groups of cells with at least 20 cells for each test condition from each group imaged and analyzed for these studies. *Denote values that are different from vehicle control baseline (P < 0.05). 20-HETE (diagonal hatched bars) evoked an increase in DHE fluorescence in BAECs in a manner similar in magnitude and character as that observed in BPAECs, although the peak response appears modestly delayed (at 10 rather than 5 min) compared with that in BPAECs.
NADPH oxidase inhibitors block 20-HETE-evoked increases in ROS
To investigate the contribution of NADPH oxidase to 20-HETE-evoked ROS production, we pretreated BPAECs with 1 μM apocynin (32), a chemical inhibitor of NADPH oxidase, for 30 min. This treatment largely blocked 20-HETE-induced increases in ROS production as detected by DHE (Fig. 6). These data suggest that 20-HETE may enhance ROS in these cells at least in part through activation of NADPH oxidase.
Fig. 6. NADPH oxidase inhibitor apocynin blocks 20-HETE-evoked increases in ROS.
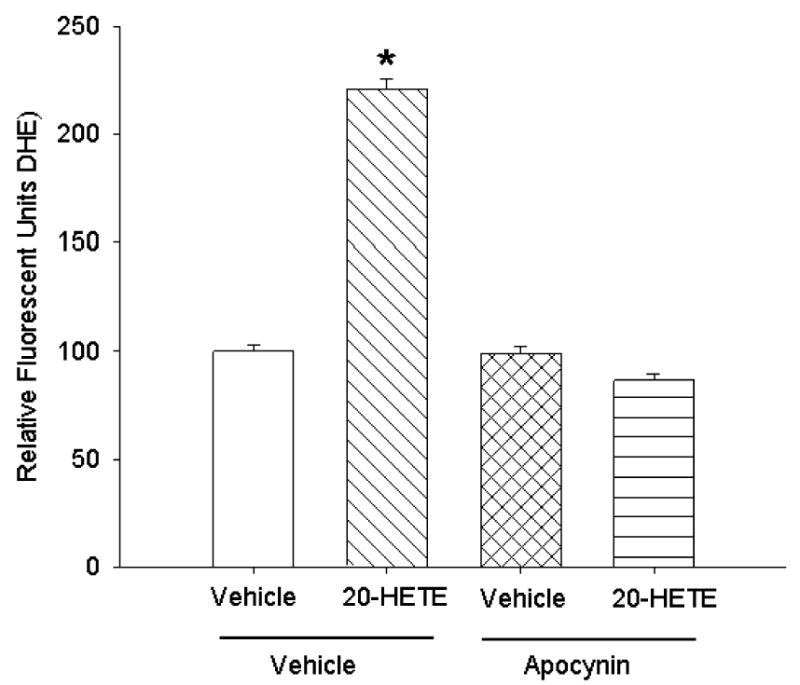
To investigate the contribution of NADPH oxidase to 20-HETE-evoked ROS, we pretreated BPAECs cells with 1 μM apocynin for 30 min (n = 3 groups of cells with more than 20 cells per group for each treatment analyzed). Apocynin pretreatment blunted 20-HETE-induced increases in ROS (*denotes P < 0.05 relative to vehicle control). These data support a contribution of NADPH oxidase to 20-HETE increases in ROS.
We also tested a second, peptide-based inhibitor of the association of p47phox to gp91 (Fig. 7; Ref. 37). Treatment with this peptide effectively blocked 20-HETE-evoked ROS production, consistent with a NADPH oxidase contribution to this endpoint. A scrambled peptide also partially blocked 20-HETE-evoked increases in ROS, suggesting that some component of the peptide-associated protection is nonspecific.
Fig. 7. Peptide-based inhibitor of NADPH oxidase blocks 20-HETE-evoked ROS.
