

Abstract
Although Hedgehog (HH) signaling plays a critical role in patterning the ventral midbrain, its role in early midbrain specification is not known. We examined the midbrains of sonic hedgehog (Shh) and smoothened (Smo) mutant mice where HH signaling is respectively attenuated and eliminated. We show that some ventral (Evx1+) cell-fates are specified in the Shh-/- mouse in a Ptc1- and Gli1-independent manner. HH-independent ventral midbrain induction was further confirmed by the presence of a Pax7-negative ventral midbrain territory in both Shh-/- and Smo-/- mice at and before E8.5. Midbrain signaling centers are severely disrupted in the Shh-/- mutant. Interestingly, dorsal markers are upregulated (Wnt1, Gdf7, Pax7), downregulated (Lfng) or otherwise altered (Zic1) in the Shh-/- midbrain. Together with the increased cell-death seen specifically in Shh-/- dorsal midbrains (E8.5-E9), our results suggest specific regulation of dorsal patterning by SHH, rather than a simple deregulation due to its absence.
Keywords: HH-independent ventral midbrain specification, Midbrain-hindbrain boundary, size regulation, roof plate, oculomotor neurons, dopaminergic neurons, Evx1
Introduction
The role of Hedgehog (HH) signaling in patterning the ventral neural tube is well established (Jacob and Briscoe, 2003). The HH family member, Sonic Hedgehog (SHH) is secreted by the floor plate, a ventral midline signaling center which extends along the length of the neuraxis (Marti et al., 1995; Ericson et al., 1996). HH signaling begins when one of the HH family members (SHH, Indian [IHH] or Desert Hedgehog [DHH]) binds to the HH receptor, patched (Marigo et al., 1996; Stone et al., 1996). In the absence of HH signaling, patched (PTC) maintains a blockade on the seven-pass transmembrane protein SMO, an obligate effector of HH signaling (Alcedo et al., 1996; Akiyama et al., 1997). HH binding to PTC lifts the blockade on SMO so that downstream signaling can occur via the GLI family of transcription factors (Ingham and McMahon, 2001).
HH signaling has been extensively studied in the ventral spinal cord where multiple classes of ventral cell-fates are specified at different distances from the HH source (Jessell, 2000). In vitro gain of function experiments have shown that lower concentrations of the HH protein specify progressively more dorsal spinal cell fates. Ventral to dorsal, these fates include the floor plate (FP), V3 interneurons, spinal motor neurons, and V2, V1 and V0 (EVX1+) interneurons (Roelink et al., 1995; Ericson et al., 1996).
Despite the sufficiency of HH in specifying all ventral cell-fates, some spinal cell-fates (V0, V1, few V2), are specified in the Shh-/- mouse, raising the issue of whether other HHs (e.g., IHH) or HH-independent signals (e.g., retinoic acid) might be involved in patterning these cell-fates (Litingtung and Chiang, 2000; Zhang et al., 2001; Wijgerde et al., 2002). Interestingly, with the exception of floor plate and V3 interneurons, all ventral spinal fates are present in the Gli2-/-:Gli3-/- and Smo-/-:Gli3-/- double knockouts where no HH signaling is likely to occur (Wijgerde et al., 2002; Bai et al., 2004). Thus, some ventral cell-fates can be specified in the spinal cord in the absence of HH signaling, if GLI3 is also simultaneously removed (Litingtung and Chiang, 2000; Wijgerde et al., 2002; Bai et al., 2004). The absence of all ventral spinal cord cell-fates in the Smo-/- mouse has led to the idea that ventral cell-fate specification in the spinal cord requires HH signaling as long as the repressive effect of GLI3 is also present (Zhang et al., 2001; Wijgerde et al., 2002). In the telencephalon, however, FGF receptor double mutants, (e.g., Ffgr1-/-:Fgfr2-/-) display severe ventral cell-fate defects despite the presence of SHH (Gutin et al., 2006). Most interestingly, the simultaneous elimination of Gli3 does not rescue ventral cell-fates in these mutants, suggesting that HH-independent pathways may play an important role in ventral specification in anterior regions of the neural tube (Gutin et al., 2006).
Considerably less attention has been focused on the patterning of the vertebrate ventral midbrain. Here, cell-fates are organized into longitudinal stripes (midbrain arcs) arrayed parallel to the midbrain/rostral floor plate (rFP) source of SHH (Agarwala et al., 2001; Sanders et al., 2002). We have previously shown that in the chick, an ectopic source of SHH can specify the entire ventral midbrain pattern of arcs in a concentration- and position- dependent manner (Agarwala et al., 2001). Several clinically important neuronal cell-types, such as the oculomotor complex (OMC), the red nucleus and the dopaminergic (MDA) neurons are midbrain constituents and develop within the context of the midbrain arc pattern (Agarwala and Ragsdale, 2002). We have previously shown that SHH overexpression is also sufficient to specify these midbrain nuclei (Agarwala and Ragsdale, 2002). Recent studies have shown that in the chick and mouse midbrain, HH signaling is not just sufficient, but also necessary for the specification of ventral cell-types (Blaess et al., 2006; Bayly et al., 2007).
In the chick, HH signaling can induce the rFP at presomitic stages (Hamburger-Hamilton stages 4-6), although Nodal signaling appears to be important for this induction as well (Hamburger and Hamilton, 1951; Patten et al., 2003). HH blockade studies in the chick corroborate an early requirement for HH, but suggest that additional factors may also be important in midbrain patterning (Bayly et al., 2007). Mouse conditional knockout studies have shown that HH signaling is required early (prior to E9.5) for ventral cell-fate specification (Ishibashi and McMahon, 2002; Blaess et al., 2006). Importantly however, the state of the ventral midbrain before this time point is not known. For example, whether the early ventralization of the midbrain and the specification of all ventral midbrain cell-fates absolutely depends upon HH signaling or requires additional signals has not yet been explored.
In the fly wing and abdominal epithelia, compartment boundaries or signaling centers are established and maintained in response to HH-mediated regulation of cell affinities (Dahmann and Basler, 1999). Recent studies have shown that the midbrain-hindbrain boundary (MHB) is perturbed in the absence of HH signaling (Aoto et al., 2002; Blaess et al., 2006; Bayly et al., 2007). However, whether HH signaling is important in establishing or maintaining the MHB is not known. Furthermore, whether HH signaling regulates other boundary regions of the midbrain has also not been explored.
