

Abstract
Expression of the α6β4 integrin is associated with poor patient prognosis and reduced survival in a variety of human cancers. In recent years, a limited number of in vivo studies have examined the contribution of this integrin receptor to cancer progression and they have revealed that the α6β4 integrin plays a multifaceted role in regulating tumor development and progression. In the current study, we investigated the mechanism by which one tyrosine residue in the β4 subunit cytoplasmic domain, Y1494, contributes to the tumor-promoting functions of the α6β4 integrin in vivo. We demonstrate that Y1494 participates in the stimulation of diverse signaling pathways that promote α6β4-dependent tumor growth and invasion. Mutation of Y1494 inhibits the ability of the α6β4 integrin to support anchorage independent growth in vitro and tumor development and angiogenesis in vivo, a result that mimics the loss of total expression of the β4 subunit. Our results support the hypothesis that Y1494 regulates α6β4-dependent anchorage independent growth through activation of the ERK1/2 signaling pathway, and invasion through the combined activation of PI3K and Src. Collectively, our results identify Y1494 as a major regulatory site for signaling from the α6β4 integrin to promote tumor development and progression.
Keywords: beta4 integrin, tumor growth, signal transduction, invasion, angiogenesis
Introduction
The α6β4 integrin is a receptor for the laminin family of extracellular matrix proteins and its expression is associated with poor patient prognosis and reduced survival in a variety of human cancers (1-5). The β4 integrin subunit was initially identified as a tumor-related antigen expressed in metastatic cancer and, for this reason, many of the early in vitro studies that investigated the function of α6β4 in cancer focused on its potential contribution to later tumor events involved in tumor progression (6). In contrast with its function in regulating stable adhesion through the formation of hemidesmosomes in normal epithelial cells, these studies revealed that the α6β4 integrin promotes motility and invasion in carcinoma cells (7). For example, we demonstrated that expression of α6β4 increases the invasive potential of breast carcinoma cells, a finding that has been confirmed for other tumor cell types as well (8-10). Moreover, suppression of α6β4 expression by RNAi diminishes invasive potential (11). The ability of the α6β4 integrin to sustain the survival of carcinoma cells in stressful environments is also likely to contribute to tumor progression (12-16). In more recent years, in vivo studies have confirmed the contribution of α6β4 to tumor progression to metastasis, but have also revealed that the α6β4 integrin can impact tumor development and primary tumor growth as well (17-20). Specifically, tumor incidence and growth is significantly reduced in breast carcinoma cells when the expression of α6β4 is suppressed by RNAi targeting of the β4 subunit, and also in the Her2/Neu transgenic model when a portion of the β4 cytoplasmic domain is deleted (16, 19, 21). The ability of α6β4 to regulate VEGF expression and promote tumor cell survival is a likely mechanism by which it functions to promote expansion of primary tumors (16). Finally, a role for the α6β4 integrin in tumor initiation has been demonstrated in a Ras-induced squamous cell carcinoma mouse model (22). The multifaceted role of α6β4 in cancer underscores the importance of understanding the mechanism of its action in tumors.
The α6β4 receptor is distinct from other integrin receptors because the β4 subunit contains a 1000 amino acid cytoplasmic domain (23). This cytoplasmic domain is essential for coupling α6β4 to the cytoskeleton, as well as for its ability to activate intracellular signaling pathways (9, 24). It is the latter function in regulating signal transduction that is thought to be the major mechanism by which the α6β4 receptor contributes to cancer. It has been hypothesized that the α6β4 integrin becomes competent for signaling in response to signals that disrupt hemidesmosomes, resulting in the release of the α6β4 receptor from interactions with the cytokeratin cytoskeleton and de novo interaction with the actin cytoskeleton and signaling molecules (25). In this signaling competent state, the α6β4 integrin can also cooperate with growth factor receptors and other surface molecules to amplify intracellular signaling pathways through the β4 subunit cytoplasmic domain (17, 26-28). We, and others, have shown that the β4 cytoplasmic domain can be phosphorylated by both tyrosine and serine/threonine kinases and that these phosphorylation events play a critical role in the function of the α6β4 integrin by regulating its interactions with intracellular cytoskeletal and signaling intermediates (29-33). Although some progress has been made in identifying key sites in the β4 subunit cytoplasmic domain that are critical for activating downstream signaling pathways, the specific contributions of these sites to α6β4 function in tumors has not been investigated in vivo (30, 31).
A site in the β4 cytoplasmic domain that we believe may play an essential role in α6β4-dependent tumor cell functions is tyrosine 1494 (30). In our previous studies, we established that one mechanism by which the α6β4 integrin promotes breast carcinoma cell invasion and survival is through its ability to promote PI3K activation (9, 14). Mutation of Y1494 to phenylalanine inhibited the ability of the α6β4 integrin to activate PI3K, as well as to promote breast carcinoma cell invasion and survival (30). In fact, mutation of Y1494 significantly inhibited overall tyrosine phosphorylation of the β4 subunit in response to α6β4 ligation, suggesting that this site could be a master regulator of α6β4 phosphorylation and signaling (30). Y1494 is located within a consensus-binding motif for the SH2 domain containing tyrosine phosphatases SHP-1 and SHP-2, which have been shown to regulate signaling from a number of growth factor receptors both positively and negatively (34). Recent studies by our group and others have demonstrated that SHP-2 can bind directly to the β4 cytoplasmic domain and that Y1494 contributes to this binding (35, 36). Moreover, SHP-2 activity is required for activation of Src by the α6β4 integrin (35).
In our current study, we investigated the contribution of Y1494 to α6β4 function in vivo. Our results reveal that signals intiated through Y1494 are essential for α6β4-dependent tumor development and stimulation of angiogenesis. Moreover, multiple, distinct signaling pathways are activated through Y1494 to regulate anchorage independent tumor cell growth and invasion.
Materials and Methods
Cells, antibodies and reagents
MDA-MB-435 human breast carcinoma cells expressing wildtype and mutant β4 subunits were generated previously (9, 30). MDA-MB-231 human breast carcinoma cells were obtained from the Lombardi Breast Cancer Depository (Georgetown University). The mouse monoclonal antibody that recognizes the human β4 subunit (UM-A9) was obtained from Ancell. The mouse monoclonal antibody that recognizes the human α6 subunit (2B7) was a gift from Arthur Mercurio (UMass Medical School). Antibodies that recognize phospho-AKT, phospho-ERK1/2, total ERK1/2, phospho-MEK1/2 and total MEK1/2 were obtained from Cell Signaling, Inc. Phospho-SrcY418 and phospho-SrcY529 antibodies were obtained from Biosource and total c-Src antibodies were obtained from Santa Cruz Biotechnology Inc. Matrigel was purchased from BD Biosciences (cat. #354234). The pMCL-MKK1-R4F plasmid (MEK-DD) was kindly provided by Natalie Ahn (University of Colorado) and the p110-CAAX and SrcY527F plasmids were generously provided by Alex Toker (Harvard Medical School). pLPCX was obtained from Clontech.
Integrin clustering
Cells were deprived of serum overnight in RPMI medium containing 25mM Hepes and 0.1% heat inactivated BSA (RH/BSA). Cells were trypsinized and resuspended in RH/BSA at a concentration of 106 cells/ml and incubated for 30 minutes with integrin-specific antibodies (2 μg/ml) or in buffer alone. Washed cells were resuspended in RH/BSA and added to plates that had been coated overnight at 4°C with anti-mouse IgG (100 μg/10 cm plate) or EHS laminin. After incubation at 37°C for 30 minutes, the cells were solubilized at 4°C for 10 minutes in a 20 mM Tris buffer, pH 7.4, containing 0.14 M NaCl, 1% NP-40, 10% glycerol, 1 mM sodium orthovanadate, and protease inhibitors (Complete Mini Tab; Roche).
Immunoblotting
Cell extracts containing equivalent amounts of total protein were resolved by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked for an hour using a 50mM Tris buffer, pH 7.5, containing 0.15M NaCl and 0.05% Tween-20 (TBST) and 5% (w/v) Carnation dry milk. Membranes were incubated overnight at 4°C in the same buffer containing primary antibodies. Proteins were detected by enhanced chemiluminescence (Pierce). For phospho-immunoblots, the blocking buffer for the primary antibodies contained 5% (w/v) BSA.