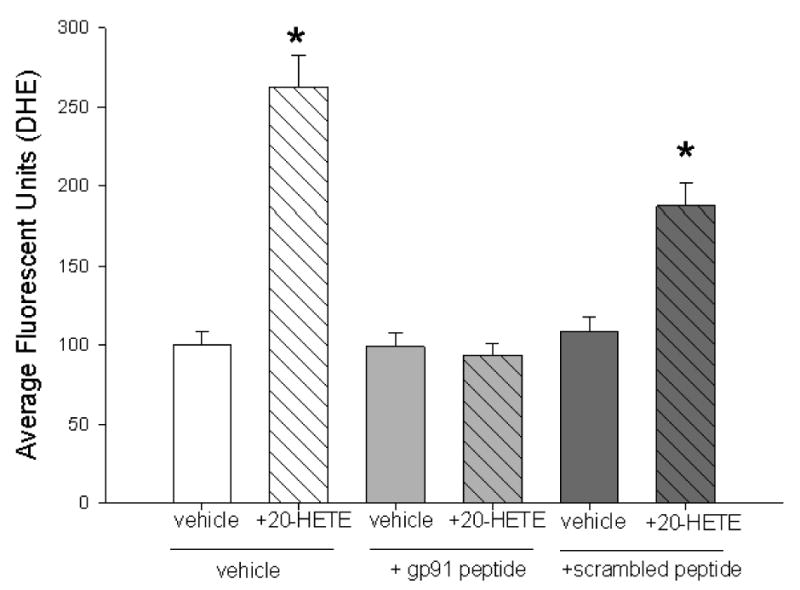
We tested the action of a mechanistically distinct inhibitor or NADPH oxidase, a peptide that blocks the interaction of p47phox with gp91, on 20-HETE-induced fluorescence of DHE in BPAECs. In these studies, pretreatment with 50 μM peptide alone for 30 min at 37°C (light gray filled bar indicates +gp91 peptide below) caused no change in fluorescence over vehicle alone. However, treatment with this peptide effectively prevented the 20-HETE (diagonal cross hatched pattern)-induced increase in DHE fluorescence. In contrast, treatment with the scrambled peptide (dark gray fill, bar below indicates scrambled peptide) partially blocked 20-HETE-induced increases in DHE signal (n = 3 groups of cells and >60 cells imaged for each test condition; *P < 0.05 relative to vehicle control). Together with the data from cells treated with apocynin, these observations provide evidence of NAPDH oxidase participation in 20-HETE-evoked ROS production.
Rac1/2 contributes to 20-HETE-induced ROS production
Test groups of BPAECs were pretreated for 30 min with 20 μM Rac1 inhibitor, which impairs ability of Rac1 to interact with intracellular effectors (NSC23766; Ref. 17), or vehicle and then loaded with DHE and imaged after treatment with 20-HETE or vehicle for this lipid. Cells treated with the Rac1 inhibitor exhibited no increase in ROS production as tracked by DHE, whereas those treated with vehicle for this compound did (Fig. 8). These data support a role for Rac1 signaling in 20-HETE-induced ROS production of BPAECs.
Fig. 8.
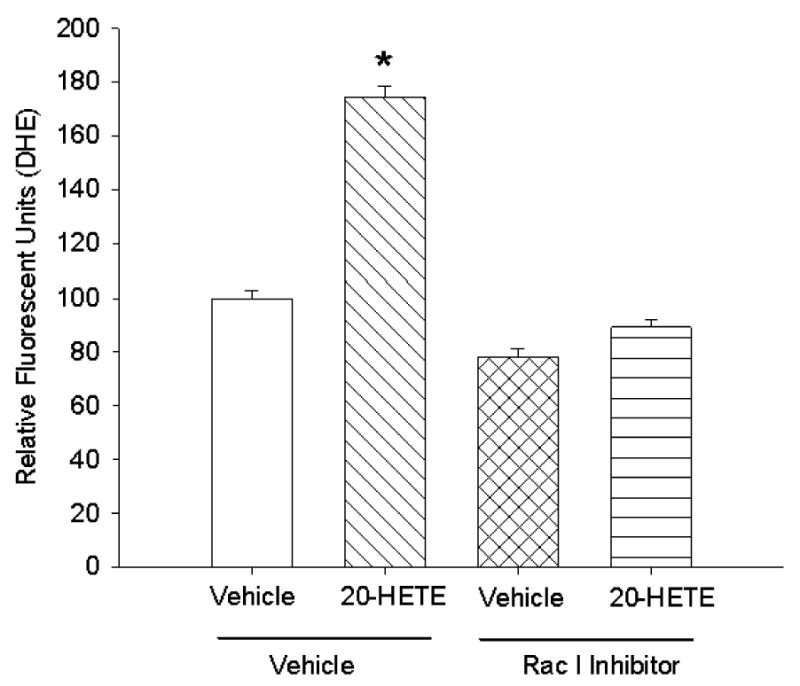
To test the contribution of Rac1/2 to 20-HETE-induced DHE fluorescence, we pretreated cells with 20 μM Rac1 inhibitor NSC23766 (17) or vehicle for 30 min before loading with DHE and imaging for 20-HETE-induced fluorescence. Pretreatment with this compound effectively blocked 20-HETE-evoked increases in fluorescence, consistent with activation of this signaling pathway in ROS production associated with this lipid (n = 4 groups of cells with >60 cells per test condition imaged and analyzed; *P < 0.05 relative to vehicle control).
Treatment with 20-HETE translocates and phosphorylates p47phox
Pulmonary vascular endothelia express at least two forms of NADPH oxidase, Nox2 and Nox4. Nox2 is activated by the recruitment of cytosolic component p47phox to the membrane (25, 33) and phosphorylation of serine (23) and tyrosine (16) residues. To further examine the mechanism of 20-HETE-induced activation of NADPH oxidase in BPAECs, we studied the effect of 1 μM 20-HETE on the translocation and phosphorylation of p47phox-associated tyrosine. Treatment with 20-HETE increased membrane-associated p47phox 10 min after application of the lipid over that of vehicle (ethanol) alone (Fig. 9).
