

Abstract
Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are the leading cause of genetically inherited Parkinson’s disease (PD). While this multi-domain protein has been shown to have both GTPase and kinase activities through the Roc and MAPKKK domains, respectively, the protein-protein interactions and pathways involved in LRRK2-mediated signaling remain elusive. Utilizing a combination of protein pull-down assays, mass spectrometry, Western blotting and immunofluorescence microscopy, this study identifies and describes the interaction between LRRK2 and microtubules. The Roc or GTPase-like domain of LRRK2 is sufficient for interaction with α/β-tubulin heterodimers. This interaction occurs in a guanine-nucleotide independent manner, suggesting that tubulin may not be an effector of the LRRK2 GTPase domain. The R1441C pathogenic mutation, located within the Roc domain, retains interaction with α/β-tubulin heterodimers, suggesting that disruption of this interaction is not likely the mechanism whereby the R1441C mutation leads to disease. At a subcellular level, endogenous LRRK2 protein was found to colocalize with α/β-tubulin in primary hippocampal neurons. These findings are significant because they link LRRK2 with microtubules, a structural component of the cell that is critically involved in the pathogenesis of several neurodegenerative diseases, including PD.
Keywords: Parkinson’s disease, leucine-rich repeat kinase 2, R1441C, tubulin, neurodegeneration
Introduction
Parkinson’s disease (PD)1 is the second most prevalent neurodegenerative disorder. This disease is pathologically characterized by selective loss of dopaminergic neurons in the substantia nigra, which results in impaired motor function (Tanner and Ben-Shlomo 1999). The etiology of PD remains somewhat elusive and while most cases occur sporadically, mutations in several genes have been linked to rare familial forms of the disease (Moore et al., 2005).
Mutations in LRRK2 are the leading cause of autosomal dominantly inherited PD (Goldwurm et al., 2005; Hernandez et al., 2005). LRRK2, a member of the ROCO family, is a 286kDa protein with five putative functional domains; a N-terminal leucine-rich repeat (LRR) domain, a Roc (Ras of complex protein) domain that shares sequence homology to the Ras-related GTPase superfamily, a COR (C-terminal of Roc) domain, a mitogen-activated protein kinase kinase kinase (MAPKKK) domain and C-terminal WD40 repeats (Bosgraaf and Van Haastert 2003). Currently, there are several known pathogenic mutations within LRRK2, including three different amino acid substitutions at the same position within the Roc domain (R1441C/G/H) (Berg et al., 2005; Farrer et al., 2005; Khan et al., 2005; Mata et al., 2005).
Dysfunction of the microtubule-dependent transport system is emerging as a contributing factor in several neurodegenerative diseases, including PD (Crosby 2003). The cytoskeleton is critical in maintaining the asymmetrical shape and structural polarity of neuronal cells, making this cell type particularly susceptible to an abnormally assembled cytoskeleton, which can lead to impairment of neurotransmission and subsequent neuronal toxicity (Benitez-King et al. 2004). Interestingly, genes involved in microtubule-dependent transport have been shown to be downregulated in dopaminergic cells under conditions of oxidative stress and in the substantia nigra of PD patients compared to controls (Grunblatt et al., 2004; Kim et al., 2006; Noureddine et al., 2005; Yoo et al., 2003).
Several lines of evidence suggest that the accumulation of misfolded tubulin may be involved in the pathogenesis of PD (Burke et al., 1989; Weinstein and Solomon 1990). For example, both ubiquitin and tubulin are major components of Lewy bodies, suggesting that ubiquitinated tubulin may be present in this pathological feature of PD (Galloway et al., 1992; Lowe et al., 1988). Also, the dopaminergic neurotoxin 1-methyl-4-phenypyridinium (MPP+) and the agricultural pesticide rotenone both cause microtubule depolymerization and induce PD-like motor symptoms (Betarbet et al., 2000; Brinkley et al., 1974; Cappelletti et al., 2005; Langston et al., 1983; Przedborski and Jackson-Lewis 1998).
Numerous reports on genetic parkinsonism support the idea that microtubules may be a player in the neurodegenerative process. For example, α-synuclein interacts with α-tubulin and this interaction promotes α-synuclein fibrillogenesis, which may accelerate α-synuclein aggregation and subsequent formation of Lewy bodies (Alim et al., 2002). Also, α-synuclein overexpression leads to disruption of the microtubule network, impairment of microtubule-dependent trafficking and neuronal degeneration (Lee et al., 2006). Parkin, a protein-ubiquitin E3 ligase, strongly binds α/β-tubulin heterodimers to prevent microtubule depolymerization and ubiquitinates toxic free tubulin monomers to accelerate their degradation. These effects are abolished by PD-associated mutations of parkin (Burke et al., 1989; Ren et al., 2003; Yang et al., 2005). Tauopathies display filamentous deposits of hyperphosphorylated tau, an abundant neuronal specific microtubule-associated protein. Hyperphosphorylated tau loses its ability to bind to and stabilize microtubules, which causes deficits in microtubule-dependent trafficking and subsequent neuronal toxicity (Garcia and Cleveland 2001).
In this study we identify and describe the interaction between LRRK2 and microtubules. This is the first report, to our knowledge, that the Roc or GTPase-like domain of LRRK2 is sufficient for interaction with α/β-tubulin heterodimers. This interaction occurs in a guanine-nucleotide independent manner, suggesting that tubulin may not be an effector of the LRRK2 GTPase domain. The Roc domain R1441C pathogenic mutation retains interaction with α/β-tubulin heterodimers, suggesting that disruption of this interaction is not the mechanism whereby the R1441C mutation leads to disease. At a subcellular level, endogenous LRRK2 protein was found to colocalize with α/β-tubulin in primary hippocampal neurons. These findings are significant because they implicate an interaction of LRRK2 with microtubules, a structural component of the cell that is critically involved in the pathogenesis of PD.
Materials and Methods
Plasmids and cloning
Construction of LRRK2-FLAG constructs: A cDNA clone (TC124162) from a human cDNA library encoding full-length LRRK2 was isolated, sequenced and subcloned into the plasmid pCMV6-XL4 by OriGene Technologies (Rockville, MD). A C-terminal FLAG (DYKDDDDK) epitope tag was added to this cDNA by shuttling from pCMV6-XL4 into the mammalian expression plasmid pCEP4 (Invitrogen) to produce LRRK2(WT)-FLAG:pCEP4. The R1441C mutation was introduced into LRRK2(WT)-FLAG:pCEP4 by site-directed mutagenesis using the QuikChange II XL mutagenesis kit (Stratagene), according to the manufacturer’s instructions, to produce LRRK2(R1441C)-FLAG:pCEP4. The integrity of the cDNA open reading frame was confirmed by DNA sequencing (Davis Sequencing, Davis, CA).