In this study, we show that a ventral (Pax7-negative) midbrain can be initiated in Shh-/- and Smo-/- mice, although it cannot be maintained without HH signaling beyond E8.5. Furthermore, a subset of Evx1+ ventral midbrain neurons, located near the dorsoventral (DV) boundary of the midbrain, is specified in the Shh-/- mouse. Surprisingly, the expression of Ihh is ectopically upregulated in the Shh-/- mouse in tissues subjacent to the neural tube. However, the ectopic Ihh expression is not accompanied by a concurrent upregulation of Gli1 or Ptc1, both indicators of HH signaling (Marigo et al., 1996; Goodrich et al., 1997). Together, these results provide the first evidence that HH-independent mechanisms may be involved in patterning the ventral midbrain. We also show that HH signaling regulates the spatial coherence and signaling properties of multiple midbrain signaling centers, including the midbrain hindbrain boundary (MHB) and the roof plate (RP). Like the MHB, the RP also becomes broader and displays increased cell-scatter several cell diameters away from the dorsal midline. However, the broadening of the RP cannot be attributed to a mere deregulation of dorsal expression patterns due to the loss of ventral signals because dorsal markers are selectively upregulated (Wnt1, Gdf7, Pax7), downregulated (Lfng) or altered (Zic1) in the absence of SHH.
Results
Midbrain arcuate territories are arrayed parallel to the rostral Floor Plate
As with the chick, the mouse ventral midbrain pattern is composed of a set of arcuate territories arrayed parallel to the rostral floor plate (rFP) source of Shh (Fig. 1A-G; (Agarwala et al., 2001; Sanders et al., 2002). These are marked by the expression of Phox2a and Isl1 in the oculomotor complex neurons (arc 1), and more laterally by the expression of Pax6 and Evx1 (Fig. 1A-G). Corresponding to the location of V0 (Evx1+) interneurons in the spinal cord, midbrain Evx1+ neurons represent a ventral cell fate located within the Pax7-negative region of the midbrain, close to the dorsoventral (DV) boundary (Fig. 1A-D, F, G; Fig. 8; (Agarwala et al., 2001; Wijgerde et al., 2002).
Figure 1. Mouse ventral midbrain organization.

A, B, E, F. Wholemounts with rostral to the top and ventricular surface facing the viewer. C, D. Transverse sections with ventricular surface to the top. G. Embryo in sagittal view with rostral to the right and dorsal to the top. A, B. Wild-type E12.5 midbrain (A) and a schematic (B) showing the ventral midbrain stripes of cell-fates (midbrain arcs) and their relationship to Shh expression in the rostral floor plate (rFP). Here, most of dorsal midbrain (blue, B) has been removed to facilitate a viewing of ventral midbrain. Midbrain arcs are labeled medial to lateral by the expression of the homeobox (HX) genes Phox2a (1/P2), Pax6 (P6) and Evx1 (E1) and are arrayed parallel to the rFP source of Shh. The Phox2a+ oculomotor complex neurons (1/P2) are obscured in A by the ventricular expression of Shh and are shown in the schematic in B and cross sections in C and D. C, D. Cross-sections of wholemounts shown in A and B (through positions marked by the horizontal lines in A and B). The labeling conventions in A-D and the color schemes in B and D are identical. E. Isl1+ labeling marks the oculomotor complex (OMC) neurons of the first arc (1) in an E12.5 wholemount. F. Expression of homeobox genes, Pax6 (blue) and Evx1 (brown) marks arcuate territories lateral to the OMC. G. Sagittal view of E12.5 midbrain demonstrating that the Pax7-negative/Evx1+ (blue) neurons lie immediately ventral to the dorsoventral boundary of the midbrain, here identified by the expression of Pax7 (brown) in dorsal midbrain. uAbbreviations: 1: first arc; III: third ventricle; E1: Evx1; P2: Phox2a; P6: Pax6; HX: Homeobox expression of Phox2a, Pax6, Evx1; FB: forebrain; HB: hindbrain; MHB: midbrain-hindbrain boundary; OMC: oculomotor complex; rFP: rostral floor plate, TEC:Tectum/dorsal midbrain.
Figure 8. Summary of ventral midbrain specification by SHH signaling.

Schematic representation of wild-type (A, E, G) and Shh-/- (B-D, F, H) midbrains in wholemount (A-F) and in transverse section (G-H) views. Wholemounts (open book preparation) are presented with rostral to the top and the ventricular surface facing the viewer. Cross-sections are shown with dorsal to the top and ventral toward the bottom of each schematic. A. Wild-type mouse showing that by E8, Shh expression (tan) is seen at the ventral midline of the midbrain and the expression of Pax7 (blue) is dorsally restricted. B-D. The initial specification of ventral midbrain (demonstrated by the absence of Pax7), occurs in the absence of SHH (B, C) and SMO (data not shown), but becomes dependent upon HH signaling by E9 (D). The ventral-dorsal transformation of the Shh-/- midbrain (Pax7+) displays a rostral-caudal sequence of progression, with Pax7 appearing first rostrally at E8.5 (C) and then ubiquitously by E9 (D). The midbrain-hindbrain boundary (MHB) is specified and is spatially coherent between E8.5-E9 in the Shh-/- midbrain. The mutant midbrain is significantly smaller than the wild-type by E9 (see text). E. At E12.5, the ventral midbrain of wild-type mice is organized into a series of arcuate territories labeled medial to lateral by the expression of Phox2a (arc1: pink) Pax6 (purple) and Evx1 (red). Midbrain arcs are arrayed parallel to Shh (tan) expression in the rostral floor plate (rFP). The expression of Pax7 (blue) continues to be restricted to dorsal midbrain (tectum, tec). The position of the MHB is indicated by Wnt1 (yellow) and Fgf8 (green) expression. F. E12.5 wholemount demonstrating the continued presence of Pax7 throughout the Shh-/- midbrain. The MHB is severely perturbed, displaying a complete absence of Fgf8 and severely reduced and disrupted Wnt1 expression. Midbrain size is dramatically reduced. G. Cross-section through the wholemount shown in E demonstrates the relationship between ventral midbrain arc-specific cell-fates (color coded as in E), the dorsal midbrain expression of Pax7 and Shh. Note that all Evx1+ neurons are located in the mantle (differentiated) layer of ventral (Pax7-negative) midbrain. Note also the spatially restricted expression of Wnt1 in the roof plate (RP). H. Cross-section through the E12.5 Shh-/- midbrain shown in F, displaying the complete absence of Phox2a and Pax6 and the sparing of 25% of the normal number of Evx1+ neurons. The spared Evx1+ neurons are obscured from view in the wholemount view shown in F by the ventricular layer expression of Pax7. The RP is broadened and disrupted. Abbreviations: MHB: midbrain-hindbrain boundary; rFP: rostral floor plate; RP: roof plate; TEC: tectum.