Fig. 9. 20-HETE promotes membrane translocation of p47phox in BPAECs.
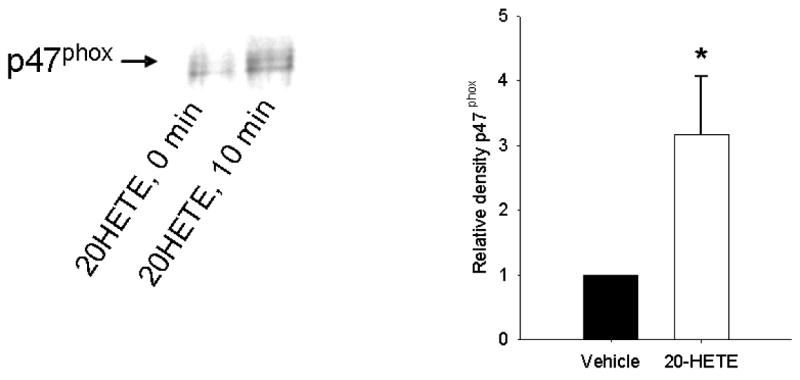
20-HETE (1 μM) or vehicle was added to BPAECs passages 3–8. Ten minutes later, cells were harvested, lysed, and centrifuged per protocol in MATERIALS AND METHODS for enhancement of membrane-associated proteins. Membrane fractions were separated by SDS-PAGE and then probed with a primary antibody to p47phox (cat. no. sc-17844, Santa Cruz). A representative example appears on the left with averaged data from 5 experiments 10 min after application of 20-HETE on the right. 20-HETE increased the density of p47phox immunospecific protein over that of vehicle-treated controls (n = 4 isolates of cells; *P < 0.05 relative to vehicle control).
Next, we immunoprecipitated BPAEC lysates with a polyclonal antibody to p47phox and then probed for phosphorylated tyrosine (as well as p47phox using a monoclonal antibody for an internal control). 20-HETE increased tyrosine phosphorylation of immunoprecipitated p47phox in a manner that peaked ∼10 min after exposure to 20-HETE (Fig. 10).
Fig. 10. 20-HETE increase phosphorylation of p47phox in BPAECs.
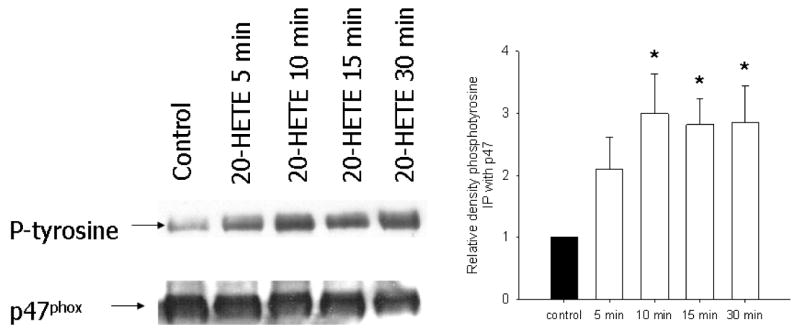
Because 1 mechanism of NADPH oxidase activation includes phosphorylation of p47phox, we tested the potential of 20-HETE to modify this endpoint. p47phox Was immunoprecipitated from BPAECs treated with vehicle or 1 μM 20-HETE for 5, 10, 15, or 30 min and then probed with a primary antibody to phosphotyrosine (n = 4 isolates of cells; see MATERIALS AND METHODS for sources of antibodies). A representative experiment appears on the left. In the graph on the right, open bars indicate data from cells treated with 20-HETE, filled bar represents baseline control, and * indicates increase over vehicle control P < 0.05. In cells treated with 20-HETE, tyrosine phosphorylation of p47phox was elevated over that of control at 10 min.
Rac1/2 activation increases with 20-HETE
We examined the effect of 20-HETE on GTP-bound Rac1/2 in BPAECs with the hypothesis that Rac1/2 activation may promote 20-HETE-induced ROS production. 20-HETE increased the density of GTP-bound Rac in BPAECs. This response peaked between 10–20 min after exposure to the lipid (Fig. 11). These observations are consistent with reports by Ushio-Fukai et al. (39, 40) and Bedard and Krause (7) that associate activation of NADPH oxidase with activation and recruitment of Rac1/2 to the membrane in vascular cells.
Fig. 11. 20-HETE increases Rac1/2 in BPAECs.