Construction of glutathione S-transferase (GST)-Roc construct: The isolated Roc domain of LRRK2 was constructed by PCR using LRRK2(WT)-FLAG:pCEP4 as a template with the following primers; 5′-GCA GGA TCC ATG CGA ATG AAA CTT ATG ATT GTG GGA-3′ and 5′-TAA CCC GGG TCA GAA ATT AAG GCT CTC-3′. This region corresponds to amino acids 1334-1512 of LRRK2 (GenBank accession no. AY792511). The cDNA encoding the Roc domain of LRRK2 was subcloned C-terminally into the GST-fusion bacterial expression plasmid pGEX-KT to produce GST-Roc:pGEX-KT (Smith and Johnson 1988). The integrity of the PCR product was confirmed by DNA sequencing (Davis Sequencing, Davis, CA).
Expression of GST-Roc recombinant protein
Recombinant protein expression was induced in BL21 codon plus DES (RIL) Escherichia coli (Stratagene) with 1mM isopropyl β-D-thiogalactoside (IPTG). Bacteria were pelleted at 5,000×g for 10min at 4°C and lysed on ice in lysis buffer (100mM Hepes, pH 7.4; 2.5mM MgCl2; 0.1mM DTT; 100μM GTP; protease inhibitor cocktail (PIC) (Sigma)) by sonication. Sonicated lysates were pelleted at 10,000×g for 15min at 4°C and the supernatants filtered through 0.22μm filters resulting in a cleared protein lysate that was stored with 20% glycerol at −80°C. GST-Roc recombinant protein expression was verified by resolving on 12.5% Tris-glycine SDS-PAGE, electrophoretically transferring to polyvinylidine difluoride (PVDF) membrane (Millipore) and analyzing by Western blot with GST antibody (Novagen).
Cell culture, transfection and lysate preparation
Mouse fibroblast NIH-3T3 and human embryonic kidney 293T (HEK293T) cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Human neuroblastoma SH-SY5Y cells were cultured in Opti-MEM (Invitrogen) supplemented with 10% FBS and 1% penicillin-streptomycin. All cells were incubated at 37°C in 5% CO2. HEK293T cells were transiently transfected with LRRK2-FLAG:pCEP4 constructs using Lipofectamine Plus (Invitrogen) according to the manufacturer’s instructions. Cells were plated approximately 20hrs prior to transfection at a density of 800,000 cells/100mm dish and harvested 40–48hrs post-transfection. Mammalian cell lysates were prepared by washing and harvesting cells in Dulbecco’s phosphate buffered saline (DPBS), without calcium and magnesium. Cells were then homogenized in ice-cold lysis buffer (100mM Tris-HCl, pH 7.4; 0.1mM GDP; 0.1mM GTP; 1mM MgCl2; 50mM NaF; 1mM PMSF; PIC) for 2min followed by centrifugation at 500×g for 10min at 4°C to pellet nuclei and unbroken cells. Cleared mammalian cell lysates were then utilized in pull-down and co-precipitation assays as described below.
GST-Roc pull-down assays
GST-Roc recombinant protein was purified from bacterial lysates using glutathione (GSH) agarose beads (Sigma) according to the manufacturer’s instructions. Briefly, GSH-agarose was prepared by washing three times with 100mM DTT, once with 5mM DTT and resuspended in phosphate buffered saline (PBS). GST-Roc recombinant protein lysate was incubated with GSH-agarose for 2.5hrs at 4°C with rotation followed by washing twice with Buffer A (20mM Tris, pH 7.5; 100mM NaCl; 1mM EDTA; 0.5% NP-40; 0.5% dry milk; PIC) and washing twice with Buffer B (20mM Hepes, pH 7.9; 10% (v/v) glycerol; 60mM NaCl; 1mM DTT; 6mM MgCl2; 1mM EDTA; PIC). GST-Roc recombinant protein bound to the beads was then incubated with approximately 500μg of NIH-3T3 or SH-SY5Y cell lysate for 16hrs at 4°C with rotation. Mammalian cell lysates were prepared as described above. The beads were then washed twice with Buffer A and twice with Buffer B. For Western blot analysis, proteins were eluted from the beads with 10mM GSH, separated by 12.5% Tris-glycine SDS-PAGE and electrophoretically transferred to PVDF membrane. Membranes were analyzed by Western blotting with α-tubulin antibody (Santa Cruz), stripped with Stripping Buffer (50mM Tris-HCL, pH 2.3; 150mM NaCl), Western blotted with β-tubulin antibody (Sigma), stripped again and Western blotted with GST antibody. Western blots were quantified by computer-assisted densitometry using Quantity One software (Bio-Rad, Hercules, CA). To determine guanine nucleotide dependence for the interaction between GST-Roc and tubulin, GST-Roc protein was incubated with either 10mM GTP or 10mM GDP in the presence of 10mM EDTA, to unload already bound nucleotide, for 20min at 23°C with rotation prior to the pull-down assay. To stabilize the bound nucleotide, 20mM MgCl2 was subsequently added (Guo et al., 2007).
Protein sequencing by mass spectrometry
To identify proteins interacting with the GST-Roc domain of LRRK2, proteins from the GST-Roc pull down assays were eluted from GSH-agarose by boiling with Laemmli SDS sample buffer, separated by 12.5% Tris-glycine SDS-PAGE and visualized using the Silver Bullit kit (Amresco) according to the manufacturer’s instructions. Protein bands of interest on the silver stained gel were subjected to protein sequencing by liquid chromatography-electrospray ionization mass spectrometry (LC-ESI-MS) at the Lerner Research Institute Proteomics Laboratory at the Cleveland Clinic Foundation (Dr. Michael Kinter, Director). Protein bands of interest were cut from the gel, washed and destained in 50% ethanol, 5% acetic acid and reduced and alkylated with DTT and iodoacetamide. Gel pieces were then dehydrated in acetonitrile and dried in a Speed-Vac. In-gel proteolytic digestion was accomplished by adding 5μl of 20ng/μl trypsin in 50mM ammonium bicarbonate and incubated overnight at room temperature. Tryptic peptides were extracted from the polyacrylamide in two aliquots of 30μl in 50% acetonitrile with 5% formic acid. Extracts were then combined and evaporated to <10μl in a Speed-Vac and resuspended in 1% acetic acid to a final volume of 30μl. Identification of tryptic peptides was accomplished by LC-ESI-MS analysis on a Finnigan LCQ ion trap mass spectrometer system coupled with a capillary HPLC column (8cm × 75μm i.d.) packed with Jupiter C18 reversed-phase resin. The extracts (2 μl) were injected and the peptides eluted from the column by an acetonitrile/50mM acetic acid gradient at a flow rate of 0.3μl/min and were introduced into the source of the mass spectrometer on-line. The microelectrospray ion source was operated at 2.5kV. The peptides were analyzed by tandem MS using the data dependent multi-task capability of the instrument acquiring full scan mass spectra, to determine peptide molecular weights, and product ion mass spectra through collisionally induced dissociation (CID), to determine amino acid sequence in successive instrument scans. This mode of tandem MS analysis produces approximately 2500 CID spectra of ions ranging in abundance over several orders of magnitude. The data were then analyzed using all digitized CID mass spectra collected to search the National Center for Biotechnology Information (NCBI) non-redundant database with the search program Mascot (Matrix Sciences, London, UK) using a mammalian taxonomy filter. All mass spectra matching protein sequences in the NCBI database were verified by manual interpretation and by additional searches using the programs Sequest (ThermoElectron, San Jose, CA) and Blast (NCBI, Bethesda, MD) as needed.