HH signaling is necessary for cell-fate specification in the ventral midbrain
Unlike the wildtype mouse, Isl1+ oculomotor neurons of the first midbrain arc were absent from the Shh-/- mutant (Fig. 2A-D). However, a dorsal Isl1+ cell population (Fig. 2A-D), likely to represent mesencephalic trigeminal sensory neurons was expanded rostrally and ventrally in the Shh-/- mouse (Fedtsova and Turner, 2001; Blaess et al., 2006). The complete absence of the oculomotor neuron marker Phox2a, which is expressed exclusively in the ventral midbrain, confirmed that the Isl1+ neurons seen in the Shh-/- mouse constitute an expanded population of dorsally-derived mesencephalic trigeminal sensory neurons (Fig. 2E-H).
Figure 2. Residual ventral cell-fate specification in the Shh-/- midbrain.

Columns 1, 2: embryos shown in sagittal view with rostral to the right. Columns 3, 4: Midbrain cross-sections with dorsal to the top. Arrowheads in column 2 delineate the midbrain from the hindbrain. The midbrain-hindbrain transition in Shh-/- mouse in this and subsequent figures was determined by the expression of Wnt1, Otx2 and Gbx2, the changed shape of the ventricle, and the presence of the physical constriction delineating midbrain from hindbrain. A-D. Ventrally located Isl1+ OMC neurons present in the controls (arrowhead, A, C) are absent from the Shh-/- midbrain (B, D). Dorsal Isl1+ trigeminal sensory neurons (arrows A, C) are expanded ventrally and rostrally (arrows, B, D). Broken line in B marks the plain of section shown in D. E-H. E10.5 embryos showing that Phox2a+ expression, present exclusively in the OMC (E, G), is completely missing in the Shh-/- midbrain (F, H). The cross-section in H demonstrates that the faint labeling seen in F (*) is outside the midbrain. I-L. E10.5 embryos showing that Pax6+ cells (arrow in I, K) are missing from the Shh-/- midbrain (J, L). Arrow in J marks the residual diencephalon in the Shh-/- embryos. M-P. Some Evx1+ neurons are present in the E12.5 Shh-/- midbrain (N, P). Control emrbyos are shown in M, O. Abbreviations: FB; forebrain; HB: hindbrain; MB: midbrain; MHB: midbrain-hindbrain boundary; OMC: oculomotor complex.
In the spinal cord, some V1 and V0 (Evx1+) interneurons, normally present near the DV boundary, continue to be specified in the Shh-/- mouse (Litingtung and Chiang, 2000). We therefore asked if more intermediate and lateral ventral cell-fates (Pax6+, Evx1+) might also be spared in Shh-/- midbrain. Interestingly, although no Pax6+ neurons were seen, a small number of Evx1+ cells (∼25% of normal) were present in the Shh-/- midbrain (p<0.01; Fig. 2I-P; Table 1). These results suggest that as with the Shh-/- spinal cord, a subpopulation of Evx1+ neurons normally located at the lateral periphery of the ventral midbrain, can be specified in the midbrain in the absence of SHH (Litingtung and Chiang, 2000).
Table 1. Total numbers of TUNEL+ and Evx1+ neurons in wild-type and Shh-/- midbrains.
| Embryonic Age | Mean Cell Count/brain (WT) | Mean Cell Count/brain (Shh-/-) | % Difference* | p Value** | |
|---|---|---|---|---|---|
| TUNEL+ cells | E8.5 | 86.7 ± 5.5 | 196.7 ± 16.0 | 126.9% | <0.01 7 |
| E9 | 113.3 ± 11.6 | 218.0 | 92.4% | <0.023 | |
| Evx1+ cells | E12.5 | 11,631.4 ± 1,587.2 | 3055.9 ± 283.7 | -73.7% | <0.014 |
Percent Difference was calculated as follows: (wild-type total cell number - Shh-/- total cell number)/wild-type midbrain x 100. (For details, see Materials and Methods)
determined by Student’s t test.
Initial and transient specification of ventral midbrain in the absence of SHH
In the normal spinal cord, low levels of HH activity are thought to be required to restrict Pax7 expression to the dorsal neural tube (Ericson et al., 1996). Possibly due to the presence of gut-or node- derived IHH (Zhang et al., 2001), a small Pax7-negative ventral region is seen in the Shh-/- spinal cord and is thought to generate the spared V0 and V1 ventral interneurons (Litingtung and Chiang, 2000). We asked if a similar (Pax7-negative) ventral region was present in the Shh-/- midbrain from which the spared Evx1+ neurons might emerge.
The onset of Shh expression in the axial mesoderm underlying the midbrain occurs between E7.25-E8, prior to somite formation (Echelard et al., 1993). By 8 somites, Shh expression can be seen within the midbrain rFP and the subjacent mesoderm underlying the midbrain (Echelard et al., 1993; Marti et al., 1995). Based on the Shh-dependent dorsal restriction of Pax7 in the spinal cord and the early expression of Shh in the midbrain, we expected that Pax7 expression in the wildtype midbrain might be dorsally restricted by E8 and that this restriction would be compromised in the Shh-/- mutant (Ericson et al., 1996). Surprisingly, we found that Pax7 expression was dorsally restricted at E8 (6 somites) in both the control and the Shh-/- midbrains (Fig. 3A, B). The absence of any Gli1 or Ptc1 in the Shh-/- midbrain at E8 further confirmed that the dorsal confinement of Pax7 can occur in the absence of any HH signaling (Fig. 3A-D; (Goulding et al., 1991; Goulding et al., 1993).
Figure 3. Ventral midbrain is specified but cannot be maintained in the Shh-/- mouse.