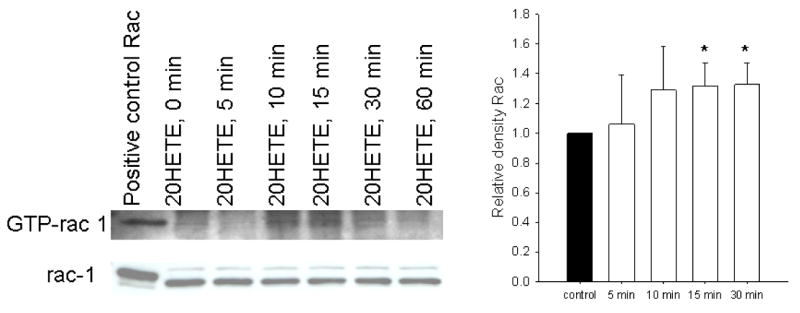
Lysates of BPAEC 80–95% confluence in passages 4 and 5 were separated using an epitope protein that binds to the PAK-PBD domain (Rac effector protein, p21 activated kinase 1-p21 binding domain) to capture activated Rac associated with GTP. After capture, proteins in Western blots were probed with a primary polyclonal antibody to Rac1/2. The positive control for these experiments is histidine-tagged recombinant Rac1. To determine the effect of 20-HETE on total Rac1/2, crude cell lysates from cells treated identically to those used for capture of activated Rac were probed with the primary antibody to Rac1/2 provided in the kit. A representative image from these experiments appears here. Exposure to 20-HETE increased activated Rac1/2 in a manner that peaked within 10 min (n = 4 isolates of cells studied through 30 min; *P < 0.05 relative to vehicle control), and values remained elevated above vehicle control for 30 min. Total Rac1/2 is not affected by 20-HETE treatment in this time frame.
Discussion
Our studies confirm, for the first time, acute 20-HETE induced time- and concentration-dependent enhanced production of superoxide in BPAECs. This conclusion is supported by 20-HETE-induced signals of DHE- and DCF-derived fluorescent product(s), which peaked within 5 min of application of the lipid, and blunting of the signal by pretreatment of the cells with PEG-SOD and PEG catalase, respectively. DHE signal increased in cells treated with 20-HETE but not vehicle or a structurally similar analog that acts as an antagonist in renal or cerebral vessels (4, 38). The signal detected by DHE should represent superoxide (44), but other metabolites are almost surely generated as well. Moreover, hydrogen peroxide rather than superoxide may be the effector ROS product stimulated by 20-HETE. Because we did not explore further the chemical nature of 20-HETE-induced ROS products, we refer in this work to the products promoted by 20-HETE simply as ROS.
Regulation of NADPH oxidase in renal arteries by CYP4 was recently reported by Wang et al. (41). In that study, overexpression of CYP4A using adenoviral vectors carrying the CYP4A construct (administered via intravenous administration) in Sprague-Dawley rats increased synthesis of 20-HETE in renal interlobar arteries as determined by gas chromatography-mass spectrometry (GC/MS) analysis. Rats overexpressing CYP4A demonstrate increased generation of superoxide and increased expression of gp91 (41). The contribution of endothelium to ROS production, or the acute effect of 20-HETE itself (as opposed to CYP4 overexpression), was not examined. We are particularly interested in the influence of 20-HETE on pulmonary artery endothelial cell ROS production because CYP4 is uniquely expressed in these cells, whereas it has not been reported to be so in endothelium from other vascular beds. Our studies are the first to report enhanced ROS production in BPAECs treated with 20-HETE.
This work also shows that enhanced production of ROS by 20-HETE is not a nonspecific effect of lipids and that 20-HETE stimulates ROS in aortic endothelial cells from the same species in a similar magnitude as endothelium from small pulmonary arteries. CYP4A protein is not expressed in BAECs, but vascular smooth muscle cells from many systemic, as well as pulmonary, circulations express CYP isoforms that are capable of metabolizing AA to 20-HETE (38). Moreover, our data imply that BAECs as well as BPAECs have the cell signaling mechanisms to increase ROS in response to 20-HETE. Therefore, it is possible that smooth muscle-derived 20-HETE acts as a paracrine factor, stimulating ROS release in subjacent endothelial cells. There is precedent for paracrine actions of endothelial and smooth muscle-derived factors: NO decreases the state of activation of cerebral and renal arteries through inactivation with covalent interaction of the prosthetic heme group to CYP4A enzymes (e.g., Ref. 5).
We also performed some experiments to examine the source of 20-HETE-evoked ROS production in BPAECs. Although ROS may be produced from several cellular sources, including mitochondrial electron transport chain, xanthine oxidase, CYP, uncoupled NOS, and NADPH oxidases (36), the dominant sources in endothelium are believed to be mitochondrial electron transport and NADPH oxidase (36). Indeed, there is excellent evidence of “cross talk” between mitochondrial and NADPH oxidase sources of ROS in pulmonary artery endothelial cells, suggesting that there may not be a simple or single contributor to enhanced ROS production in any cell or tissue type. For example, hyperoxia induces ROS from mitochondrial sources in pulmonary capillary endothelial cells but subsequently activates NADPH oxidase through calcium-dependent and Rac1-dependent mechanisms (9). Our data demonstrate that inhibition of NADPH oxidase by two mechanistically distinct agents (1 peptide-based inhibitor and 1 chemical) significantly blunted 20-HETE-evoked increases in ROS in BPAECs. There is evidence that NADPH oxidase may be a particularly important form of ROS in BPAECs. In support of this view, Al-Medhi et al. (2) have identified endothelial NADPH oxidase as a primary source of ROS in ischemic lungs. Gupte and colleagues (27) have reported that bovine pulmonary arteries generate 60–80% more superoxide than similarly sized bovine coronary arteries, and >50% of this signal was abolished by the NADPH oxidase inhibitor, apocynin, in pulmonary arteries.