LRRK2 co-precipitation assays
Mammalian cell lysates were prepared from HEK293T cells transiently transfected with LRRK2 constructs as described above. Cell lysates were incubated for 8hrs at 4°C with anti-LRRK2 antibody p268 (Novus Biologicals) conjugated to tosylactivated M-280 Dynabeads (Invitrogen) in IP Buffer (1mM PMSF and PIC in PBS). Beads were washed four times with ice-cold IP Buffer and proteins were eluted by boiling with Laemmli SDS sample buffer. Proteins were resolved by 6% Tris-glycine SDS-PAGE, electrophoretically transferred to PVDF membrane and analyzed by Western blot with FLAG antibody (Sigma) for LRRK2 overexpression and with α-tubulin and β-tubulin antibodies. Western blots were quantified by computer-assisted densitometry using Quantity One software.
Immunofluorescence microscopy of rat hippocampal neurons
Neurons from embryonic day 18 (E18) rat hippocampus (BrainBits) were seeded on Lab-Tek II CC2 chamber slides (Nalge Nunc International). At DIV (days in vitro) 7, the primary neuronal cells were washed with pre-warmed PBS and then fixed with freshly made 4% paraformaldehyde for 45min at room temperature. Cells were then permeabilized with 0.5% Triton X-100 in PBS for 30min and subsequently blocked with 10% normal goat serum (NGS) in PBS for 30min at room temperature. Cells were then incubated with primary antibodies (α-tubulin, mouse monoclonal, 1:500 (Upstate), β-tubulin, mouse monoclonal, 1:1000 (Sigma) and anti-LRRK2 p267, rabbit polyclonal, 1:50 (Novus Biologicals)) in PBS containing 1% NGS at 4°C overnight. After washing with PBS, cells were incubated in 10% NGS for 10min and then with goat-anti-mouse (GAM) or goat-anti-rabbit (GAR) FITC-conjugated secondary antibodies (Invitrogen) for 1hr at 37°C in the dark. Cells were then washed three times with PBS and mounted onto slides with ProLong anti-fade medium (Invitrogen). Fluorescence images were captured with a Zeiss LSM 510 inverted laser-scanning confocal fluorescence microscope. Confocal images of red fluorescence were collected using 543nm excitation light from an argon laser and a 560nm long pass filter. Confocal images of green fluorescence were collected using 488nm excitation light from an argon laser and a 500–550nm band pass barrier filter.
Results
The isolated Roc domain of LRRK2 selectively interacts with α/β-tubulin heterodimers
While the identity of LRRK2-interacting proteins remains elusive, determining such protein-protein interactions may help identify LRRK2-mediated signaling pathways. The studies described herein sought to identify proteins that interact selectively with the Roc or GTPase-like domain of LRRK2. This domain was isolated and cloned into a bacterial GST-tagging plasmid for GST-fusion recombinant protein expression and GST-Roc pull-down assays were performed from mammalian cell lysates, as a source of potential interacting proteins. Fig. 1A shows a representative silver stained gel of a GST-Roc pull-down assay. Three protein bands of interest were seen in the experimental lane (lane 4) that were not evident in any of the control lanes (lanes 1–3). These indicated bands of interest were excised from the gel, subjected to proteolytic digestion with trypsin and peptide sequences were determined by tandem mass spectrometry. Three LRRK2 Roc domain-interacting proteins were identified by this approach, including α/β-tubulin and two ribosomal proteins, S8 and L23 (arrows, Fig. 1A). Fig. 1B shows the tryptic peptides identified by LC-ESI-tandem MS, corresponding to the amino acid sequences of α-tubulin and β-tubulin isolated from the GST-Roc pull-down assay. A total of 17 tryptic peptides (Fig. 1B, bold) from the in-gel digested α-tubulin were identified by tandem MS, representing 51% protein coverage. A total of 20 tryptic peptides (Fig. 1B, bold) from the in-gel digested β-tubulin were also identified, representing 66% protein coverage. These results suggest that α/β-tubulin and several other proteins are potential binding partners of the Roc domain of LRRK2.
Fig. 1. The isolated Roc domain of LRRK2 selectively interacts with α/β-tubulin heterodimers.

(A) Identification of proteins interacting with the Roc domain of LRRK2. NIH-3T3 cell lysate was used as a source of interacting proteins in a pull-down assay with the recombinant GST-Roc domain of LRRK2 immobilized on GSH-agarose beads. Proteins bound to the beads were resolved by 12.5% Tris-glycine SDS-PAGE and silver-stained. Lanes 1–3 of the silver stained gel indicate negative controls while lane 4 represents the experimental condition. Indicated bands of interest (boxed) were excised, trypsinized and subjected to protein sequencing by tandem MS. Proteins identified by MS are labeled. The silver stained gel shown is representative of three independent experiments. (B) Summary of tryptic peptides identified by tandem MS within the amino acid sequences of α-tubulin and β-tubulin. A protein band of approximately 50kDa from a pull-down assay of the GST-Roc domain of LRRK2 (as shown in A, lane 4) was excised, in-gel digested and subjected to LC-ESI-MS analysis as detailed in Materials and Methods. Peptides identified by tandem MS for α-tubulin (Blast GI no. 13436317) and β-tubulin (Blast GI no. 18088719) are indicated in bold. (C) Western blot analysis of GST-Roc pull-down assay. Proteins present in NIH-3T3 and SH-SY5Y cell lysates that co-precipitate with the isolated GST-Roc domain of LRRK2 were resolved by 12.5% Tris-glycine SDS-PAGE, electrophoretically transferred to PVDF and Western blotted with GST, α-tubulin and β-tubulin antibodies. The Western blots shown are representative of three independent experiments.