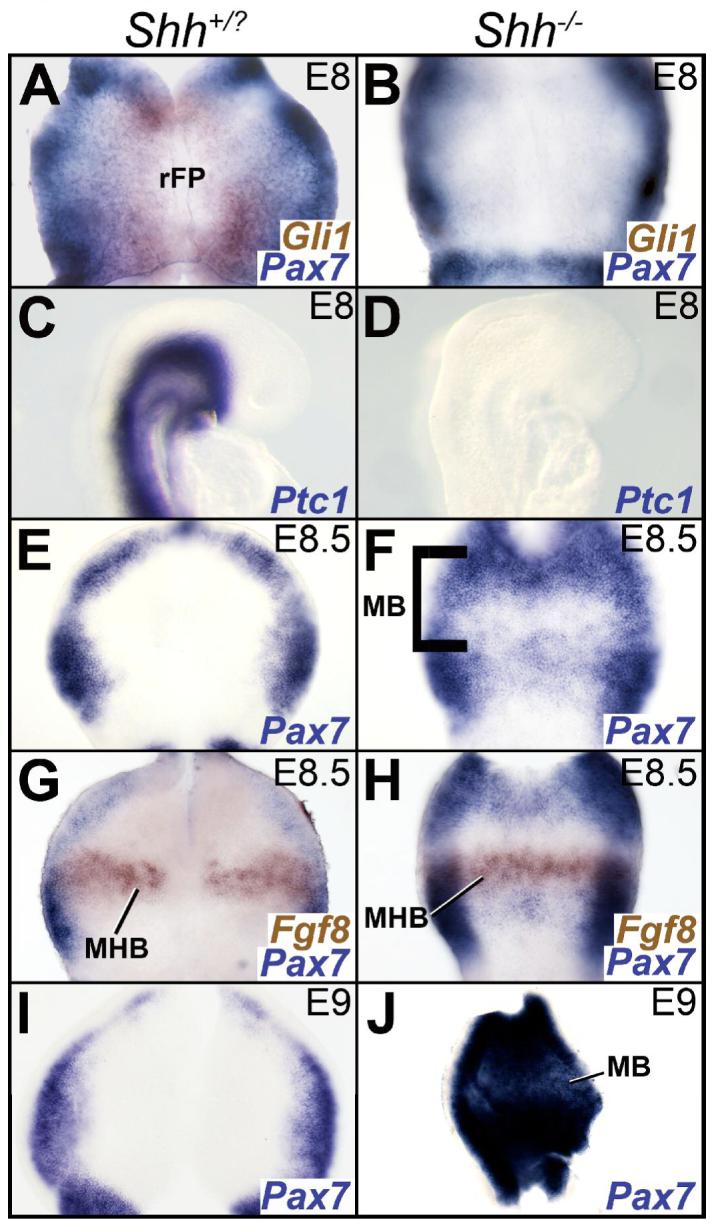
A, B, E-J. Wholemounts oriented as in 1A. C, D. Embryos in sagittal view, rostral to the right. A, B. The expression of Pax7 (blue) is restricted dorsally in the midbrain of the control (A) and Shh-/- mouse (B) at E8 (6 somites). Gli1 expression (brown), present in A and missing in B, identifies the Shh-/- mouse. C, D. E8 (6 somites) embryos showing the complete loss of Ptc1 expression from the Shh-/- midbrain (D) compared to its control littermate (C). E, F. Pax7 expression in E8.5 (10 somites) controls is restricted to the dorsal midbrain (E). Increased Pax7 expression in the rostral, but not the caudal portion of the Shh-/- midbrain (F) at E8.5 (10 somites). The bracket in F marks the rostrocaudal extent of the midbrain. G, H. Fgf8 (brown) expression in the MHB and Pax7 (blue) expression in the midbrain in an E8.5 wild-type (9 somites; G) and Shh-/- mouse (10 somites; H). Note that the Pax7-negative territory is rostral to the Fgf8 expression domain and therefore within the midbrain. I, J. Pax7 expression demonstrating that compared to controls (I), the Shh-/- midbrain (J) is completely dorsalized by E9 (15 somites). Abbreviations: rFP: rostral floor plate; MB: midbrain; MHB: midbrain-hindbrain boundary.
By E8.5, Pax7 expression in the rostral half of the Shh-/- ventral midbrain was upregulated in comparison to the wild-type (Fig. 3E, F). At this age, the caudal limit of Otx2 expression lies anterior to that of Wnt1, although their caudal limits become coincident a day later at E9.5 (Bally-Cuif et al., 1995; Millet et al., 1996). Thus, an Otx2-, Wnt1+, Fgf8-region marks the caudal midbrain anlage at this age (Fig. 3G, H; Fig. 6A-D; (Bally-Cuif et al., 1995; Millet et al., 1996; Zervas et al., 2004). Interestingly, despite the dorsalization of the anterior half of the midbrain, this caudal (Otx2-, Wnt1+, Fgf8-) midbrain region remained ventral (Pax7-negative) in the absence of SHH activity at E8.5 (Fig. 3E-H, data not shown; see also Fig. 4G, H). However, in the absence of SHH, this ventral (Pax7-negative) state could not be maintained, and Pax7 expression was ubiquitously seen throughout the midbrain by E9 (Fig. 3I, J). These results confirm the early requirement for SHH signaling in the midbrain (Blaess et al., 2006; Bayly et al., 2007). They also suggest that a ventral midbrain can be initiated in the absence of SHH signaling prior to E9. However, this ventral state is transient and cannot be maintained without HH activity beyond E9.
Figure 6. The MHB is initially established but can not be sustained without SHH.

A, B E9 Embryos shown in sagittal view with rostral to the right. The non-overlapping expression of Otx2 (blue) and Gbx2 (brown) demonstrates that a spatially coherent MHB is initially established in the Shh-/- mouse (B) and its wild-type littermate (A). C-J. Wholemounts with rostral to the top, ventricular surface facing the reader. C, D. Wnt1 (blue) and Fgf8 (brown) domains are correctly established and segregated in the Shh-/- mouse (D) and its wild-type littermate (C) at E9. Note that the continuous expression of Wnt1 and Fgf8 across the ventral midline reflects a loss of ventral midline structures by this age. E, F. Wnt1 expression at the MHB is broadened in the E10.5 Shh-/- mouse (F) compared to the wild-type (E). Inset shows Wnt1+ cells scattered several cell diameters (arrow) away from the MHB. G-J. Wnt1 (G, H) and Fgf8 (I, J) expression in control (G, I) and Shh-/- brains (H, J) demonstrating that the MHB in the Shh-/- mouse is severely disrupted and displays a near complete loss of Wnt1 (H) and a complete loss of Fgf8 by E12.5 (J). A morphological constriction can be seen between the midbrain and hindbrain in the Shh-/- mutant in J (arrowhead). K-N. E12.5 embryos shown in sagittal view with rostral to the right. Otx2 (K, L) and Gbx2 (M, N) expression in the midbrain showing that the sharp boundary formed by Otx2 at the wildtype MHB (K) is maintained in the Shh-/- mouse (L) and that Gbx2+ hindbrain cells are never seen in the midbrain of wildtype (M) or the Shh-/- (N) mouse. Arrowheads in J, L and N mark the boundary between the midbrain and hindbrain. Abbreviations: HB: hindbrain; MB: midbrain; MHB: midbrain-hindbrain boundary; rFP: rostral floor plate.
Figure 4. Ventral midbrain induction in the Shh-/- and Smo-/- midbrains is likely to be HH-independent.