Guo et al. (26) recently reported enhanced DHE fluorescence in human umbilical vein and dermal microvascular endothelial cells treated with 20-HETE and an analog of this compound, WIT003. Our results stand in contrast to those of these investigators (26) in the dependence of 20-HETE-stimulated ROS on NADPH oxidase in endothelial cells. Most of the experiments of Guo et al. (26) were performed with 20-HETE analog, WIT003, at concentrations higher than those of 20-HETE we used, so we cannot exclude differences in signaling pathways based on these factors. Also important may be the different origin of endothelial cells investigated in the two studies. Human dermal microvascular endothelial cells may well express different Nox isoforms and/or activate signaling pathways distinct from those of pulmonary artery endothelial cells. Finally, the experimental approach employed by Guo et al. (26) was different from ours, focusing on 20-HETE-mediated signaling on VEGF-induced ROS production and signaling in endothelial cells rather than effect of 20-HETE on components of the NADPH oxidase as we did.
To obtain further insight, we investigated mechanisms through which 20-HETE may activate NADPH oxidase. In its activated form, NAPDH oxidase is a multimeric protein consisting of at least three cytosolic subunits, including p47phox, p67phox, p40phox, either Rac1 or Rac2, and a membrane-associated cytochrome reductase complex consisting of gp91 and p22phox (e.g., Refs. 16, 33). Activation of vascular NADPH oxidase (Nox1 and Nox2) is characterized by recruitment of p47phox from cytosolic to membrane pools and phosphorylation of at least three amino acid residues (33). Phosphorylation of p47phox from human neutrophils occurs at multiple serine residues (23). However, Src-mediated tyrosine phosphorylation of p47phox is associated with activation of NADPH oxidase and generation of ROS in human PAECs (16). Results of the present studies demonstrate that 20-HETE promotes translocation of p47phox to the membrane and enhances tyrosine phosphorylation of p47phox in BPAECs. Moreover, activation of Nox1 and Nox2 is promoted by binding of Rac1 to Noxa1 or other regulatory subunits (e.g., Refs. 7, 15, 39). Our data show that 20-HETE increases GTP-bound Rac1/2 in BPAECs, another feature characteristic of NADPH oxidase activation. Activation of Rac1 is necessary for 20-HETE-induced ROS production in our BPAECs in that inhibition of this pathway blocks HETE-evoked fluorescence. Thus, in addition to blunted 20-HETE-evoked ROS production in BPAECs by NADPH oxidase and Rac1/2 inhibition, we show for the first time that, in BPAECs, 20-HETE acts on three endpoints in the signaling pathway of NADPH activation: translocation and tyrosine phosphorylation of p47phox and activation of Rac1/2. Based on the sum of these observations, we speculate that one mechanism through which 20-HETE increases ROS production in BPAECs is through activation of NADPH oxidase. It is interesting that the peak in 20-HETE-evoked DHE fluorescence appears within ∼5 min, whereas phosphorylation and Rac activation in stimulated cells peaks 5–10 min later. It is possible that crude temporal resolution accounts for this difference in peak responses. Also, some mechanisms of NADPH oxidase activation other than the endpoints examined (and there are many) or cross talk between mitochondrial and NADPH oxidase-stimulated ROS production may be responsible for this difference (3). If so, 20-HETE may be as important in sustaining or amplifying stimulated ROS production as initiating it.
Whereas both Nox2 and Nox4 are expressed in pulmonary artery endothelial cells, only Nox2 is activated by binding of p47phox (31). Therefore, given 20-HETE-induced translocation and tyrosine phosphorylation of p47phox and Rac1/2 activation in BPAECs and apocynin- and peptide-based inhibition of NADPH oxidase on evoked ROS production, we believe that 20-HETE affects at least Nox2 isoforms in these cells.
What might be the implications of stimulated ROS production in BPAECs? ROS including superoxide and hydrogen peroxide signal a host of processes in endothelial cells including stimulation of growth and migration and activation of transcription factors and protein kinases including ERK, p38 MAPK, and Akt (20, 28). Uncontrolled release of particularly mitochondrial-derived ROS has been associated with pathologies such as diabetes and Alzheimer's. Impaired pulmonary arterial endothelial dysfunction (blunted acetylcholine-induced relaxation) in mice exposed to hyperoxia depends on gp91phox, which appears to trigger rather than directly mediate impaired NO-dependent relaxation (24). Thus some actions of increased ROS production and activation of NADPH oxidase in pulmonary vascular cells promote dysfunction.