Based on the involvement of microtubules in the neurodegenerative process we pursued characterization of the GST-Roc/tubulin interaction. To verify that this interaction exists in mammalian cells, Western blot analysis of GST-Roc pull-down assays was performed. As shown in Fig. 1C, α/β-tubulin, from lysates of both non-neuronal (NIH-3T3) and neuronal (SH-SY5Y) cell lines, co-precipitated with the isolated GST-Roc domain of LRRK2. The inability of α/β-tubulin to co-precipitate with the GST negative control demonstrates that this protein-protein interaction is specific for the Roc domain (Fig. 1C). These data indicate that the isolated Roc domain of LRRK2 selectively interacts with α/β-tubulin heterodimers from mammalian cells.
The Roc domain/tubulin interaction is guanine nucleotide independent
Ras-related GTPases serve as molecular switches to regulate diverse cellular functions by cycling between GTP-bound and GDP-bound conformations. It is in the GTP-bound conformation that GTPases are active and can bind to effector proteins to signal downstream cellular effects. Hydrolysis of GTP to GDP results in a conformational change that disrupts interaction with effector proteins, thereby terminating downstream signaling (Takai et al., 2001). Our group has recently shown that LRRK2 functions as an authentic GTPase, able to bind GTP and undergo intrinsic GTP hydrolysis, and that the isolated Roc domain is sufficient for these activities (Guo et al., 2007). It was, therefore, of interest to determine if α/β-tubulin heterodimers preferentially interact with GTP-bound GST-Roc versus GDP-bound GST-Roc.
To determine if a guanine nucleotide dependence for the GST-Roc/tubulin interaction existed, GST-Roc recombinant protein was pre-loaded with either GTP or GDP prior to performing pull-down assays with SH-SY5Y cell lysate as previously described (Guo et al., 2007). Western blot analysis demonstrates that α/β-tubulin co-precipitates similarly with GTP-bound GST-Roc compared to GDP-bound GST-Roc (Fig. 2A). The percent α/β-tubulin bound was quantified after normalizing to the amount of GST-Roc expressed (Fig. 2B). There is no statistically significant difference between the amount of α/β-tubulin co-precipitated with GTP-bound GST-Roc compared to that co-precipitated with GDP-bound GST-Roc (Fig. 2B). These data suggest that the Roc domain/tubulin interaction occurs in a guanine nucleotide independent manner.
Fig. 2. The Roc domain/tubulin interaction is guanine nucleotide independent.
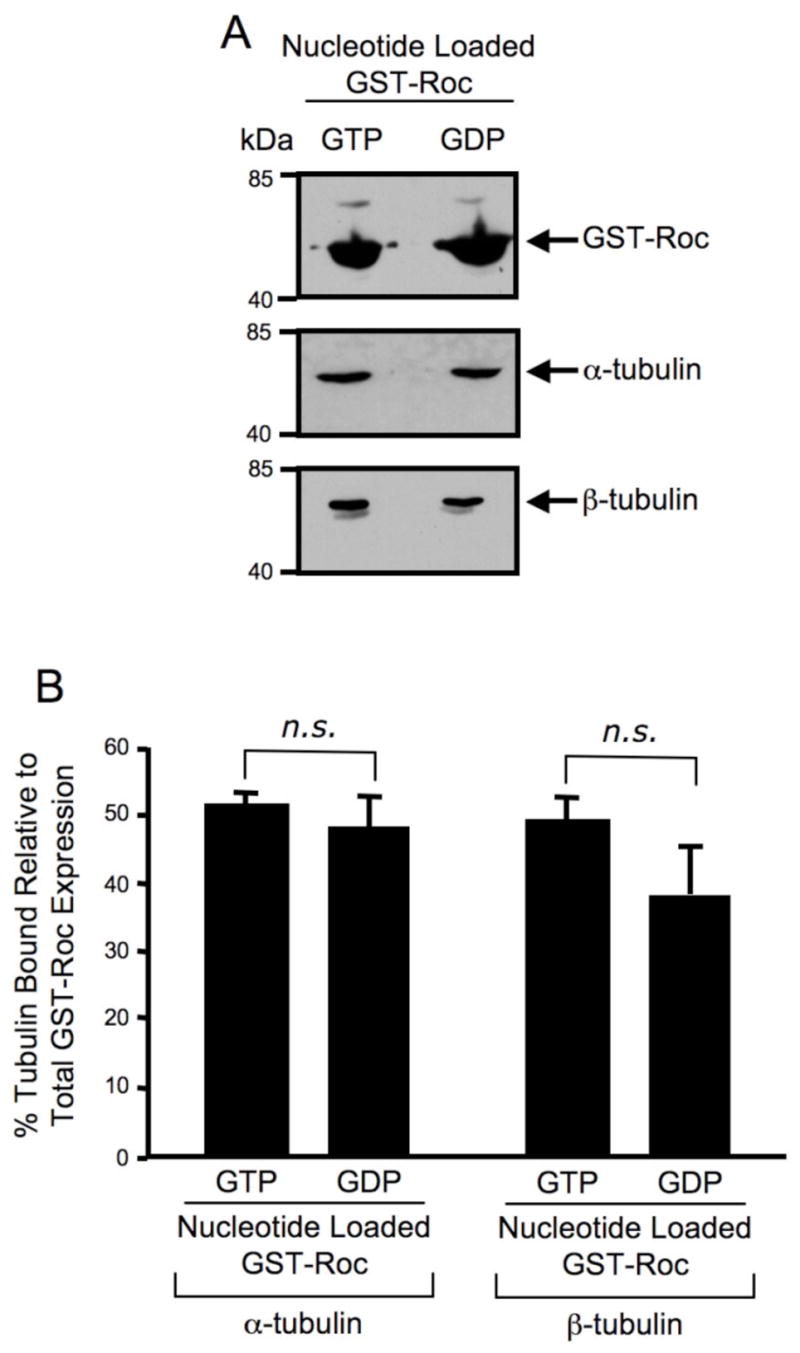
(A) Western blot analysis of guanine nucleotide loaded GST-Roc pull-down assay. GST-Roc recombinant protein was pre-loaded with either GTP or GDP prior to a pull-down assay with SH-SY5Y cell lysate. Precipitating proteins were resolved by 12.5% Tris-glycine SDS-PAGE, electrophoretically transferred to PVDF and Western blotted with GST, α-tubulin and β-tubulin antibodies. The Western blots shown are representative of three independent experiments. (B) Quantification of Western blot analysis of guanine nucleotide loaded GST-Roc pull-down assay. GST, α-tubulin and β-tubulin Western blots were quantified by densitometry and the percentage of α/β-tubulin bound was normalized to GST-Roc protein levels. Error bars represent standard error of the mean (SEM) for three independent experiments. n.s. is non-significant as assessed by a two-tailed unpaired Student’s t-test.