Invasion assays
Matrigel invasion assays were performed as described previously using 6.5 mm Transwell chambers (8 μm pore size) (9, 30). After 5 hours, the cells that had invaded to the lower surface of the filters were fixed in methanol for 10 minutes. The fixed membranes were mounted on glass slides using Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA). Invasion was quantified by counting the number of stained nuclei in five independent fields in each Transwell.
Orthotopic in vivo assays
Cells were trypsinized, washed five times with sterile PBS, and resuspended in 35 μL phenol red-free Matrigel immediately prior to injection. Female immunocompromised mice (nu/nu; National Cancer Institute) at 7 to 9 weeks of age were anesthetized briefly with halothane and cells were injected into the #3 mammary fat pad (1 × 106 cells per injection, one injection site per mouse). The mice were palpated every 2-3 days to detect the onset of mammary tumor development and tumors were measured using calipers weekly until 15 weeks of age or 1.5 cm tumor burden. Tumor volume was determined using the following formula: volume = (4/3)(π)(1/2 × smaller diameter)2(1/2 × larger diameter) (37). Portions of the tumors were either snap frozen or fixed in 10% buffered formalin for histologic processing.
Immunohistochemistry
Tissue sections were deparaffinized and rehydrated before endogenous peroxidase activity was quenched in 3% H2O2 for 5 min. To detect angiogenesis, tissue sections were stained with rat anti-mouse CD31 (2.5 μg/ml; BD Biosciences) as described previously (37). CD31 staining was quantitated using NIH Image 1.61 software. To detect apoptotic cells, tissue sections were stained using the ApopTag Plus peroxidase in situ apoptosis detection kit according to the manufacturer's instructions (Chemicon, Temecula, CA).
3D Matrigel assay
A base layer of Matrigel (BD Biosciences, Bedford, MA; 200 μl/well) was overlaid in duplicate wells of a 24-well dish with 1.0 × 104 cells suspended in 300 μL of a 2:1 mixture of PBS and Matrigel. The Matrigel was overlaid with complete serum-containing medium (0.5 ml/well), which was changed every 3 days. Images were captured with SPOT image analysis software (Molecular Diagnostics). Single-cell suspensions were recovered from the Matrigel at the end of the incubation period by dispase treatment (500 μl/well; BD Biosciences) for 2 hours at 37°C followed by trypsin digestion for 5 minutes at 37°C. The recovered cells were labeled with either Annexin V-PE or 7-AAD (BD Biosciences). All labeled cells were analyzed by flow cytometry. To determine cell viability in situ, cells were stained with 2μM calcein AM and 4μM Ethidium homodimer-1 (LIVE/DEAD Viability/Cytotoxicity Kit; Invitrogen Molecular Probes, OR) according to manufacturer's protocol.
Soft agar assay
Cells (1.0 × 103 cells) were suspended in serum-containing medium (2 ml) containing 0.3% agar (Life Technologies) and overlaid on a 1 ml base layer of 0.75% agar in 6-well plates. The soft agar was overlaid with complete medium (0.5 ml/well), which was changed every 3 days. After a 2-week incubation, the total number of colonies was quantified by counting 50 fields per well using bright-field optics. Images were captured by SPOT image analysis software and colonies with diameter of more than 2 mm were counted.
Results
Y1494 in the β4 subunit cytoplasmic domain is required for α6β4-dependent tumor development in vivo
The α6β4 integrin promotes carcinoma cell survival and invasion in vitro and Y1494 in the β4 subunit cytoplasmic domain plays a role in regulating these functions (9, 30). To assess the role of Y1494 in α6β4-dependent tumor development and progression in vivo, stable subclones of MDA-MB-435 cells transfected with empty pcDNA3 vector (Mock), complete human β4 cDNA (wt-β4) or the β4 subunit containing a point mutation to phenylalanine at Y1494 (Y1494F-β4) were injected into the mammary fat pad of immunocompromised mice. The viability of the cell lines prior to injection was equivalent as determined by trypan blue staining (data not shown). Cells expressing vector alone or the wildtype β4 subunit formed tumors efficiently in 90-95% of the mice, respectively (Table 1). In contrast, tumors developed in only 25% of the mice injected with Y1494F-β4 cells (Table 1). This marked reduction in tumor incidence was similar to that observed when total expression of the β4 subunit was suppressed in SUM-159PT breast carcinoma cells, suggesting that Y1494 plays a vital role in α6β4-dependent regulation of tumor development in vivo (16). Morphologically, the tumors that formed from cells expressing Y1494F-β4 had a more well-differentiated, glandular morphology when compared with the solid morphology of tumors formed by Mock and wt-β4 expressing cells (Supplemental Fig. 1). In addition, evidence of tumor cell invasion into the muscle layer was observed more often in the wt-β4 expressing tumors when compared with Mock or Y1494F-β4 tumors (Supplemental Fig. 1).
Table 1.
Contribution of Y1494 to α6β4 dependent tumor development, angiogenesis and survival in vivo. Mock, wt-β4 and Y1494F-β4 cells were injected orthotopically into the #3 mammary fat pad of immunocompromised mice and tumor development was monitored for 15 weeks or 1.5 cm tumor burden.
| Cell lines | Mock | wt-β4 | Y1494F-β4 |
|---|---|---|---|
| Tumor Incidence (%) | 90%
(18/20) |
95%
(19/20) |
25%
(5/20) |
| Vascular Density1 (mm2) | 4.89 ± 0.33 | 16.39 ± 0.71* | 4.04 ± 0.24 |
| Apoptotic Cells2 (%) | 16.90 ± 1.16 | 4.44 ± 0.37* | 35.22 ± 1.79* |
Mean area of CD31 immunohistochemical staining (± SEM).
Mean percentage of cells (± SEM) that stained positively for Apoptag reagent.
p < .01 compared with Mock.
The α6β4 integrin has been reported to promote tumor progression in part through the regulation of VEGF expression and stimulation of tumor angiogenesis (16, 38). To determine if Y1494 contributes to α6β4-dependent regulation of tumor angiogenesis, microvascular density was assessed in the tumors by staining for the endothelial cell specific marker CD31 (37). Confirming previous studies, mean tumor vasculature density was 4-fold greater in the tumors that expressed wildtype α6β4 when compared with tumors that express α6β1 as the only α6-containing integrin (Mock) (Table 1, p < 0.01). However, mutation of Y1494 inhibited the α6β4-dependent increase in tumor angiogenesis. The mean vasculature density of the Y1494F-β4 tumors was similar to that observed for mock tumors, and these tumors were also highly necrotic. In addition to promoting angiogenesis, the α6β4 integrin also signals to inhibit apoptosis to promote tumor cell survival in stressful environments (14-16). To analyze the role of Y1494 in tumor cell survival, apoptotic cells in the tumors were quantified by staining with Apoptag reagent. The percentage of apoptotic cells in the tumors was increased 2-fold in Y1494F-β4 tumors when compared to Mock tumors, whereas wt-β4 tumors had a 4-fold decrease in apoptosis (Table 1, p <0.01). Taken together, these immunohistochemical data demonstrate that Y1494 plays a critical role in α6β4-dependent tumor cell survival and promotion of tumor angiogenesis.
To dissect further the contribution of Y1494 to tumor development, the ability of Y1494F-β4 cells to grow in an anchorage-independent manner in vitro was examined. As was observed for tumor development in vivo, both Mock and wt-β4 expressing cells grew efficiently as colonies in soft agar (Fig. 1A). In contrast, cells expressing Y1494F-β4 formed few colonies, and those colonies that did form were approximately 3-fold smaller in size when compared with the colonies formed by Mock and wt-β4 expressing cells.
Figure 1.