A-F. Embryos shown in sagittal view, with rostral to the right. G-L. Wholemounts oriented as in 1A. Columns 1, 3: wild type controls; Columns 2, 4. Shh-/- or Smo-/- mutants. A, B. Upregulation of Ihh in the foregut (FG; B) of the Shh-/- mouse. Note that Ihh expression in the controls (A) is confined to the hindgut (HG). C-F. Gli1 (C) and Ptc1 (E) expression in control midbrains flanks the Shh+ region of rFP (data not shown; (Aglyamova and Agarwala, 2007). Note the normal Gli1 and Ptc1 expression domains in the gut. A complete loss of Gli1 (D) and Ptc1 (F) expression from the midbrain of the Shh-/- mouse. Note the continued expression of Gli1 (D) and Ptc1 (F) in the mid- (Ptc1) and hind- gut (Ptc1, Gli1). G, H. The expression of Pax7 is restricted dorsally in the midbrain of the control (G) and Smo-/- mouse (H) at E8 (7 somites). I, J. Pax7 expression is dorsally restricted in the Smo+/? (I) midbrain and is rostrally and dorsally restricted in the Smo-/- midbrain (J) at E8.5 (8 somites). Compare G-J with Fig. 3 A, B, E-H. The bracket in J marks the rostrocaudal extent of the midbrain. K, L. Ubiquitous Gli3 expression in the E8.5 Shh-/- midbrain (L) compared to its restricted expression in control littermates (K). Abbreviations: FG: foregut; HB: hindbrain; HG: hindgut; MB: midbrain; MG: midgut; rFP: rostral floor plate.
Evx1+ neuron specification and the initial induction of the ventral midbrain in the Shh-/- midbrain are likely to be HH-independent
In the spinal cord, node-or gut-derived IHH and HH-independent mechanisms have both been implicated in specifying the residual intermediate (V0, V1) cell-fates in the Shh-/- mouse (Pierani et al., 1999; Zhang et al., 2001). The gut region briefly underlies the midbrain early in mouse development and Ihh derived from the gut region could help to initiate ventral midbrain specification and specify the spared Evx1+ neurons of the Shh-/- midbrain (Jacobson and Tam, 1982). Surprisingly, ectopic expression of Ihh was noted in the foregut region of the Shh-/- mutant at E10.5 (Fig. 4A, B). However, no concurrent upregulation of Ptc1 or Gli1 expression was noted in either the foregut or the midbrain at E8 or E10.5 (Fig. 3A-D; Fig. 4C-F).
To fully rule out HH signaling as the mediator of the residual ventral specification seen in the Shh-/- mouse, we analyzed the Smo-/- midbrain at E8-8.5, when the ventral midbrain is still fully (E8) and partially (E8.5) Pax7-negative in the Shh-/- mouse (Fig. 3A, B, E-H). We reasoned that if SHH-independent, but HH-dependent mechanisms were involved in restricting Pax7 expression to the dorsal midbrain, Pax7 expression would fully encompass the entire rostrocaudal extent of the ventral midbrain in the Smo-/-, but not the Shh-/- midbrain by E8.5 (Fig. 3E-H). Surprisingly, Pax7 expression in the E8 and E8.5 Smo-/- midbrains was identical to that seen in the Shh-/- midbrains at corresponding ages and did not extend to the caudal half of the midbrain at E8.5 (Compare Fig. 4G-J to Fig. 3A, B, E-H). These results suggest that the dorsal restriction of Pax7 in the midbrain can be initiated in the absence of all HH signaling. The ubiquitous ectopic expression of Gli3 in the midbrain at E8.5, (typically a negative indicator of HH signaling), further implicates HH-independent mechanisms in transiently ventralizing the midbrain prior to E9 (Fig. 4K, L). Taken together, these results suggest that the initiation of the ventral midbrain and the specification of the residual Evx1 neurons in the Shh-/- and Smo-/- mice are likely to be HH-independent.
Midbrain size reduction in the Shh-/- mouse is primarily due to increased early dorsal midbrain cell death
HH signaling regulates cell proliferation and survival in the developing neural tube and its absence results in a profound reduction in midbrain size (Dahmane et al., 1997; Ishibashi and McMahon, 2002; Blaess et al., 2006). Here we established a time line for the reduction in midbrain size and asked whether it was due to increased cell death, reduced proliferation or both. In particular, we asked whether the loss of the ventral midbrain territory in the Shh-/- mutant between E8-E9 was a result of increased cell death, reduced proliferation or a conversion of ventral cell-fates to dorsal phenotypes.
At E8, no substantial differences in cell death or midbrain areas were noted between the controls and the Shh-/- midbrains (data not shown). Thereafter, a progressive and dramatic reduction in midbrain size was noted, such that compared to controls, the Shh-/- midbrains were 24.6% (p<0.04), 45.7% (p<0.02) and 71.3% (p<0.001) smaller at E8.5, E9 and E12.5 respectively (Table 2).
Table 2. Progressive reduction in the size of the midbrain in the absence of SHH.
| Embryonic Age | WT (mm2) | Shh-/- (mm2) | % Difference* | p Value** |
|---|---|---|---|---|
| E8.5 | 0.097 ± 0.009 | 0.073 ± 0.006 | -24.6% | <0.040 |
| E9 | 0.432 ± 0.023 | 0.235 ± 0.017 | -45.7% | <0.018 |
| E12.5 | 7.167 ± 0.472 | 2.060 ± 0.227 | -71.3% | <0.001 |
Percent Difference was calculated as follows: (wild-type midbrain area - Shh-/- midbrain)/wild-type midbrain x 100. (For details, see Materials and Methods)
determined by Student’s t test.
At E8.5, concentrated TUNEL labeling was seen along the dorsal midline of the wild-type and Shh-/- midbrain, the MHB, rhombomere1 and the diencephalon, particularly at the diencephalic-midbrain boundary (DMB; Fig. 5A, B). A 127% increase in midbrain cell death was noted in the Shh-/- mouse compared to the wild type controls at this age (p<0.02; Table 1; Fig. 5A-D). Notably however, apoptotic cells were all concentrated dorsally and were not seen in the ventral midbrain rudiment of the mutant at E8.5 (Fig. 5C, D).
Figure 5. Increased cell death does not account for the loss of the ventral midbrain rudiment in the Shh-/- mouse.

A, B. E8.5 (10 somites) embryos (shown in sagittal view with rostral to the right) demonstrating that compared to the wild-type (A), cell death is increased the Shh-/- midbrain (B) along the dorsal midline, the DMB, MHB and rhombomere 1 (see Table 1). C, D. Transverse sections through the midbrain of E8.5 embryos demonstrating the absence of apoptotic cells within the ventral midbrain rudiment of the mutant (D) and wild-type (C). E-H. Sagittal views (E, F) and transverse sections (G, H) of the midbrain showing that compared to controls (E, G), increased cell death is seen particularly at the DMB in the Shh-/- neural tube at E9 (F). Dashed lines in G and H mark the ventral limit of the midbrain. I-P. Cyclin B2 expression and BrdU labeling demonstrates normal cell proliferation in the midbrain of the Shh-/- mouse at E8.5, E9 and E10.5 (J, L, N, P) compared to wild-type littermates (I, K, M, O). I, J. wholemounts, with rostral to the top. K, L. Embryos shown in sagittal views with rostral to the right. M-P. Midbrain cross-sections with dorsal to the top. The strong BrdU labeling (M-P, arrowhead in M) along the pial surface of the brain is meningeal. Abbreviations: DMB: diencephalic-midbrain boundary; HB: hindbrain; MB: midbrain; MHB: midbrain-hindbrain boundary; rFP: rostral floor plate; RP: roof plate.