Wild-type and the R1441C pathogenic mutant of LRRK2 selectively interact with α/β-tubulin heterodimers
The data described thus far demonstrate that the isolated Roc domain of LRRK2 interacts with α/β-tubulin heterodimers and that this interaction occurs in a guanine nucleotide independent manner. It was next confirmed that this interaction occurs with full-length LRRK2. Briefly, wild-type and R1441C mutant LRRK2 were overexpressed in HEK293T cells and proteins were immunoprecipitated from cell lysates utilizing anti-LRRK2 coupled to Dynabeads. Western blot analysis demonstrates that α/β-tubulin co-precipitates similarly with both wild-type and R1441C mutant LRRK2 (Fig. 3A). The inability of α/β-tubulin to co-precipitate from control (untransfected) cells demonstrates that this interaction is specific for LRRK2 (Fig. 3A). The percent α/β-tubulin bound was quantified after normalizing to the amount of LRRK2-FLAG protein overexpressed (Fig. 3B). There is no statistically significant difference between the amount of α/β-tubulin co-precipitated with wild-type LRRK2 compared to that co-precipitated with LRRK2(R1441C) (Fig. 3B). These data indicate that LRRK2 selectively interacts with α/β-tubulin heterodimers in mammalian cells and that the R1441C pathogenic mutant of LRRK2 retains association with tubulin.
Fig. 3. Wild-type and the R1441C pathogenic mutant of LRRK2 selectively interact with α/β-tubulin heterodimers.
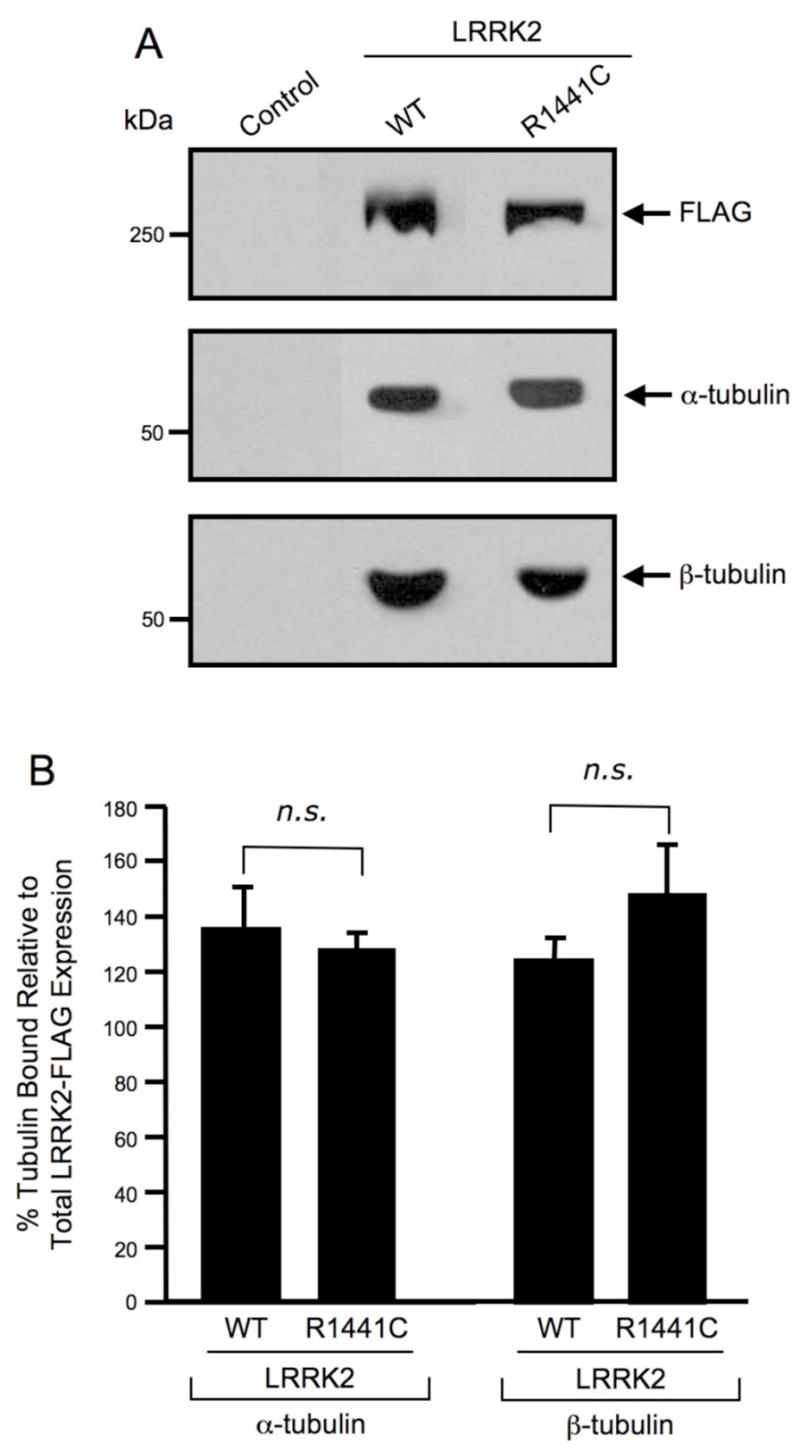
(A) Western blot analysis of LRRK2 co-precipitation assay. FLAG-tagged wild-type and mutant LRRK2 proteins were overexpressed in HEK293T cells and immunoprecipitated with anti-LRRK2 coupled to Dynabeads. Immunoprecipitates were separated by 6% Tris-glycine SDS-PAGE, electrophoretically transferred to PVDF and Western blotted with FLAG, α-tubulin and β-tubulin antibodies. Control refers to untransfected cells. The Western blots shown are representative of three independent experiments. (B) Quantification of Western blot analysis of LRRK2 co-precipitation assay. FLAG, α-tubulin and β-tubulin Western blots were quantified by densitometry and the percentage of α/β-tubulin bound was normalized to LRRK2-FLAG protein expression levels. Error bars represent SEM for three independent experiments. n.s. is non-significant as assessed by a two-tailed unpaired Student’s t-test.