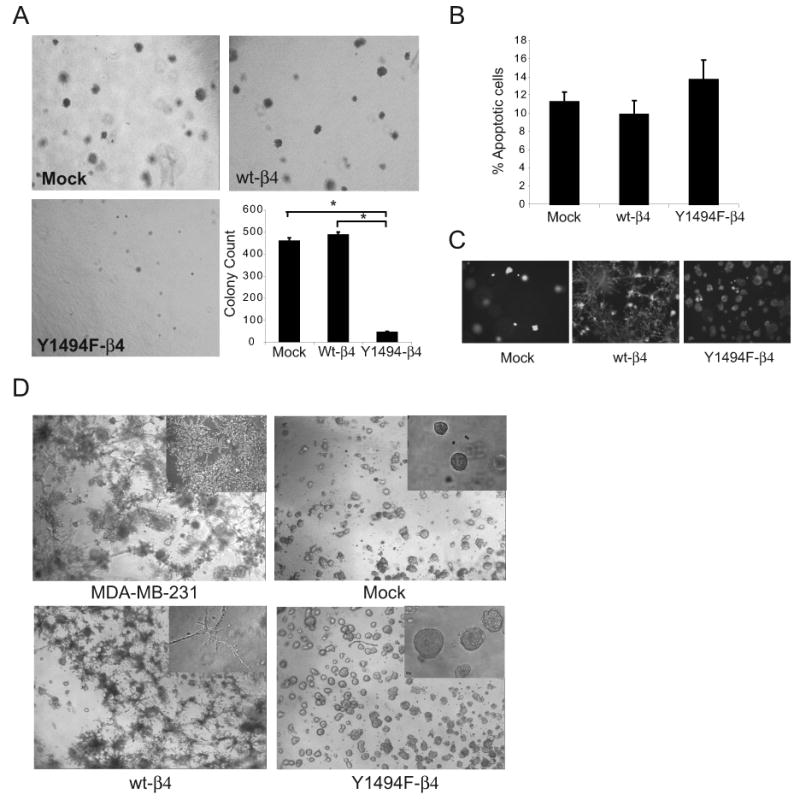
Y1494 in the β4 integrin subunit is required for α6β4 integrin-dependent anchorage independent growth and invasion of tumor cells. (A) Representative bright-field images captured at 4X magnification of Mock, wt-β4 and Y1494F-β4 transfected MDA-MB-435 cell lines grown in 0.3% agar with complete growth medium for 2 weeks. The graph shows the mean (± SEM) of three representative experiments performed in triplicate. 50 fields per well were counted for each assay. *p < 0.01 (B) Cells were recovered from 3D Matrigel after 10 days by dispase treatment and apoptosis was measured by flow cytometry after staining with Annexin V-PE and 7-AAD. The data shown represent the mean percentage (± SEM) of Annexin V-PE+, 7-AAD-cells from three independent experiments performed in duplicate. p > 0.05. (C) Representative fluorescence images captured at 10X magnification of Mock, wt-β4 and Y1494F-β4 transfected MDA-MB-435 cells that were stained with calcien dye after 10 days of growth in 3D Matrigel cultures to detect viable cells. (D) Representative images captured at 4X magnification of MDA-MB-231 cells and Mock, wt-β4 and Y1494F-β4 transfected MDA-MB-435 cells grown for 10 days in 3D Matrigel culture. (inset magnification = 20X).
Y1494 in the β4 subunit is required for α6β4-mediated invasion, but not growth in 3D Matrigel culture
Previous studies that have evaluated the contribution of α6β4 to tumor development and progression have demonstrated that this integrin promotes tumor cell survival in 3-dimensional (3D) Matrigel culture (15, 16). To investigate if Y1494 contributes to α6β4 integrin-dependent survival in 3D matrix, cells were embedded in Matrigel and cultured for 10 days. The cells were recovered from the 3D-Matrigel cultures and stained for Annexin V-PE and 7-AAD to identify apoptotic cells. All of the cell lines grew efficiently in Matrigel and there were no significant differences in apoptosis for Mock, wt-β4 or Y1494F-β4 cells (Fig. 1B). The viability of cells in situ in 3D Matrigel culture was also analyzed by staining with the polyanionic dye calcein or with ethidium homodimer (EthD-1). Live cells produce an intense green fluorescence with calcein and dead cells produce a red fluorescence with EthD-1. All of the cell lines stained positively for calcein (Fig. 1C), but little if any detectable EthD-1 staining was observed for any of the cells (data not shown), confirming that Y1494 is not required for α6β4-dependent viability in 3D Matrigel culture.
Although all of the cell lines survived and grew in 3D Matrigel, marked differences in the morphology of the colonies that formed were observed. Mock transfected cells grew as relatively round, non-invasive colonies, similar to their appearance in soft agar. In contrast, colonies formed by cells expressing wt-β4, either exogenously (MDA-MB-435/wt-β4 cells) or endogenously (MDA-MB-231 cells), exhibited a stellate morphology commensurate with an invasive phenotype (Fig. 1D). Mutation of Y1494 suppressed the β4-dependent invasive behavior and colonies formed by these cells were similar in morphology to those formed by the Mock transfected cells. These findings complement results from our previous studies using 2-dimensional (2D) Transwell chamber Matrigel invasion assays that revealed a role for Y1494 in regulating α6β4-dependent invasion (30).
Analysis of α6β4 integrin dependent signaling pathways regulated by Y1494
The α6β4 integrin regulates cellular behavior through both adhesive and signaling mechanisms and the β4 subunit cytoplasmic domain has been implicated in both functions. Signaling molecules that have been shown to be activated through the α6β4 integrin include PI3K, Src and the ERK mitogen activated protein kinases (MAPKs) (9, 35, 39). To investigate further the mechanism by which Y1494 contributes to α6β4-dependent tumor development in vivo and anchorage independent growth in vitro, we initially assessed the status of ERK signaling because this pathway has been widely implicated in promoting tumor cell proliferation and growth (40, 41). Cells expressing wt-β4 or Y1494F-β4 were either maintained in suspension or allowed to adhere to laminin-coated plates to engage the α6β4 receptor in a ligand-dependent manner. As shown in Fig. 2A, mutation of Y1494 markedly inhibited α6β4-mediated phosphoryation of p42/44 ERK (ERK1/2). This inhibition of ERK1/2 activation was also observed when cells expressing Y1494F-β4 were ligated directly with either α6- or β4-specific antibodies (Fig. 2B). To investigate further the involvement of MAPK signaling in α6β4-dependent tumor cell functions, cells were recovered from 3D Matrigel cultures by dispase digestion and analyzed for activation of MAPK signaling. When compared with Mock cells, wt-β4 transfected cells exhibited a 2-fold increase in ERK1/2 activation. However, cells transfected with Y1494F-β4 exhibited a 3-fold decrease in activation of ERK1/2, as well as a 6-fold decrease in activation of the upstream regulator MEK1 (Fig. 2C).
Figure 2.