Compared to the wild type, a 92% increase in TUNEL-labeled cells was seen in the E9 Shh-/- mouse, again concentrated primarily along the dorsal midline, particularly at the DMB (Fig. 5E-H; p<0.02; Table 1). As with the E8.5 brains however, no cell death was noted along the ventral region of the neural tube at E9 (Fig. 5G, H; (Ishibashi and McMahon, 2002; Blaess et al., 2006). As reported before, by E10.5 no substantial differences in cell death were noted between the controls and the mutants (data not shown; (Blaess et al., 2006). These results suggest that HH signaling is required primarily for dorsal midbrain cell survival before E10.5. Importantly, the absence of cell death in the residual ventral midbrain between E8.5-E9 suggests that increased cell death cannot account for the loss of the ventralized (Pax7-negative) region of the midbrain.
Labeling with Cyclin B2 (a marker of the G2-M transition of the cell cycle), Cyclin D1 (G1-S transition) and BrdU (a marker of the S-phase) showed no differences in cell proliferation between the wild-type controls and the Shh-/- midbrains at E8.5, E9 and E10.5 (Fig. 5I-P, data not shown; (Ishibashi and McMahon, 2002; Blaess et al., 2006). Taken together with our cell death data, these results suggest that early dorsal cell death, and not reduced proliferation, is the principal cause for the dramatic reduction in midbrain size. Furthermore, the absence of any ventral cell death at all 3 ages examined suggests, that the most likely explanation for the loss of the ventral progenitors in the Shh-/- midbrain, is that they are unable to maintain their ventral status and turn on dorsal cell-fates (Pax7+) in the absence of SHH (Fig. 3I, J).
The Midbrain-Hindbrain Boundary is correctly initiated, but cannot be maintained in the absence of SHH
Previous studies have shown that disruptions of HH signaling perturb the MHB (Aoto et al., 2002; Blaess et al., 2006; Bayly et al., 2007). We asked whether this was due to a defect in the establishment or the maintenance of the MHB. The mutual repression of Otx2 and Gbx2 results in their non-overlapping expression patterns and a properly positioned MHB at their interface (Zervas et al., 2004). The mutually exclusive expression of Otx2 and Gbx2 at E9 suggests that a spatially restricted, sharply defined MHB is initially specified in the Shh-/- mouse (Fig. 6A, B). However, by E10.5, the MHB is broadened and Wnt1+ cells are found scattered at a distance from the MHB (Fig. 6E, F). By E12.5, Wnt1 expression within the MHB is severely reduced while Fgf8 expression in caudal MHB is completely lost (Fig. 6G-J). Taken together, these results suggest that HH signaling is required late in development (between E10-E12.5) for the maintenance, but not for the early initiation of the MHB.
Interestingly, the severe perturbation of Wnt1 and Fgf8 expression occurs even as the caudal limit of Otx2 expression remains sharp at the MHB, suggesting that the caudal restriction of the Otx2 domain at this age is unlikely to depend upon MHB-derived signals or SHH (Fig. 6K, L). The expression of Gbx2 is uninformative with regard to the MHB at E12.5, but is nevertheless excluded from the Shh-/- midbrain, suggesting that the restriction of Otx2 and Gbx2 to the midbrain and hindbrain respectively may be mutually independent at this age (Fig. 6M, N).
Perturbed regulation of the roof plate and dorsal gene expression in the midbrain in the absence of SHH
In the absence of HH signaling, the failure to maintain the MHB could be due to a deregulation of cell affinities (Rodriguez and Basler, 1997; Lawrence et al., 1999; Bayly et al., 2007). If so, other midbrain’s boundaries might also be disrupted. Expanded Gdf7 and Wnt1 expression suggested an expansion of the RP in the Shh-/- mouse (Fig. 7A-F). Similar to the MHB at E10.5 (Fig. 6E, F), we noted scattered Wnt1+ cells several cell diameters away from the RP (Fig. 7E, F). The expansion of the RP could be a result of the deregulation of dorsal cell-fates in the absence of SHH activity. If so, other dorsal markers should be similarly deregulated and expanded. An expansion of dorsal Isl1+ trigeminal sensory neurons and increased Pax7 expression has already been noted before (Fig. 2A-D; Fig. 3E-J (Fedtsova and Turner, 2001).
Figure 7. Disruption of the roof plate and dorsal midbrain gene expression patterns in the absence of SHH.

A, B, E, F. Free floating embryos shown in dorsal view with rostral to the top. C, D. Midbrain cross-sections with dorsal to the top. G-J: wholemounts in sagittal view, rostral to the right. A-F. Broadening and disruption of the RP markers, Gdf7 (A-D) and Wnt1 (E, F) in the Shh-/- mouse (B, D, F) compared to wild-type littermates (A, C, E). Dashed lines in C and D indicate the ventral outline of the midbrain. Arrows in F point to ectopically scattered Wnt1+ cells. G, H. Expression of Lfng at E9.5 in the Shh-/- midbrain (H) is reduced compared to control littermate (G). I, J. Increased spread and reduced intensity of the dorsal marker Zic1 in the E10.5 Shh-/- mouse (J), compared to controls (I). Arrowheads in H and J delineate the midbrain from the hindbrain in the Shh-/- mice. Abbreviations: DMB: diencephalic-midbrain boundary; HB: hindbrain; MB: midbrain; MHB: midbrain-hindbrain boundary; RP: roof plate.
Examination of multiple dorsal gene expression patterns suggested that a simple deregulation in the absence of opposing ventral signals does not occur in the Shh-/- midbrain. In contrast to the expansion of Wnt1, Gdf7, Isl1 and Pax7 reported above, we noted a reduction in the expression of Lfng and Lim2 in the Shh-/- mouse at E9.5 and 10.5 (Fig. 7G, H; data not shown). This pattern contrasted sharply with the lowered intensity, but increased range of Zic1, a marker of dorsal neural fates and neural crest cells (Fig. 7I, J). Taken together with the previously reported loss of tectal Dbx1 in the Shh-/- midbrain (Ishibashi and McMahon, 2002), these results suggest that in the absence of SHH, dorsal gene expression patterns are specifically upregulated or downregulated, and do not exhibit a generic upregulation due to a derepression of dorsal signals.