Endogenous LRRK2 co-localizes with α/β-tubulin in primary hippocampal neurons
Immunofluorescence confocal microscopy of primary embryonic rat hippocampal neurons was performed to determine the localization of endogenous LRRK2 and α/β-tubulin. Neurons were fixed, permeabilized and co-stained with LRRK2 and α-tubulin or β-tubulin antibodies, followed by labeling with fluorophore-tagged secondary antibodies. Filamentous α/β-tubulin staining can be seen throughout the cell body and along dendritic and axonal processes (Fig. 4A and 4D). Endogenous LRRK2 demonstrated cytosolic localization and can also be seen along dendritic and axonal processes (Fig. 4B and 4E). The merge images (Fig. 4C and 4F) reveal co-localization of LRRK2 and α/β-tubulin in both the cell body and along neuronal processes. These data suggest that LRRK2 co-localizes with microtubules in primary neuronal cells.
Fig. 4. Endogenous LRRK2 co-localizes with α/β-tubulin in primary hippocampal neurons.

Cultured embryonic rat hippocampal neurons were used for immunofluorescence microscopy. Neuronal cells were immunostained with respective primary antibodies followed by labeling with fluorophore-tagged secondary antibodies. Fluorescence images were analyzed by inverted laser-scanning confocal microscopy. (A). α-tubulin mouse monoclonal antibody primary staining followed by Alexa Flour GAM-488 secondary staining. (B) LRRK2 rabbit polyclonal antibody primary staining followed by Alexa Flour GAR-543 secondary staining. (C) Digitally merged image of Panels A and B. (D). β-tubulin mouse monoclonal antibody primary staining followed by Alexa Flour GAM-488 secondary staining. (E) LRRK2 rabbit polyclonal antibody primary staining followed by Alexa Flour GAR-543 secondary staining. (F) Digitally merged image of Panels D and E. Small white boxes in Panels A-F indicate zoomed area as shown in Panel insets. (G) A field of cultured embryonic rat hippocampal neurons viewed by differential interference contrast (DIC). (H-I) Staining with fluorophore-labeled secondary antibodies alone. Scale bars are as indicated.
Discussion
LRRK2 mutations are the most common susceptibility determinant of PD identified to date, however, very little is known about the protein-protein interactions associated with LRRK2 signaling. Discovering LRRK2-interacting proteins and elucidating LRRK2-mediated signaling pathways will be crucial in understanding and combating the pathogenesis of PD. In this study we have identified and described the interaction between LRRK2 and microtubules. This is the first report, to our knowledge, that the Roc or GTPase-like domain of LRRK2 is sufficient for interaction with α/β-tubulin heterodimers (Fig. 1). This interaction occurs in a guanine nucleotide independent manner (Fig. 2), suggesting that tubulin is likely not an effector of the LRRK2 GTPase domain. The R1441C pathogenic mutant of LRRK2, which occurs within the Roc domain, retains interaction with α/β-tubulin heterodimers, suggesting that disruption of this interaction is likely not the mechanism whereby this mutation leads to PD (Fig. 3). Endogenous LRRK2 and α/β-tubulin were found to co-localize in primary hippocampal neurons (Fig. 4), suggesting that this specific interaction occurs in vivo. These findings are significant because they implicate LRRK2 as a binding partner with microtubules, a structural component of the cell that is critically involved in neurodegenerative processes.
There is a growing body of evidence linking LRRK2 with components of the cytoskeleton, including microtubules. For example, GFP-tagged LRRK2 co-localizes with β-tubulin in HEK293T cells, but fails to overlap with the actin cytoskeleton or with intermediate filaments in this cell line (Gloeckner et al., 2006). The G2019S PD-associated mutant of LRRK2 leads to tau-positive inclusions in neurons and co-localizes with tau within these inclusions, demonstrating interaction with this neuronal-specific microtubule-associated protein (MacLeod et al., 2006). Similar to the results presented here, endogenous LRRK2 co-localizes with β-tubulin in primary rat cortical neurons and was also found to co-localize with Golgi transport vesicles, endosomes, lysosomes and mitochondria, suggesting a possible role for LRRK2 in vesicular transport and/or microtubule-dependent transport (Biskup et al., 2006). LRRK2 also immunoprecipitates with several cytoskeletal and trafficking proteins such as clathrin heavy chain, vimentin and Sec1 family domain containing protein 1 (Dachsel et al., 2007). The present study demonstrates that the Roc domain of LRRK2 is sufficient for interaction with α/β-tubulin heterodimers.
Due to the extreme polarity and long processes characteristic of neurons, microtubule-dependent transport is particularly important in maintaining neuronal structure and function. Supporting evidence for this is that several neurodegenerative diseases are credited to mutations in components of the microtubule system. For example, mutations in the motor protein KIF1Bβ and in the dynactin subunit p150glued cause Charcot-Marie-Tooth disease and a form of lower motor neuron disease, respectively (Puls et al., 2003; Zhao et al., 2001). Also, mutations in the MAP1B interacting protein gigaxonin and in the neuronal-specific microtubule-associated protein tau cause giant axonal neuropathy and frontotemporal dementia with parkinsonism linked to chromosome 17, respectively (Ding et al., 2002; Hutton et al., 1998). If LRRK2 is involved in microtubule-dependent transport, PD-associated mutations may disrupt this transport and lead to neuronal toxicity. Our studies indicate that the R1441C pathogenic mutant of LRRK2 retains interaction with α/β-tubulin heterodimers, but this does not eliminate the possibility that this mutant may disrupt a microtubule related function.
It is interesting to note that LRRK2 is not the first member of the ROCO protein family to be associated with components of the cytoskeleton. For example, another human ROCO protein, death-associated protein kinase (DAPk), is a Ca2+/calmodulin-regulated ser/thr kinase that positively influences apoptosis. DAPk phosphorylates myosin light chain kinase, which results in membrane blebbling, a process that requires cytoskeletal rearrangements. Interestingly, DAPk associates with actin microfilaments and this interaction is essential for its death-promoting activity. Analogous to the results presented here, the Roc domain of DAPk is found to be essential and sufficient for interaction with the actin cytoskeleton (Bialik et al., 2004; Cohen et al., 1997). GbpC, a Dictyostelium ROCO protein, is critically involved in cell polarity and chemotaxis through, at least in part, activation of myosin II light chain phosphorylation (Bosgraaf et al., 2002). Pats1, another Dictyostelium ROCO protein, plays a role in cytokinesis by regulating the localization of myosin heavy chain to the cleavage furrow (Abysalh et al., 2003). In the future, it will be interesting to determine if association with and/or regulation of cytoskeletal components is a general feature of ROCO family proteins.