However, these oxygen products are also essential to physiological signaling in vascular cells (e.g., Refs. 36, 42). Hydrogen peroxide functions as a dilator of pulmonary arteries (10, 11). Furthermore, hydrogen peroxide in nanomolar-to-micromolar concentrations stimulates proliferation and migration of BAECs (43). Human coronary and dermal microvascular endothelial cells require ROS derived from NADPH oxidase for proliferation and migration (1). Moreover, hydrogen peroxide transactivates mitochondrially derived growth factors and downstream signaling in human umbilical vein endothelial cells, further supporting a role for ROS in mediating endothelial cell growth and proliferation (14). Angiogenesis induced by VEGF-suffused subcutaneously implanted sponges is deficient in Nox2 knockout mice, implicating both NADPH oxidase and ROS in this process (40). Although the implications of our observations remain to be determined, the above detailed roles of ROS in vascular cells raise the possibility that CYP4/20-HETE-stimulated ROS in BPAECs may serve to promote vascular repair after acute lung injury, severe pneumonia, or other conditions of injury as well as modulate pulmonary vascular tone.
Acknowledgments
We gratefully acknowledge the participation and input from investigators in the Free Radical Research Center and the Cardiovascular Center at the Medical College of Wisconsin. We also thank Laurel Dunn for help in processing the manuscript. Financial support was provided by NIH Grants HL49294 (ER Jacobs), HL68627 (ER Jacobs), HL069996 (M Medhora), GM 31278 (JR Falck) and the Robert A. Welch Foundation (JR Falck).
References
- 1.Abid MR, Kachra Z, Spokes KC, Aird WC. NAPDH oxidase activity is required for endothelial cell proliferation and migration. FEBS Letter. 2000;486:252–256. doi: 10.1016/s0014-5793(00)02305-x. [DOI] [PubMed] [Google Scholar]
- 2.Al-Medhi AB, Zhao G, Dodia C, Tozawa K, Costa K, Muzykantov V, Ross C, Blecha F, Dinauer M, Fisher AB. Endothelial NADPH oxidase as the source of oxidants in lungs exposed to ischemia or high K+ Circ Res. 1998;83(7):730–7. doi: 10.1161/01.res.83.7.730. [DOI] [PubMed] [Google Scholar]
- 3.Aley PK, Porter KE, Boyle JP, Kemp PJ, Peers C. Hypoxic modulation of Ca2+ signaling in human venous endothelial cells. Multiple roles for reactive oxygen species. J Biol Chem. 2005;280(14):13349–54. doi: 10.1074/jbc.M413674200. [DOI] [PubMed] [Google Scholar]
- 4.Alonso-Galicia M, Falck JR, Reddy KM, Roman RJ. 20-HETE agonists and antagonists in the renal circulation. Am J Physiol. 1999;277:F790–F796. doi: 10.1152/ajprenal.1999.277.5.F790. [DOI] [PubMed] [Google Scholar]
- 5.Alonso-Galicia M, Hudetz AG, Shen H, Harder DR, Roman RJ. Contribution of 20-HETE to vasodilator actions of nitric oxide in the cerebral microcirculation. Stroke. 1999;30:2727–2734. doi: 10.1161/01.str.30.12.2727. [DOI] [PubMed] [Google Scholar]
- 6.Amaral SL, Maier KG, Schippers DN, Roman RJ, Greene AS. CYP4A metabolites of arachidonic acid and VEGF are mediators of skeletal muscle angiogenesis. Am J Physiol Heart Circ Physiol. 2003;284(5):H1528–35. doi: 10.1152/ajpheart.00406.2002. [DOI] [PubMed] [Google Scholar]
- 7.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidase: physiology and pathophysiology. Phys Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 8.Bilski P, Belanger AG, Chignell CF. Photosensitized oxidation of 2′,7′-dichlorofluorescin: singlet oxygen does not contribute to the formation of fluorescent oxidation product 2′,7′-dichlorofluorescein. Free Radic Biol Med. 2002;33:938–46. doi: 10.1016/s0891-5849(02)00982-6. [DOI] [PubMed] [Google Scholar]
- 9.Brueckl C, Kaestle S, Kerem A, Habazettl H, Krombach F, Kuppe H, Kuebler WM. Hyperoxia-induced reactive oxygen species formation in pulmonary capillary endothelial cells in situ. American Journal of Respiratory Cell and Molecular Biology. 2006;34:453–463. doi: 10.1165/rcmb.2005-0223OC. [DOI] [PubMed] [Google Scholar]
- 10.Burke-Wolin T, Abate C, Wolin M, Gurtner GH. Hydrogen peroxide-induced pulmonary vasodilation: role of guanosine 3′5′-cyclin monophosphate. Am J Phys. 1991;261(Lung Cell Mol Physiol 5):L393–L398. doi: 10.1152/ajplung.1991.261.6.L393. [DOI] [PubMed] [Google Scholar]