Discussion
A summary of our results is schematically represented in Fig. 8. We show that a ventral midbrain territory is present in the Shh-/- and Smo-/- midbrains between E8-8.5 (Fig. 8A-C). In the absence of SHH, this territory becomes progressively dorsalized (Pax7+) in a rostral-caudal sequence between E8-E9 (Fig. 8B-D). Early ventral midbrain progenitors do not die in the absence of SHH, but fail to maintain their ventral (Pax7-negative) status beyond E9 (Fig. 8A-F). Although the midbrain arc pattern is severely disrupted in the Shh-/- mutant, at least one ventral cell-fate (Evx1+) is specified in the absence of all HH signaling (Fig. 8E-H). We show that although midbrain signaling centers (MHB, RP) can be established, they cannot be correctly maintained in the absence of HH signaling (Fig. 8E-H). Finally, dorsal gene expression patterns are upregulated (Wnt1, Gdf7, Pax7, Isl1), downregulated (Lfng, Lim2) or altered (Zic1) in the Shh-/- mutant in a manner inconsistent with a simple deregulation of dorsal cell fates due to the absence of opposing ventral signals (Fig. 8G, H).
The ventral midbrain can be induced in the absence of HH signaling
Contrary to expectation, we found that Pax7 expression was dorsally restricted in both the control and the Shh-/- and Smo-/- midbrains at E8, suggesting that ventral midbrain induction can be initiated in the absence of HH activity (Fig. 3A-H; Fig. 4G-J). However, maintenance of this ventral midbrain territory is dependent upon SHH, with the Shh-/- midbrain ubiquitously expressing Pax7 by E9 (Fig. 3I, J).
The molecular nature of such ventral influences and whether they operate in the normal animal is not yet understood. Exposure to IHH derived from the gut or the node has been proposed as a mechanism for residual ventral specification in the Shh-/- ventral spinal cord (Zhang et al., 2001). Interestingly, we observed ectopic Ihh expression in the Shh-/- foregut, but it was not accompanied at either E8 or E10.5 by the simultaneous upregulation of Gli1 or Ptc1, both positive indicators of HH signaling (Fig. 3A-D; Fig. 4C-F; (Goodrich et al., 1996; Marigo et al., 1996). The upregulation of Gli3 by E8.5 makes it further unlikely that the ventral midbrain induction in the Shh-/- midbrain is dependent upon a functional HH signal (Fig. 4K, L). Most definitively, the dorsal restriction of Pax7 in the Smo-/- midbrain prior to E9 rules out the involvement of SHH-independent, HH-dependent mechanisms in the initial ventral specification of the midbrain, although the presence of non-canonical HH signaling remains a theoretical possibility (Fig. 4G-J; (Krishnan et al., 1997; Bourikas et al., 2005).
HH-independent mechanisms (e.g., retinoic acid) have also been proposed in the specification of some ventral cell-fates (V0, V1) in the spinal cord (Pierani et al., 1999). Interestingly, multiple ventral cell fates are present in the Gli2-/-:Gli3-/- and Smo-/-:Gli3-/- spinal cords, where HH signaling is unlikely to occur (Wijgerde et al., 2002; Bai et al., 2004). These results have led to the suggestion that in the absence of any repressive GLI3 activity, ventral cell fates can be specified without HH activity. However, in the telencephalon, ventral cell-fate specification is severely disrupted in the Fgfr1-/-:Fgfr2-/- double knockouts despite the presence of Shh and Gli1 (Gutin et al., 2006). Furthermore, when Gli3 is removed from the Fgfr1-/-:Fgfr2-/- double knockouts, no rescue of ventral cell-fates is seen in the triple mutants (Gutin et al., 2006). Taken together, these results provide evidence that some ventral cell-fate specification can occur in the absence of HH signaling in multiple regions of the ventral neural tube.
Lateral, but not intermediate ventral midbrain fates are specified in the absence of SHH
The results of this study show that although medial and intermediate ventral cell-fates (OMC, MDA, Pax6+ neurons) are lost, ∼25% of the normal numbers of Evx1+ neurons are specified in the Shh-/- mouse (Fig. 2; (Fedtsova and Turner, 2001; Blaess et al., 2006). Unlike the V0 (Evx1+) interneurons of the spinal cord however, the Evx1+ neurons in the Shh-/- mutant midbrain are seen within the mantle (differentiated) layer of the midbrain, subjacent to a fully dorsalized (Pax7+) progenitor layer (Fig. 1; Fig. 2; data not shown). This raises questions as to the origin (dorsal or ventral) of the signals that specify the Evx1+ cell fate in the midbrain. Evx1 expression is first seen in the midbrain at E12.5 (Fig. 1A). Thus, we do not know whether the residual Evx1+ neurons are specified in the Shh-/- midbrain prior to E9 when SHH-independent ventral signals might be present or whether a fraction of Evx1+ neurons can be specified by dorsally-derived cues. In the chick midbrain, at least a subset of EVX1+ neurons are allocated early and have an absolute requirement for HH signaling (Bayly et al., 2007). However, this study did not rule out the possibility that a fraction of EVX1+ could also be dorsally derived. An analogous requirement for ventral signals has been demonstrated for the ventral-most dorsal (dI6; DBX+) spinal interneurons (Wijgerde et al., 2002).
An interesting feature of the spared Evx1+ neurons in the Shh-/- midbrain is their absence of spatial coherence (Fig. 2O, P). A similar absence of spaital coherence within discretely organized neuronal clusters has been previously noted following HH perturbations and has been attributed to HH-mediated regulation of cell-affinities (Agarwala et al., 2005; Bayly et al., 2007). Furthermore, given the perturbations of the MHB, a lineage restriction boundary in the mouse, it remains possible that the Evx1+ cells seen in the Shh-/- midbrain are interlopers from the hindbrain (Zervas et al., 2004; Blaess et al., 2006). However, we think this is unlikely since the expression of Otx2 remains sharp caudally in the mutant (Fig. 6K, L) and the expression of Gbx2, though uninformative with regard to the MHB at E12.5, is never seen within the Otx2+ midbrain territory (Fig. 6M, N; data not shown).