The studies described herein indicate that association of LRRK2 with α/β-tubulin heterodimers occurs to a similar extent regardless of whether the Roc or GTPase-like domain of LRRK2 is in a GTP or GDP-bound conformation. While the physiological significance of the LRRK2-tubulin interaction is currently unknown, these data suggest that tubulin in not an effector of the LRRK2 GTPase domain. However, this interaction may serve a structural role or possibly be involved in controlling proper localization of LRRK2. While it is unlikely that tubulin is a signaling partner of the LRRK2 GTPase domain, this does not eliminate the possibility that tubulin, or other microtubule associated proteins, may be targets of LRRK2 kinase activity. Determining if LRRK2 phosphorylates microtubules or microtubule associated proteins will be important in elucidating the functional significance of this interaction. As the protein-protein interactions that enable LRRK2-mediated signaling are elucidated, determination of the functional significance of the LRRK2-tubulin interaction will likely be facilitated. Since LRRK2 and microtubules are both critical players implicated in neurodegenerative processes, further characterization of the interaction between these two cellular components may lead to improved understanding of Parkinson’s disease pathogenesis.
Footnotes
This work was supported in part by the National Institutes of Health (grant 1R21AG028797), National Parkinson Foundation (Mega Research Grants Program), National Science Foundation (Advance Institutional Transformation Grant SBE-0245054) and Johnson & Johnson Corporate Office of Science and Technology (Focused Funding Program).
Abbreviations
PD, Parkinson’s disease; LRRK2, leucine-rich repeat kinase 2; GST, glutathione S-transferase; PCR, polymerase chain reaction; IPTG, isopropyl β-D-thiogalactoside; PIC, protease inhibitor cocktail; PMSF, phenylmethylsulphonyl fluoride; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; PVDF, polyvinylidine difluoride; HEK293T, human embryonic kidney 293T cell line; DMEM, Dulbecco’s modified Eagle’s medium; FBS, fetal bovine serum; DPBS, Dulbecco’s phosphate buffered saline; GSH, glutathione; DTT, dithiothreitol; PBS, phosphate buffered saline; EDTA, ethylenediamine tetraacetic acid; LC-ESI-MS, liquid chromatography-electrospray ionization mass spectrometry; HPLC, high performance liquid chromatography; CID, collisionally induced dissociation; NCBI, National Center for Biotechnology Information; DIV, days in vitro; NGS, normal goat serum; GAM, goat-anti-mouse; GAR, goat-anti-rabbit; DIC, differential interference contrast; SEM, standard error of the mean.
References
- Abysalh JC, Kuchnicki LL, Larochelle DA. The identification of pats1, a novel gene locus required for cytokinesis in Dictyostelium discoideum. Mol Biol Cell. 2003;14(1):14–25. doi: 10.1091/mbc.E02-06-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alim MA, Hossain MS, Arima K, Takeda K, Izumiyama Y, Nakamura M, Kaji H, Shinoda T, Hisanaga S, Ueda K. Tubulin seeds alpha-synuclein fibril formation. J Biol Chem. 2002;277(3):2112–2117. doi: 10.1074/jbc.M102981200. [DOI] [PubMed] [Google Scholar]
- Benitez-King G, Ramirez-Rodriguez G, Ortiz L, Meza I. The neuronal cytoskeleton as a potential therapeutical target in neurodegenerative diseases and schizophrenia. Curr Drug Targets CNS Neurol Disord. 2004;3(6):515–533. doi: 10.2174/1568007043336761. [DOI] [PubMed] [Google Scholar]
- Berg D, Schweitzer K, Leitner P, Zimprich A, Lichtner P, Belcredi P, Brussel T, Schulte C, Maass S, Nagele T. Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson’s disease. Brain. 2005;128(Pt 12):3000–3011. doi: 10.1093/brain/awh666. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3(12):1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Bialik S, Bresnick AR, Kimchi A. DAP-kinase-mediated morphological changes are localization dependent and involve myosin-II phosphorylation. Cell Death Differ. 2004;11(6):631–644. doi: 10.1038/sj.cdd.4401386. [DOI] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RL, Emson PC, Torp R, Ottersen OP, Dawson TM, Dawson VL. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60(5):557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- Bosgraaf L, Russcher H, Smith JL, Wessels D, Soll DR, Van Haastert PJ. A novel cGMP signalling pathway mediating myosin phosphorylation and chemotaxis in Dictyostelium. Embo J. 2002;21(17):4560–4570. doi: 10.1093/emboj/cdf438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosgraaf L, Van Haastert PJ. Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 2003;1643(1–3):5–10. doi: 10.1016/j.bbamcr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Brinkley BR, Barham SS, Barranco SC, Fuller GM. Rotenone inhibition of spindle microtubule assembly in mammalian cells. Exp Cell Res. 1974;85(1):41–46. doi: 10.1016/0014-4827(74)90210-9. [DOI] [PubMed] [Google Scholar]
- Burke D, Gasdaska P, Hartwell L. Dominant effects of tubulin overexpression in Saccharomyces cerevisiae. Mol Cell Biol. 1989;9(3):1049–1059. doi: 10.1128/mcb.9.3.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappelletti G, Surrey T, Maci R. The parkinsonism producing neurotoxin MPP+ affects microtubule dynamics by acting as a destabilising factor. FEBS Lett. 2005;579(21):4781–4786. doi: 10.1016/j.febslet.2005.07.058. [DOI] [PubMed] [Google Scholar]
- Cohen O, Feinstein E, Kimchi A. DAP-kinase is a Ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. Embo J. 1997;16(5):998–1008. doi: 10.1093/emboj/16.5.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby AH. Disruption of cellular transport: a common cause of neurodegeneration? Lancet Neurol. 2003;2(5):311–316. doi: 10.1016/s1474-4422(03)00383-1. [DOI] [PubMed] [Google Scholar]