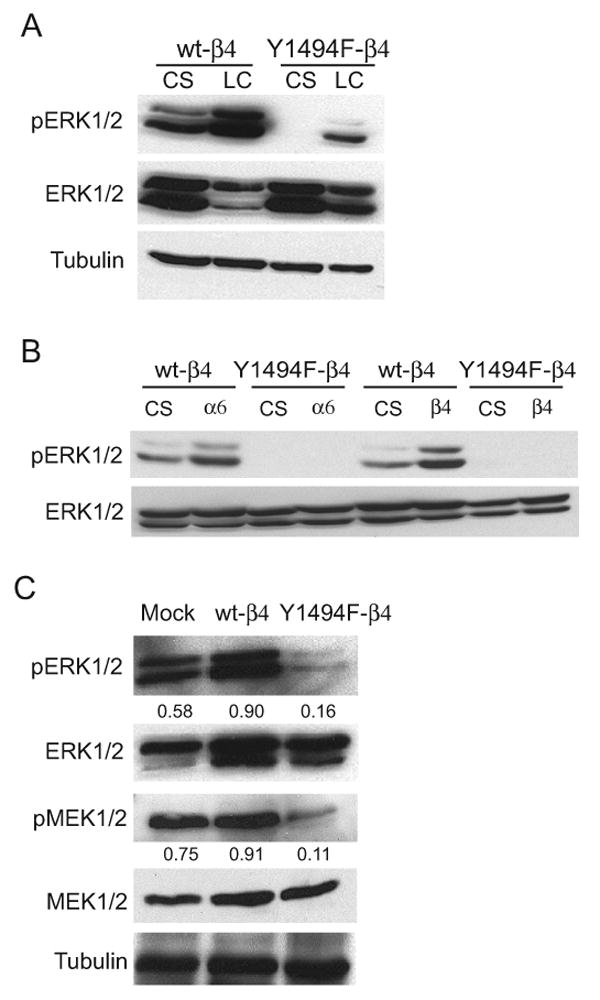
Y1494 in the β4 integrin subunit is required for α6β4 integrin-dependent activation of ERK1/2. (A) wt-β4 and Y1494F-β4 transfected MDA-MB-435 cells were maintained in suspension or allowed to adhere to laminin-1 coated plates for 30 minutes. Aliquots of cell extracts that contained equivalent amounts of total protein were resolved by SDS-PAGE (10%) and then immunoblotted with the indicated antibodies. (B) wt-β4 and Y1494F-β4 transfected MDA-MB-435 cells were maintained in suspension or incubated with either α6- or β4-specific antibodies and allowed to adhere to anti-mouse IgG-coated plates for 30 minutes. (C) Mock, wt-β4 and Y1494F-β4 transfected MDA-MB-435 cells were recovered from 3D-Matrigel after 10 days by dispase treatment. Aliquots of cell lysates containing equivalent amounts of total protein were immunoblotted with antibodies specific for phospho-ERK1/2, phospho-Ser217/221 of MEK1/2 or tubulin. The immunoblots for the phospho-specific antibodies were stripped and reprobed for total expression levels. Densitometry measurements shown in (C) are the respective ratio of phospho-protein to total protein levels. CS, cells maintained in suspension; LC, cells adherent to laminin-1; α6, cells clustered with a α6-specific antibody (2B7); β4, cells clustered with a β4-specific antibody (UM-A9).
Constitutively active MEK rescues α6β4-dependent anchorage independent growth, but not invasion
To investigate a role for ERK1/2 signaling through Y1494 in the ability of the α6β4 integrin to promote anchorage independent growth and tumor cell invasion, cells expressing Y1494F-β4 were stably transfected with a constitutively active MEK mutant (MEK-DD) (42). Expression of MEK-DD enhanced the activation of ERK1/2 2-fold when compared with Y1494F-β4 cells expressing an empty vector (Fig. 3A). Transfected cells were assayed for their ability to grow in an anchorage independent manner in soft agar and to invade using both 2D and 3D Matrigel invasion assays. As shown in Fig. 3B, constitutively active MEK restored the anchorage independent growth of cells expressing Y1494F-β4. In contrast, MEK-DD expression did not rescue the invasive potential of these cells in 3D Matrigel, even though a 2-fold increase in ERK1/2 activation was maintained in these cells in 3D culture (Figs. 3C and D). The colonies formed in 3D Matrigel by the Y1494F-β4 cells that expressed MEK-DD were similar in morphology to those that were transfected with vector alone, with no evidence of invasive outgrowth (Fig. 3C). Furthermore, MEK-DD did not enhance the invasive potential of the cells expressing Y1494F-β4 when assayed using a Matrigel Transwell 2D invasion assay (Fig. 5C). Taken together, these data reveal that α6β4 signals through Y1494 to activate ERK1/2 to promote anchorage independent growth. However, the inability of MEK-DD to restore α6β4-dependent invasion to the Y1494F-β4 cells provides evidence that additional signaling pathways are activated through Y1494 to promote invasive behavior.
Figure 3.
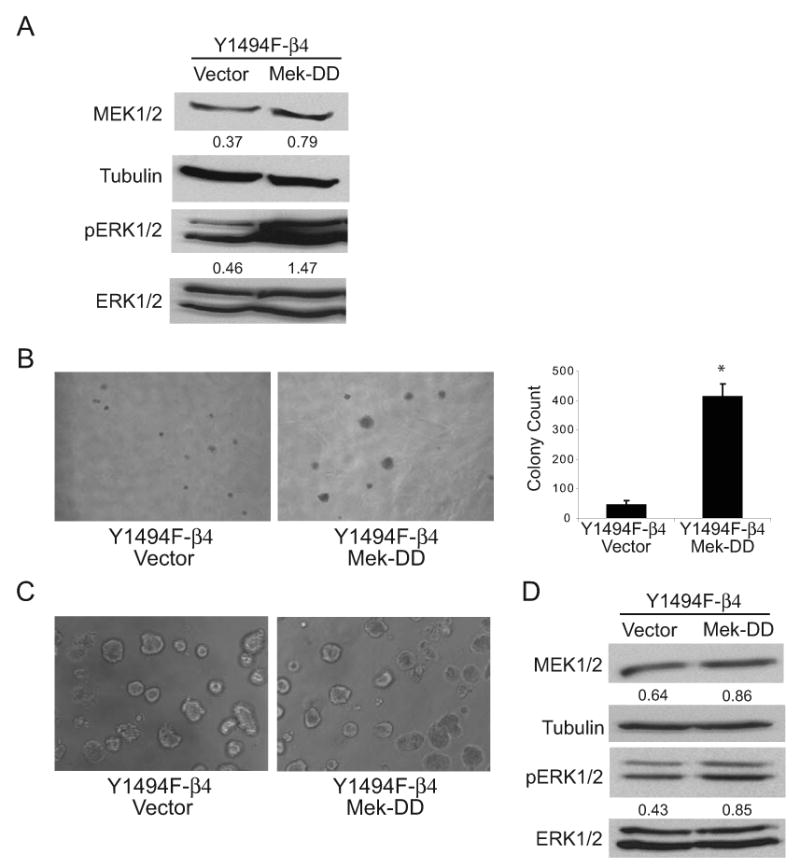
Constitutively active MEK rescues Y1494F-β4 integrin-dependent anchorage independent growth in soft agar but not invasion in 3D Matrigel. Y1494F-β4 cells were co-transfected with either pMCL-MKK1-R4F (constitutively active MEK-DD) or empty vector and pLPCX vector for puromycin selection. (A) Aliquots of cell lysates containing equivalent amounts of total protein were immunoblotted with antibodies specific for MEK1/2, phospho-ERK1/2, or tubulin. Densitometry measurements shown below each panel are the respective ratio of MEK1/2 to tubulin and phospho-ERK1/2 to total ERK1/2 levels. (B) Representative bright-field images captured at 4X magnification of empty vector or MEK-DD-transfected Y1494F-β4 cells grown in 0.3% agar with complete growth medium for 2 weeks. Quantitation of the soft agar assays is shown in the graph on the right. The data represent the mean (± SEM) of three representative experiments performed in triplicate. 50 fields per well were counted for each assay. *p < 0.01 (C) Representative bright-field images at 10X magnification of empty vector or MEK-DD-transfected Y1494F-β4 cells grown in 3D Matrigel for 10 days. (D) Cells were recovered from the 3D Matrigel cultures by dispase treatment and aliquots of cell lysates containing equivalent protein were lysed and immunoblotted with antibodies specific for MEK1/2, phospho-ERK1/2, or tubulin. Densitometry measurements shown below each panel are the respective ratio of MEK1/2 to tubulin and phospho-ERK1/2 to total ERK1/2 levels.
Figure 5.