- 11.Burke-Wolin T, Wolin M. H2O2 and cGMP may function as an O2 sensor in the pulmonary artery. Journal Applied Phys. 1989;66(1):167–170. doi: 10.1152/jappl.1989.66.1.167. [DOI] [PubMed] [Google Scholar]
- 12.Casado JA, Merino J, Cid J, Subira ML, Sanchez-Ibarrola A. Simultaneous evaluation of phagocytosis and Fc gamma R-mediated oxidative burst in human monocytes by a simple flow cytometry method. J Immunol Methods. 1993;159:173–176. doi: 10.1016/0022-1759(93)90155-z. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y, Medhora MM, Falck JR, Pritchard KA, Jacobs ER. Mechanisms of activation of eNOS by 20-hydroxyeicosatetraenoic acid and VEGF in bovine pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;291(3):L378–85. doi: 10.1152/ajplung.00424.2005. [DOI] [PubMed] [Google Scholar]
- 14.Chen P, Guo M, Wygle D, Edwards PA, Falck JR, Roman RJ, Scicli AG. Inhibitors of cytochrome P450 4A suppress angiogenic responses. Am J Pathol. 2005;166(2):615–24. doi: 10.1016/S0002-9440(10)62282-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng G, Diebold BA, Hughes Y, Lambeth JD. Nox-1 dependent reactive oxygen generation is regulated by Rac1. J Biol Chem. 2006;281:17718–26. doi: 10.1074/jbc.M512751200. [DOI] [PubMed] [Google Scholar]
- 16.Chowdhury AK, Watkins T, Parinandi NL, Saatian B, Kleinberg ME, Usatyuk PV, Natarajan V. Src-mediated tyrosine phosphorylation of p47phox in hyperoxia-induced activation of NADPH oxidase and generation of reactive oxygen species in lung endothelial cells. J Biol Chem. 2005;280(21):20700–11. doi: 10.1074/jbc.M411722200. [DOI] [PubMed] [Google Scholar]
- 17.Desire L, Bourdin J, Loiseau N, Peillon H, Picard V, De Oliveira C, Bachelot F, Leblond B, Taverne T, Beausoleil E, Lacombe S, Drouin D, Schweighoffer F. RAC1 inhibition targets amyloid precursor protein processing by gamma-secretase and decreases Abeta production in vitro and in vivo. J Biol Chem. 2005;280(45):37516–25. doi: 10.1074/jbc.M507913200. [DOI] [PubMed] [Google Scholar]
- 18.Dhanasekaran A, Al-Saghir R, Lopez B, Zhu D, Gutterman DD, Jacobs ER, Medhora MM. Protective Effects of Epoxyeicosatrienoic Acids (EETs) on Human Endothelial Cells from the Pulmonary and Coronary Vasculature. Am J Physiol Heart Circ Physiol. 2006;291(2):H517–31. doi: 10.1152/ajpheart.00953.2005. [DOI] [PubMed] [Google Scholar]
- 19.Dhanasekaran A, Kotamraju S, Kalivendi SV, Matsunaga T, Shang T, Keszler A, Joseph J, Kalyanaraman B. Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. J Biol Chem. 2004;279(36):37575–87. doi: 10.1074/jbc.M404003200. [DOI] [PubMed] [Google Scholar]
- 20.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 21.Drouin A, Thorin-Trescases N, Hamel E, Falck JR, Thorin E. Endothelial nitric oxide synthase activation leads to dilatory H2O2 production in mouse cerebral arteries. Cardiovasc Res. 2007;73(1):73–81. doi: 10.1016/j.cardiores.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Fernandes DC, Wosniak J, Jr, Pescatore LA, Bertoline MA, Liberman M, Laurindo FR, Santos CX. Analysis of DHE-derived oxidation products by HPLC in the assessment of superoxide production and NADPH oxidase activity in vascular systems. Am J Physiol Cell Physiol. 2007 Jan;292(1):C413–22. doi: 10.1152/ajpcell.00188.2006. [DOI] [PubMed] [Google Scholar]
- 23.Fontayne A, Dang PM, Gougerot-Pocidalo MA, El-Benna J. Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: effect on binding to p22phox and on NADPH oxidase activation. Biochemistry. 2002;41(24):7743–50. doi: 10.1021/bi011953s. [DOI] [PubMed] [Google Scholar]
- 24.Fresquet F, Pourageaud F, Leblais V, Brandes RP, Savineau JP, Marthan R, Muller B. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. British Journal of Pharmacology. 2006;148(5):714–23. doi: 10.1038/sj.bjp.0706779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griendling KK, Sorescu D, Ushio-Fukai M. NADPH oxidase: role in cardiovascular biology and disease. Circulation Research. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 26.Guo AM, Arbab AS, Falck JR, Chen P, Edwards PA, Roman RJ, Scicli AG. Activation of vascular endothelial growth factor through reactive oxygen species mediates 20-hydroxyeicosatetraenoic acid-induced endothelial cell proliferation. J Pharmacol Exp Ther. 2007;321(1):18–27. doi: 10.1124/jpet.106.115360. [DOI] [PubMed] [Google Scholar]