- Dachsel JC, Taylor JP, Mok SS, Ross OA, Hinkle KM, Bailey RM, Hines JH, Szutu J, Madden B, Petrucelli L, Farrer MJ. Identification of potential protein interactors of Lrrk2. Parkinsonism Relat Disord. 2007 doi: 10.1016/j.parkreldis.2007.01.008. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Liu JJ, Kowal AS, Nardine T, Bhattacharya P, Lee A, Yang Y. Microtubule-associated protein 1B: a neuronal binding partner for gigaxonin. J Cell Biol. 2002;158(3):427–433. doi: 10.1083/jcb.200202055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer M, Stone J, Mata IF, Lincoln S, Kachergus J, Hulihan M, Strain KJ, Maraganore DM. LRRK2 mutations in Parkinson disease. Neurology. 2005;65(5):738–740. doi: 10.1212/01.wnl.0000169023.51764.b0. [DOI] [PubMed] [Google Scholar]
- Galloway PG, Mulvihill P, Perry G. Filaments of Lewy bodies contain insoluble cytoskeletal elements. Am J Pathol. 1992;140(4):809–822. [PMC free article] [PubMed] [Google Scholar]
- Garcia ML, Cleveland DW. Going new places using an old MAP: tau, microtubules and human neurodegenerative disease. Curr Opin Cell Biol. 2001;13(1):41–48. doi: 10.1016/s0955-0674(00)00172-1. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O’Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15(2):223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- Goldwurm S, Di Fonzo A, Simons EJ, Rohe CF, Zini M, Canesi M, Tesei S, Zecchinelli A, Antonini A, Mariani C, Meucci N, Sacilotto G, Sironi F, Salani G, Ferreira J, Chien HF, Fabrizio E, Vanacore N, Dalla Libera A, Stocchi F, Diroma C, Lamberti P, Sampaio C, Meco G, Barbosa E, Bertoli-Avella AM, Breedveld GJ, Oostra BA, Pezzoli G, Bonifati V. The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson’s disease and originates from a common ancestor. J Med Genet. 2005;42(11):e65. doi: 10.1136/jmg.2005.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunblatt E, Mandel S, Jacob-Hirsch J, Zeligson S, Amariglo N, Rechavi G, Li J, Ravid R, Roggendorf W, Riederer P, Youdim MB. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J Neural Transm. 2004;111(12):1543–1573. doi: 10.1007/s00702-004-0212-1. [DOI] [PubMed] [Google Scholar]
- Guo L, Gandhi PN, Wang W, Petersen RB, Wilson-Delfosse AL, Chen SG. The Parkinson’s disease associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res. 2007 doi: 10.1016/j.yexcr.2007.07.007. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez D, Paisan Ruiz C, Crawley A, Malkani R, Werner J, Gwinn-Hardy K, Dickson D, Wavrant Devrieze F, Hardy J, Singleton A. The dardarin G 2019 S mutation is a common cause of Parkinson’s disease but not other neurodegenerative diseases. Neurosci Lett. 2005;389(3):137–139. doi: 10.1016/j.neulet.2005.07.044. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Khan NL, Jain S, Lynch JM, Pavese N, Abou-Sleiman P, Holton JL, Healy DG, Gilks WP, Sweeney MG, Ganguly M, Gibbons V, Gandhi S, Vaughan J, Eunson LH, Katzenschlager R, Gayton J, Lennox G, Revesz T, Nicholl D, Bhatia KP, Quinn N, Brooks D, Lees AJ, Davis MB, Piccini P, Singleton AB, Wood NW. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain. 2005;128(Pt 12):2786–2796. doi: 10.1093/brain/awh667. [DOI] [PubMed] [Google Scholar]
- Kim JM, Lee KH, Jeon YJ, Oh JH, Jeong SY, Song IS, Kim JM, Lee DS, Kim NS. Identification of genes related to Parkinson’s disease using expressed sequence tags. DNA Res. 2006;13(6):275–286. doi: 10.1093/dnares/dsl016. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219(4587):979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Khoshaghideh F, Lee S, Lee SJ. Impairment of microtubule-dependent trafficking by overexpression of alpha-synuclein. Eur J Neurosci. 2006;24(11):3153–3162. doi: 10.1111/j.1460-9568.2006.05210.x. [DOI] [PubMed] [Google Scholar]
- Lowe J, Blanchard A, Morrell K, Lennox G, Reynolds L, Billett M, Landon M, Mayer RJ. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J Pathol. 1988;155(1):9–15. doi: 10.1002/path.1711550105. [DOI] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52(4):587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Mata IF, Kachergus JM, Taylor JP, Lincoln S, Aasly J, Lynch T, Hulihan MM, Cobb SA, Wu RM, Lu CS, Lahoz C, Wszolek ZK, Farrer MJ. Lrrk2 pathogenic substitutions in Parkinson’s disease. Neurogenetics. 2005;6(4):171–177. doi: 10.1007/s10048-005-0005-1. [DOI] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Noureddine MA, Li YJ, van der Walt JM, Walters R, Jewett RM, Xu H, Wang T, Walter JW, Scott BL, Hulette C, Schmechel D, Stenger JE, Dietrich F, Vance JM, Hauser MA. Genomic convergence to identify candidate genes for Parkinson disease: SAGE analysis of the substantia nigra. Mov Disord. 2005;20(10):1299–1309. doi: 10.1002/mds.20573. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V. Mechanisms of MPTP toxicity. Mov Disord 13 Suppl. 1998;1:35–38. [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33(4):455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Ren Y, Zhao J, Feng J. Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J Neurosci. 2003;23(8):3316–3324. doi: 10.1523/JNEUROSCI.23-08-03316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67(1):31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81(1):153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Ben-Shlomo Y. Epidemiology of Parkinson’s disease. Adv Neurol. 1999;80:153–159. [PubMed] [Google Scholar]
- Weinstein B, Solomon F. Phenotypic consequences of tubulin overproduction in Saccharomyces cerevisiae: differences between alpha-tubulin and beta-tubulin. Mol Cell Biol. 1990;10(10):5295–5304. doi: 10.1128/mcb.10.10.5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Jiang Q, Zhao J, Ren Y, Sutton MD, Feng J. Parkin stabilizes microtubules through strong binding mediated by three independent domains. J Biol Chem. 2005;280(17):17154–17162. doi: 10.1074/jbc.M500843200. [DOI] [PubMed] [Google Scholar]
- Yoo MS, Chun HS, Son JJ, DeGiorgio LA, Kim DJ, Peng C, Son JH. Oxidative stress regulated genes in nigral dopaminergic neuronal cells: correlation with the known pathology in Parkinson’s disease. Brain Res Mol Brain Res. 2003;110(1):76–84. doi: 10.1016/s0169-328x(02)00586-7. [DOI] [PubMed] [Google Scholar]
- Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, Saito M, Tsuji S, Hayashi Y, Hirokawa N. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105(5):587–597. doi: 10.1016/s0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]