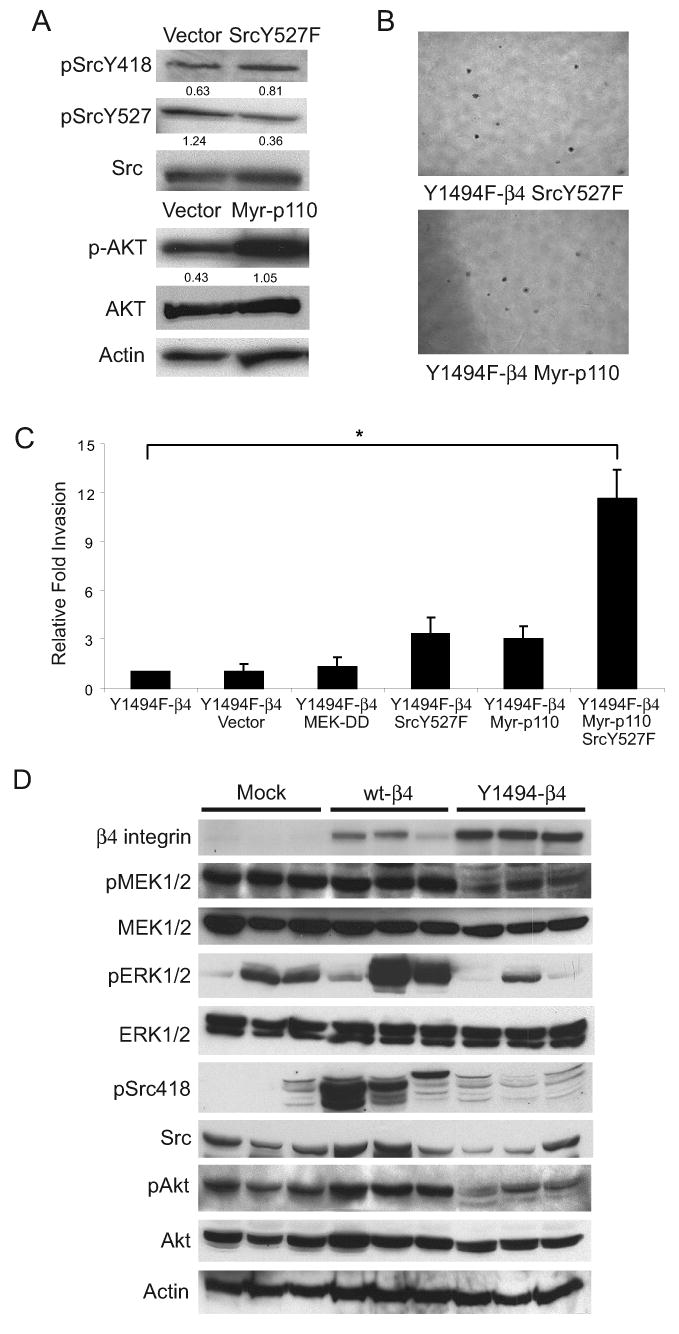
Combined activation of PI3K and Src rescues Y1494F-β4-dependent invasion. (A) Y1494F-β4 cells were stably transfected with either Src Y527F, myr-p110α PI3K, or empty vector. Aliquots of cell lysates containing equivalent amounts of total protein were immunoblotted with antibodies specific for phospho-SrcY418, phospho-SrcY527, phospho-Akt-S473, or actin. The phospho-immunoblots were stripped and reprobed for total expression levels. Densitometry measurements shown below each panel are the respective ratio of phospho- to total expression levels. (B) Representative bright-field images captured at 4X magnification of Y1494F-β4 cells transfected with Src Y527F or myr-p110 PI3K grown in 0.3% agar with complete growth medium for 2 weeks. (C) Y1494F-β4 cells were transfected transiently with empty vector, MEK-DD, SrcY527F, Myr-p110 or SrcY527F and Myr-p110 together and assayed for their invasive potential using a 2D Matrigel Transwell assay. Five independent fields/well were counted. The data shown represent the mean (±SEM) of three independent experiments performed in triplicate. *p < 0.01. (D) Aliquots of tumor extracts from Mock, wt-β4 and Y1494F-β4 derived tumors (n = 3) containing equivalent amounts of total protein were immunoblotted with antibodies specific for β4, phospho-MEK1/2, phospho-ERK1/2, phosphoY418Src, phosphoS473 of Akt and actin. The immunoblots for the phospho-specific antibodies were stripped and reprobed for total expression levels.
PI3K and Src cooperate to rescue α6β4-dependent invasion, but not anchorage independent growth
Both PI3K and Src signaling pathways have been implicated in positively regulating tumor cell invasion. In previous studies, we reported that Y1494 contributes to the α6β4-dependent activation of PI3K (9, 30). Y1494 has also been implicated in the regulation of Src family kinase activation downstream of the α6β4 integrin in response to Met receptor signaling (36). To evaluate the involvement of Y1494 in direct Src activation by α6β4 ligation, cells expressing either the wt-β4 subunit or the Y1494F-β4 mutant subunit were ligated with α6 antibodies to stimulate receptor activation. As shown previously, ligation of wt-α6β4 stimulated Src activation. However, Src activation was not observed in the cells expressing Y1494F-β4 (Fig. 4A). Total Src levels were also decreased in the cells expressing Y1494F-β4, suggesting an involvement of Y1494 in regulating Src expression. In contrast to MEK/ERK signaling, neither PI3K nor Src activation were detectable in the mock, wt-β4 or Y1494F-β4 cells when recovered from 3D Matrigel (data not shown).
Figure 4.
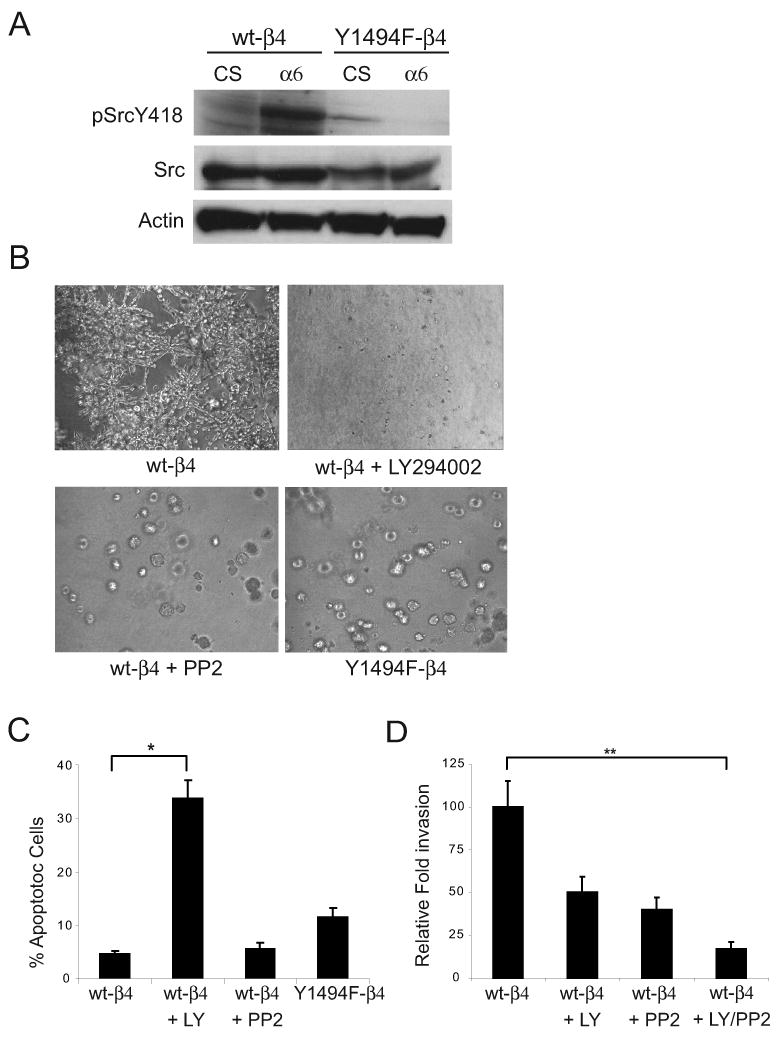
Src and PI3K activation are required for optimal promotion of invasion by the α6β4 integrin. (A) wt-β4 and Y1494F-β4 transfected MDA-MB-435 cells were maintained in suspension or incubated with α6-specific antibodies and allowed to adhere to anti-mouse IgG-coated plates for 30 minutes. Aliquots of cell lysates containing equivalent amounts of total protein were immunoblotted with antibodies specific for phospho-SrcY418 or actin. The phospho-Src immunoblot was stripped and reprobed for total Src expression levels. CS, cells maintained in suspension; α6, cells clustered with a α6-specific antibody (2B7). (B) Representative bright field images at 10X magnification of wt-β4 cells, wt-β4 cells in the presence of 50 μM LY294002 (PI3K inhibitor) or 50μM PP2 (Src inhibitor), and Y1494F-β4 cells grown for 10 days in 3D Matrigel culture. (C) Cells were recovered from Matrigel after 10 days by dispase treatment and apoptosis was measured by flow cytometry after staining with Annexin V-PE and 7-AAD. The data shown represent the mean percentage (±SEM) of Annexin V-PE+, 7-AAD- cells from three independent experiments performed in duplicate. p < 0.01. (D) Wt-β4 cells were assayed for their invasive potential in the presence of 50 μM LY294002, 50μM PP2 or both using a 2D Matrigel Transwell assay. Five independent fields/well were counted. The data shown represent the mean (±SEM) of two independent experiments performed in triplicate. **p < 0.01.