- 27.Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M, Edwards J, Wolin MS. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. AJP Heart. 2005;288:13–21. doi: 10.1152/ajpheart.00629.2004. [DOI] [PubMed] [Google Scholar]
- 28.Gutterman DD. Mitochondria and reactive oxygen species: an evolution in function. Circ Res. 2005;97(4):302–4. doi: 10.1161/01.RES.0000179773.18195.12. [DOI] [PubMed] [Google Scholar]
- 29.Jacobs ER, Zeldin DC. The lung HETEs and EETs up. Am J Physiol: Heart Circ Physiol. 2001;280(1):H1–H10. doi: 10.1152/ajpheart.2001.280.1.H1. [DOI] [PubMed] [Google Scholar]
- 30.Jacobs ER, Zhu D, Gruenloh S, Lopez B, Medhora MM. VEGF-induced relaxation of pulmonary arteries is mediated by endothelial cytochrome P450 hydroxylase. Am J Physiol Lung Cell Mol Physiol. 2006;291(3):L369–77. doi: 10.1152/ajplung.00265.2004. [DOI] [PubMed] [Google Scholar]
- 31.Lambeth JD. Nox enzymes and the biology of reactivy oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 32.Zhuowei Li, Hyseni X, Carter JD, Soukup JM, Dailey LA, Huang YT. Pollutant particles enhanced H2O2 production from NAD(P)H oxidase and mitochondria in human pulmonary artery endothelial cells. Am J Physiol Cell Physiol. 2006;291:C357–C365. doi: 10.1152/ajpcell.00365.2005. [DOI] [PubMed] [Google Scholar]
- 33.Lyle AN, Griendling KK. Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology (Bethesda) 2006:269–80. doi: 10.1152/physiol.00004.2006. [DOI] [PubMed] [Google Scholar]
- 34.Medhora M, Daniels J, Mundey K, Fisslthaler B, Busse R, Jacobs ER, Harder DR. Epoxygenase-driven angiogenesis in human lung microvascular endothelial cells. Am J Physiol Heart Circ Physiol. 2003;284(1):H215–24. doi: 10.1152/ajpheart.01118.2001. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen X, Wang M-H, Reddy KM, Falck JR, Schwartzman ML. Kinetic profile of the rat CYP4A isoforms: arachidonic acid metabolism and isoform-specific inhibitors. Am J Physiol Regul Integr Comp Physiol. 1999;276:R1691–R1700. doi: 10.1152/ajpregu.1999.276.6.R1691. [DOI] [PubMed] [Google Scholar]
- 36.Ray R, Shaw AM. NADPH oxidase and endothelial cell function. Clinical Science. 2005;109:217–226. doi: 10.1042/CS20050067. [DOI] [PubMed] [Google Scholar]
- 37.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2- and systolic blood pressure in mice. Circulation Research. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 38.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiological Reviews. 2002;82(1):131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 39.Ushio-Fukai M. Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc Res. 2006;71:226–235. doi: 10.1016/j.cardiores.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 40.Ushio-Fukai M, Tang Y, Fukai T, Dikalov SI, Ma Y, Fujimoto M, Quinn MT, Pagano PJ, Johnson C, Alexander RW. Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res. 2002;91(12):1160–7. doi: 10.1161/01.res.0000046227.65158.f8. [DOI] [PubMed] [Google Scholar]
- 41.Wang Ji-Shi, Singh H, Zhang F, Ishizuka T, Deng H, Kemp R, Wolin MS, Hintze TH, Abraham NG, Nasjletti A, Laniado-Schwartzman M. Endothelial dysfunction and hypertension in rats transduced with CYP4A2 adenovirus. Circulation Research. 2006;98(7):962–9. doi: 10.1161/01.RES.0000217283.98806.a6. [DOI] [PubMed] [Google Scholar]
- 42.Wilcox C, Gutterman D. Focus on oxidative stress in the cardiovascular and renal systems. AJP Heart and Circulatory. 2005;288:H3–H6. doi: 10.1152/ajpheart.00854.2004. [DOI] [PubMed] [Google Scholar]
- 43.Yasuda M, Ohzeki Y, Shimizu S, Naito S, Ohtsuru A, Yamamoto T, Kuroiwa Y. Stimulation of in vitro angiogenesis by hydrogen peroxide and the relation with ETS-1 in endothelial cells. Life Sci. 1999;64(4):249–58. doi: 10.1016/s0024-3205(98)00560-8. [DOI] [PubMed] [Google Scholar]
- 44.Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, Kalyanaraman B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc Natl Acad Sci U S A. 2005;102(16):5727–32. doi: 10.1073/pnas.0501719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu D, Zhang C, Medhora M, Jacobs ER. CYP4A mRNA, product and function in rat lungs: Novel localization in non-pregnant vascular endothelium. Journal of Applied Physiology. 2002;93:330–337. doi: 10.1152/japplphysiol.01159.2001. [DOI] [PubMed] [Google Scholar]