Cells expressing wt-β4 were embedded and grown in Matrigel in the presence of inhibitors of PI3K (LY294002) or Src (PP2) to begin to evaluate the importance of PI3K and Src signaling for Y1494-dependent invasion. Inhibition of PI3K impaired cell survival and, as a result, colonies did not develop in the presence of LY294002 (Fig 4B and C). In contrast, cells expressing wt-β4 formed colonies in 3D Matrigel in the presence of the Src inhibitor PP2 (Fig. 4B). However, the morphology of the colonies was strikingly different from the colonies formed by the cells with active Src signaling. Colonies that developed in the absence of Src signaling showed little if any invasive growth and they were similar in appearance to the colonies formed by cells expressing Y1494F-β4. Annexin V-PE and 7-AAD staining of cells recovered from Matrigel revealed that PP2 did not increase apoptosis significantly (Fig. 4C), which supports a role for Src in α6β4-dependent invasion. Short-term 2D Matrigel invasion assays were also performed in the presence of the same inhibitors used in the 3D assays to assess the relative contributions of PI3K and Src to α6β4-mediated invasion. Although inhibition of either PI3K or Src reduced invasion by approximately 50%, invasion was reduced by 80% when both pathways were blocked simultaneously (Fig. 4D). Taken together, these assays provide evidence that PI3K and Src cooperate downstream of the α6β4 integrin to promote tumor cell invasion.
To investigate further the involvement of PI3K and Src in Y1494-dependent promotion of invasion, constitutively active mutants of either PI3K or Src were expressed in the Y1494F-β4 cells to determine if activation of these pathways can rescue their invasive behavior. Stable expression of a myristylated p110α catalytic subunit of PI3K or Src-Y527F independently in the Y1494-β4 cells did not rescue invasion in 3D-Matrigel, even though a 2-3 fold increase in the activation of these pathways was observed in the transfected cells as assessed by phospho-SrcY418 and phospho-AktS473, respectively (Fig. 5A). To determine if PI3K and Src cooperate downstream of Y1494 to promote invasion, as suggested by the inhibitor studies (Fig. 4D), Y1494-β4 cells were transiently transfected with Myr-p110α and Src-Y527F individually, or together, and their ability to invade in a Matrigel Transwell assay was assessed. As was observed for the stable transfectants (data not shown), expression of the individual constructs alone promoted a small increase in invasion over that observed for parental or vector transfected cells. However, co-expression of Myr-p110α and Src-Y527F increased invasion 4-fold above that observed for the individual contructs, indicating that both of these signaling pathways are required for optimal promotion of invasion by the α6β4 integrin (Fig. 5C). Rescue of PI3K or Src activation independently did not rescue anchorage independent growth in soft agar, whereas expression of MEK-DD alone did not increase invasion (Figs. 5B and C). Therefore, distinct signaling pathways are activated downstream of Y1494 in the β4 subunit to regulate anchorage independent growth and tumor cell invasion in vitro.
Y1494 is required for α6β4-dependent activation of ERK, PI3K and Src in vivo
Our studies revealed that Y1494 plays an essential role in the ability of the α6β4 integrin to activate multiple signaling pathways and to promote anchorage independent growth and invasion in vitro. To determine if this tyrosine residue is also active in regulating these signaling pathways in vivo to promote tumor development and progression, we examined the status of ERK, Src and PI3K signaling in the Mock, wt-β4 and Y1494F-β4 tumors. For all of the pathways examined, activation was enhanced by expression of wt-β4 and suppressed by mutation of Y1494, a result that mimics our in vitro findings and supports the important contribution of Y1494 to α6β4-dependent tumor-promoting functions (Fig. 5D).
Discussion
In this study, we demonstrate that Y1494 in the cytoplasmic domain of the β4-integrin subunit participates in the stimulation of diverse downstream signaling pathways that regulate tumor cell function. Mutation of Y1494 inhibits the ability of the α6β4 integrin to support anchorage independent growth and tumor cell invasion in vitro and tumor development and angiogenesis in vivo, a result that mimics the loss of total expression of the β4 subunit and highlights the key role that this tyrosine residue plays in regulating β4 function. Our results support the hypothesis that Y1494 regulates α6β4-dependent anchorage independent growth through activation of the ERK1/2 signaling pathway. Furthermore, we show that Y1494 is also essential for mediating α6β4-dependent PI3K and Src activation, and that these two independent signaling pathways cooperate to promote optimal invasive potential. Collectively, our results identify Y1494 as a major regulatory site for signaling from the α6β4 integrin to promote tumor development and progression.
Our findings reveal that Y1494 plays a dominant role in the ability of α6β4 to promote tumor development. Mutation of this single residue in the β4-integrin subunit reduced tumor incidence by approximately 75%, a result previously observed when total β4 expression was suppressed (16). Although only a limited number of studies have examined α6β4 function in vivo, an understanding of the contributions of this receptor to tumor biology is emerging. What is becoming clear is that the α6β4 integrin plays a multifaceted role in tumor promotion (16-19, 21, 22). Furthermore, the cytoplasmic domain of the β4 subunit is required for mediating most of the tumor-promoting functions of this receptor (9, 14). Y1494 is located within the C-terminal region of the β4-cytoplasmic domain that is reported to be important for α6β4-dependent primary tumor growth and progression to metastasis (19). The decreased tumor incidence of cells that express a β4 subunit with a single mutation at Y1494 correlates with increased apoptosis, a finding that was foreshadowed by our previous data that Y1494 is required for α6β4-dependent regulation of VEGF expression and tumor cell survival in vitro (14). Our current results support the hypothesis that Y1494 likely plays a dual role in contributing to tumor development, through the activation of ERK1/2 to promote anchorage independent growth and through activation of PI3K to regulate mTor-dependent VEGF expression to promote tumor cell survival. The regulation of VEGF expression by Y1494 likely contributes to tumor progression as well by stimulating angiogenesis.
The ability of Y1494 to influence tumor development and progression probably derives from its involvement in regulating multiple key downstream signaling pathways. Specifically, mutation of Y1494 disrupts the ability of α6β4 to activate ERK1/2, PI3K and Src signaling. A possible mechanism for the activation of multiple pathways from a common site is the recruitment of a signaling intermediate that participates in the activation of several independent pathways. Y1494 is located within an ITIM motif, a consensus binding site for the tyrosine phosphatases SHP-1/2 and the inositol phosphatases SHIP-1/2 (34). SHP-2 can bind to the β4 subunit and there is evidence to suggest that Y1494 participates in this recruitment (35, 36). Moreover, we have previously demonstrated that the ability of the α6β4 integrin to activate Src requires SHP-2 phosphatase activity, placing SHP-2 as an upstream regulator of this signaling pathway (35). SHP-2 has also been implicated in regulating PI3K and MAPK signaling downstream of growth factor receptors, which would support the idea that SHP-2 could be a common regulator of signaling downstream of Y1494 (43-45). However, additional intermediates could also be recruited to this site to regulate these pathways, a possibility that is suggested by our previous identification of IRS-2 as a signaling adaptor for PI3K activation by α6β4 (30). Finally, mutation of Y1494 significantly diminishes the overall tyrosyl-phosphorylation of the β4 subunit in response to engagement of the α6β4 receptor, suggesting that Y1494 is required for regulating the phosphorylation of additional sites in the β4-subunit cytoplasmic domain (30). The fact that SHP-2 can positively regulate Src family kinase activation and Src family kinases have been reported to phosphorylate the β4 integrin lends credence to this potential mechanism (39).
Mutation of Y1494 prevents α6β4-mediated activation of ERK1/2, implicating this tyrosine residue as a key site for regulating this MAPK signaling pathway. It has been previously reported that the ability of the α6β4 integrin to activate ERK1/2 is dependent upon the recruitment of Shc to Y1546 in the β4 subunit cytoplasmic domain (31). Although we have not observed phosphorylation of Shc in response to α6β4 ligation in our model systems (data not shown), the possibility exists that Y1494 is required for regulating the phosphorylation of Y1526 and indirectly mediating the activation of ERK1/2 through pathways originating from this site. Another indirect mechanism for the involvement of Y1494 in regulating the MEK/ERK pathway was reported in a study investigating the function of the β4 subunit as a signaling adaptor for the c-Met receptor (36). In this model system, phosphorylation of the adaptor protein Gab1 in response to HGF is dependent upon the activation of Src downstream of the β4 subunit. Gab1 recruits Grb2, which leads to Ras-dependent ERK activation. This HGF-stimulated pathway is decreased upon mutation of Y1494 in the β4 subunit, which reduces SHP-2 recruitment and Src activation. The potential involvement of Y1526 in the β4 subunit cytoplasmic domain was not addressed in this adaptor model.
Our data demonstrate that the ability of α6β4 to enhance invasion involves activation of both PI3K and Src. Although activation of either pathway alone can rescue invasion partially when Y1494 is mutated, both pathways are required to restore maximal invasive potential. In previous studies, we reported that PI3K functions downstream of the α6β4 integrin to promote activation of the small GTPase Rac (9). The α6β4 integrin is localized to lamellipodia and filipodial extensions in migrating carcinoma cells and the ability of this receptor to activate Rac is required for the formation of these actin-based motility structures (46). Interestingly, PI3K and Src have been previously shown to play a cooperative role in epithelial cell migration in colon cancer upon EGF stimulation via Rac activation (47). The ability of α6β4 to activate Rac downstream of PI3K has also been implicated in promoting resistance to apoptosis through NF-kB (15). Src has been shown in many studies to promote cell motility and invasion and it does so primarily through the regulation of FAK and focal adhesion turnover (48, 49). However, given that SFKs can phosphorylate the β4 subunit cytoplasmic domain, Src could play an alternative role in regulating the recruitment and activation of additional signaling molecules downstream of α6β4 that contribute to invasion. Future studies will be needed to establish the independent contributions of these signaling pathways to α6β4-dependent tumor invasion in vivo.
In summary, we have established that Y1494 in the β4 integrin cytoplasmic domain plays a key role in regulating the tumor-promoting functions of the α6β4 integrin. We have identified multiple signaling pathways that are regulated through Y1494 and we have demonstrated their differential involvement in regulating anchorage independent growth and invasion in vitro. The requirement for Y1494 in regulating tumor development in vivo underscores the importance of these signaling pathways for cancer and the need for continued studies to dissect further the mechanism by which α6β4 regulates their activation.
Footnotes
This work was supported by American Cancer Society grant RSG-05-223-01(LMS), NIH grant CA090583 (LMS) and a Susan G. Komen Postdoctoral Fellowship PDF0503775 (UD).
References
- 1.Falcioni R, Sacchi A, Resau J, Kennel SJ. Monoclonal antibody to human carcinoma-associated protein complex: quantitation in normal and tumor tissue. Cancer Res. 1988;48(4):816–21. [PubMed] [Google Scholar]
- 2.Van Waes C, Kozarsky KF, Warren AB, et al. The A9 antigen associated with aggressive human squamous carcinoma is structurally and functionally similar to the newly defined integrin alpha 6 beta 4. Cancer Res. 1991;51(9):2395–402. [PubMed] [Google Scholar]
- 3.Falcioni R, Turchi V, Vittulo P, et al. Integrin beta 4 expression in colorectal cancer. International Journal of Oncology. 1994;5:573–8. doi: 10.3892/ijo.5.3.573. [DOI] [PubMed] [Google Scholar]
- 4.Tagliabue E, Ghirelli C, Squicciarini P, Aiello P, Colnaghi MI, Menard S. Prognostic value of alpha 6 beta 4 integrin expression in breast carcinomas is affected by laminin production from tumor cells. Clin Cancer Res. 1998;4(2):407–10. [PubMed] [Google Scholar]
- 5.Lu S, Simin K, Khan A, Mercurio AM. Analysis of integrin beta4 expression in human breast cancer: association with basal-like tumors and prognostic significance. Clin Cancer Res. 2008;14(4):1050–8. doi: 10.1158/1078-0432.CCR-07-4116. [DOI] [PubMed] [Google Scholar]
- 6.Falcioni R, Kennel SJ, Giacomini P, Zupi G, Sacchi A. Expression of tumor antigen correlated with metastatic potential of Lewis lung carcinoma and B16 melanoma clones in mice. Cancer Research. 1986;46(11):5772–8. [PubMed] [Google Scholar]
- 7.Borradori L, Sonnenberg A. Structure and function of hemidesmosomes: more than simple adhesion complexes. J Invest Dermatol. 1999;112(4):411–8. doi: 10.1046/j.1523-1747.1999.00546.x. [DOI] [PubMed] [Google Scholar]
- 8.Chao C, Lotz MM, Clarke AC, Mercurio AM. A function for the integrin alpha6beta4 in the invasive properties of colorectal carcinoma cells. Cancer Research. 1996;56(20):4811–9. [PubMed] [Google Scholar]
- 9.Shaw LM, Rabinovitz I, Wang HHF, Toker A, Mercurio AM. Activation of phosphoinositide 3-OH kinase by the alpha-6-beta-4 integrin promotes carcinoma invasion. Cell. 1997;91(7):949–60. doi: 10.1016/s0092-8674(00)80486-9. [DOI] [PubMed] [Google Scholar]
- 10.Jauliac S, Lopez-Rodriguez C, Shaw LM, Brown LF, Rao A, Toker A. The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat Cell Biol. 2002;4(7):540–4. doi: 10.1038/ncb816. [DOI] [PubMed] [Google Scholar]
- 11.Lipscomb EA, Dugan AS, Rabinovitz I, Mercurio AM. Use of RNA interference to inhibit integrin (alpha6beta4)-mediated invasion and migration of breast carcinoma cells. Clin Exp Metastasis. 2003;20(6):569–76. doi: 10.1023/a:1025819521707. [DOI] [PubMed] [Google Scholar]
- 12.Bachelder RE, Marchetti A, Falcioni R, Soddu S, Mercurio AM. Activation of p53 function in carcinoma cells by the alpha6beta4 integrin. J Biol Chem. 1999;274(29):20733–7. doi: 10.1074/jbc.274.29.20733. [DOI] [PubMed] [Google Scholar]
- 13.Bachelder RE, Ribick MJ, Marchetti A, et al. p53 inhibits alpha 6 beta 4 integrin survival signaling by promoting the caspase 3-dependent cleavage of AKT/PKB. J Cell Biol. 1999;147(5):1063–72. doi: 10.1083/jcb.147.5.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chung J, Bachelder RE, Lipscomb EA, Shaw LM, Mercurio AM. Integrin (alpha 6 beta 4) regulation of eIF-4E activity and VEGF translation: a survival mechanism for carcinoma cells. J Cell Biol. 2002;158(1):165–74. doi: 10.1083/jcb.200112015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zahir N, Lakins JN, Russell A, et al. Autocrine laminin-5 ligates alpha6beta4 integrin and activates RAC and NFkappaB to mediate anchorage-independent survival of mammary tumors. J Cell Biol. 2003;163(6):1397–407. doi: 10.1083/jcb.200302023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lipscomb EA, Simpson KJ, Lyle SR, Ring JE, Dugan AS, Mercurio AM. The alpha6beta4 integrin maintains the survival of human breast carcinoma cells in vivo. Cancer Res. 2005;65(23):10970–6. doi: 10.1158/0008-5472.CAN-05-2327. [DOI] [PubMed] [Google Scholar]
- 17.Trusolino L, Bertotti A, Comoglio PM. A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell. 2001;107(5):643–54. doi: 10.1016/s0092-8674(01)00567-0. [DOI] [PubMed] [Google Scholar]
- 18.Abdel-Ghany M, Cheng HC, Elble RC, Pauli BU. The breast cancer beta 4 integrin and endothelial human CLCA2 mediate lung metastasis. J Biol Chem. 2001;276(27):25438–46. doi: 10.1074/jbc.M100478200. [DOI] [PubMed] [Google Scholar]
- 19.Guo W, Pylayeva Y, Pepe A, et al. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell. 2006;126(3):489–502. doi: 10.1016/j.cell.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 20.Yang XH, Richardson AL, Torres-Arzayus MI, et al. CD151 accelerates breast cancer by regulating alpha 6 integrin function, signaling, and molecular organization. Cancer Res. 2008;68(9):3204–13. doi: 10.1158/0008-5472.CAN-07-2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bon G, Folgiero V, Bossi G, et al. Loss of beta4 integrin subunit reduces the tumorigenicity of MCF7 mammary cells and causes apoptosis upon hormone deprivation. Clin Cancer Res. 2006;12(11 Pt 1):3280–7. doi: 10.1158/1078-0432.CCR-05-2223. [DOI] [PubMed] [Google Scholar]
- 22.Dajee M, Lazarov M, Zhang JY, et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421(6923):639–43. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- 23.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 24.Murgia C, Blaikie P, Kim N, Dans M, Petrie HT, Giancotti FG. Cell cycle and adhesion defects in mice carrying a targeted deletion of the integrin beta4 cytoplasmic domain. Embo J. 1998;17(14):3940–51. doi: 10.1093/emboj/17.14.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lipscomb EA, Mercurio AM. Mobilization and activation of a signaling competent alpha6beta4integrin underlies its contribution to carcinoma progression. Cancer Metastasis Rev. 2005;24(3):413–23. doi: 10.1007/s10555-005-5133-4. [DOI] [PubMed] [Google Scholar]
- 26.Gambaletta D, Marchetti A, Benedetti L, Mercurio AM, Sacchi A, Falcioni R. Cooperative signaling between alpha(6)beta(4) integrin and ErbB-2 receptor is required to promote phosphatidylinositol 3-kinase-dependent invasion. J Biol Chem. 2000;275(14):10604–10. doi: 10.1074/jbc.275.14.10604. [DOI] [PubMed] [Google Scholar]
- 27.Chung J, Yoon SO, Lipscomb EA, Mercurio AM. The Met receptor and alpha 6 beta 4 integrin can function independently to promote carcinoma invasion. J Biol Chem. 2004;279(31):32287–93. doi: 10.1074/jbc.M403809200. [DOI] [PubMed] [Google Scholar]
- 28.Yang X, Kovalenko OV, Tang W, Claas C, Stipp CS, Hemler ME. Palmitoylation supports assembly and function of integrin-tetraspanin complexes. J Cell Biol. 2004;167(6):1231–40. doi: 10.1083/jcb.200404100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rabinovitz I, Toker A, Mercurio AM. Protein kinase C-dependent mobilization of the alpha6beta4 integrin from hemidesmosomes and its association with actin-rich cell protrusions drive the chemotactic migration of carcinoma cells. J Cell Biol. 1999;146(5):1147–60. doi: 10.1083/jcb.146.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaw LM. Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the alpha6beta4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol Cell Biol. 2001;21(15):5082–93. doi: 10.1128/MCB.21.15.5082-5093.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dans M, Gagnoux-Palacios L, Blaikie P, Klein S, Mariotti A, Giancotti FG. Tyrosine phosphorylation of the beta 4 integrin cytoplasmic domain mediates Shc signaling to extracellular signal-regulated kinase and antagonizes formation of hemidesmosomes. J Biol Chem. 2001;276(2):1494–502. doi: 10.1074/jbc.M008663200. [DOI] [PubMed] [Google Scholar]
- 32.Santoro MM, Gaudino G, Marchisio PC. The MSP receptor regulates alpha6beta4 and alpha3beta1 integrins via 14-3-3 proteins in keratinocyte migration. Dev Cell. 2003;5(2):257–71. doi: 10.1016/s1534-5807(03)00201-6. [DOI] [PubMed] [Google Scholar]
- 33.Rabinovitz I, Tsomo L, Mercurio AM. Protein kinase C-alpha phosphorylation of specific serines in the connecting segment of the beta 4 integrin regulates the dynamics of type II hemidesmosomes. Mol Cell Biol. 2004;24(10):4351–60. doi: 10.1128/MCB.24.10.4351-4360.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Unkeless JC, Jin J. Inhibitory receptors, ITIM sequences and phosphatases. Curr Opin Immunol. 1997;9(3):338–43. doi: 10.1016/s0952-7915(97)80079-9. [DOI] [PubMed] [Google Scholar]
- 35.Merdek KD, Yang X, Taglienti CA, Shaw LM, Mercurio AM. Intrinsic signaling functions of the beta4 integrin intracellular domain. J Biol Chem. 2007;282(41):30322–30. doi: 10.1074/jbc.M703156200. [DOI] [PubMed] [Google Scholar]
- 36.Bertotti A, Comoglio PM, Trusolino L. Beta4 integrin activates a Shp2-Src signaling pathway that sustains HGF-induced anchorage-independent growth. J Cell Biol. 2006;175(6):993–1003. doi: 10.1083/jcb.200605114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma Z, Gibson SL, Byrne MA, Zhang J, White MF, Shaw LM. Suppression of insulin receptor substrate 1 (IRS-1) promotes mammary tumor metastasis. Mol Cell Biol. 2006;26(24):9338–51. doi: 10.1128/MCB.01032-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mercurio AM, Bachelder RE, Bates RC, Chung J. Autocrine signaling in carcinoma: VEGF and the alpha6beta4 integrin. Semin Cancer Biol. 2004;14(2):115–22. doi: 10.1016/j.semcancer.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 39.Mariotti A, Kedeshian PA, Dans M, Curatola AM, Gagnoux-Palacios L, Giancotti FG. EGF-R signaling through Fyn kinase disrupts the function of integrin alpha6beta4 at hemidesmosomes: role in epithelial cell migration and carcinoma invasion. J Cell Biol. 2001;155(3):447–58. doi: 10.1083/jcb.200105017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katz M, Amit I, Yarden Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim Biophys Acta. 2007;1773(8):1161–76. doi: 10.1016/j.bbamcr.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 42.Mansour SJ, Matten WT, Hermann AS, et al. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265(5174):966–70. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 43.Zhang SQ, Tsiaras WG, Araki T, et al. Receptor-specific regulation of phosphatidylinositol 3′-kinase activation by the protein tyrosine phosphatase Shp2. Mol Cell Biol. 2002;22(12):4062–72. doi: 10.1128/MCB.22.12.4062-4072.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang SQ, Yang W, Kontaridis MI, et al. Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol Cell. 2004;13(3):341–55. doi: 10.1016/s1097-2765(04)00050-4. [DOI] [PubMed] [Google Scholar]
- 45.Ivins Zito C, Kontaridis MI, Fornaro M, Feng GS, Bennett AM. SHP-2 regulates the phosphatidylinositide 3′-kinase/Akt pathway and suppresses caspase 3-mediated apoptosis. J Cell Physiol. 2004;199(2):227–36. doi: 10.1002/jcp.10446. [DOI] [PubMed] [Google Scholar]
- 46.Rabinovitz I, Mercurio AM. The integrin alpha6beta4 functions in carcinoma cell migration on laminin-1 by mediating the formation and stabilization of actin- containing motility structures. J Cell Biol. 1997;139(7):1873–84. doi: 10.1083/jcb.139.7.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dise RS, Frey MR, Whitehead RH, Polk DB. Epidermal growth factor stimulates Rac activation through Src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G276–85. doi: 10.1152/ajpgi.00340.2007. [DOI] [PubMed] [Google Scholar]
- 48.Cohen LA, Guan JL. Mechanisms of focal adhesion kinase regulation. Curr Cancer Drug Targets. 2005;5(8):629–43. doi: 10.2174/156800905774932798. [DOI] [PubMed] [Google Scholar]
- 49.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18(5):516–23. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]