Size reduction in the Shh-/- midbrain is primarily due to early dorsal midbrain cell death
In this study, we show that midbrain size is dramatically reduced in the Shh-/- mouse between E8.5-E12.5, progressing from being ∼75% of the normal size at E8.5 to ∼30% of the normal size at E12.5 (Table 2). We show that this is accompanied by a progressive increase in cell death between E8.5 and E9 (Fig. 5; Table 1; data not shown). Interestingly, the increased cell death seen in the Shh-/- mutant is not just temporally restricted to occur predominantly prior to E10.5; it is also spatially restricted to the dorsal midbrain. Most notably, cell death is never seen in the ventral midbrain rudiment between E8-E9 (Fig. 5A-H). Taken together with the absence of significant changes in cell proliferation in the Shh-/- midbrain between E8.5 and 10.5 (Fig. 5I-P; (Ishibashi and McMahon, 2002; Blaess et al., 2006), our data suggest that a primary cause of the altered midbrain size in the Shh-/- mouse is the death of dorsal midbrain progenitors. Furthermore, they suggest that the most likely explanation for the loss of ventral progenitors in the Shh-/- midbrain specified prior to E9 is that they fail to maintain their ventral identities and take on dorsal identities by turning on Pax7 expression.
HH regulates midbrain’s boundaries with adjacent tissues
In this study we show that although Wnt1 and Fgf8 expression in the MHB are correctly initiated at the MHB, they fail to be maintained in the absence of HH signaling. Deletion (Wnt1) and conditional (Ffg8) mutant studies have shown that WNT1 and FGF8 are essential for the specification and/or maintenance of the midbrain and MHB and their absence results in severe early (E8-E9.5) cell death (McMahon and Bradley, 1990; Chi et al., 2003). In the Shh-/- mouse however, the loss of Wnt1 and Ffg8 expression begins relatively late (∼E10.5) and is not accompanied by the severe cell death (E8-E9.5) seen in the Wnt1-/- mouse and the conditional Fgf8-/- mutant where Fgf8 expression is extinguished at 7-10 somites (∼E8-E8.75; (Chi et al., 2003). Taken together with our data, these results suggest that SHH, FGF8 and WNT1 are all required for midbrain cell survival early in development. However, cell survival is independent of all three signaling molecules by E10.5 when Fgf8 and Wnt1 expression is downregulated at the MHB due to the absence of SHH. Finally, results of this and previous studies have shown that HH-dependent cell-fate specification in the midbrain occurs very early in the chick and mouse midbrain although Shh continues to be expressed well beyond this time point (Blaess et al., 2006; Bayly et al., 2007). We propose that one important function of the continued expression of SHH by the rFP therefore is the maintenance of the orthogonal midbrain signaling center, the MHB.
Interactions and mutual regulation between SHH and FGF8 emanating from orthogonal signaling centers has been demonstrated in multiple embryonic systems. In the vertebrate limb, SHH (from the Zone of Polarizing Activity) and FGF8 (from the Apical Ectodermal Ridge) maintain each other’s expression, albeit indirectly via BMP-mediated blockade by Gremlin (Martin, 1995; Khokha et al., 2003). In the midbrain and caudal diencephalon, the expression of Fgf15 and Fgf receptors 2 and 3 cannot be maintained in the Shh-/- mouse and SHH and FGF cooperate in the specification of dopaminergic neurons (Ye et al., 1998; Ishibashi and McMahon, 2002).
An intriguing finding of this study was the broadening and disruption of the Shh-/- MHB and the RP (Fig. 6E, F; Fig. 7A-F; (Bayly et al., 2007). This was marked for example, by the ectopic expression of Wnt1 several cell diameters away from the MHB and the RP (Fig. 6F; Fig. 7E). This phenotype can be attributed to either the ectopic expression of RP and MHB markers in the Shh-/- midbrain or to a disruption of midbrain boundaries. A loss of the spatial coherence of pattern and poorly defined tissue boundaries following HH perturbations have been previously reported in the fly wing and the vertebrate neural tube, including the chick midbrain (Dahmann and Basler, 1999; Lawrence et al., 1999; Bai et al., 2004; Agarwala et al., 2005; Bayly et al., 2007). This disruption has been attributed to the regulation of cell-affinities by HH signaling (Dahmann and Basler, 1999; Jarov et al., 2003; Schlichting et al., 2005). We therefore conclude that the most likely explanation for the cell scatter seen at the MHB and RP is a disruption of cell-affinities, although we cannot definitively rule out the ectopic expression of RP and MHB markers in midbrain cells away from the MHB and the RP in the absence of HH signaling.
Interestingly, disruptions of the MHB are not accompanied by concurrent disruptions of Otx2 and Gbx2 expression patterns in the Shh-/- mouse (Fig. 6K-M). Differential regulation of the Otx2 and Gbx2 has also been reported in the Lmx1b-/- mouse where Fgf8 expression is never initiated and Wnt1 expression is severely reduced (Guo et al., 2007). In these mutants, the anterior hindbrain expression of Gbx2 is reduced but Otx2 expression remains normal at E12.5. Thus, the maintenance of Otx2 and Gbx2 expression beyond E10.5 is unlikely to depend either upon SHH or upon MHB-derived signals (e.g., FGF8).
Dorsal Patterning by SHH
HH signaling clearly influences dorsal midbrain patterning. In the absence of SHH, midbrain size is aberrant, the inferior colliculus is missing, the RP is disrupted and dorsal markers are upregulated (Wnt1, Gdf7, Pax7, Isl1), reduced (Lfng, Dbx1, Lim2), or display altered domains and intensity of labeling (Zic1; Fig. 7; data not shown; (Ishibashi and McMahon, 2002; Blaess et al., 2006). The specificity of gene regulation and the spatially and temporally selective cell death seen in the dorsal Shh-/- midbrain make direct dorsal patterning by SHH a distinct possibility (Fig. 5; Fig. 7; (Ishibashi and McMahon, 2002; Blaess et al., 2006). Interestingly, PAX7 and GLI1 expression overlap extensively in the Hamburger-Hamilton Stage 10 chick dorsal spinal cord and HH signaling is required for the specification of the ventral-most (DBX+) dorsal spinal interneurons (Wijgerde et al., 2002; Aglyamova and Agarwala, 2007). Finally, although PTC1 and PTC2 are seen never in the chick dorsal midbrain between Hamburger-Hamilton Stage 5-E5, members of the HH signal transduction machinery (including putative receptors, SMO, GLI genes and others) are expressed and could directly transduce HH signals in the dorsal midbrain (Aglyamova and Agarwala, 2007).
Materials and Methods
Shh-/- and Smo-/- mice
The generation of the Shh-/- and Smo-/- mice was carried out as described previously (Chiang et al., 1996; Zhang et al., 2001). Homozygous Shh-/- and Smo-/- mice were recovered between E8 and E12.5 and identified either by morphological differences or the absence of the HH-target genes, Gli1 and Ptc1 (Chiang et al., 1996; Zhang et al., 2001). At early ages, embryos were staged by somite number as well as by morphological features (Theiler, 1989; Downes and Davies, 1993). At least three mutants and three control (+/?) littermates were analyzed for each gene expression pattern displayed.
In situ hybridization
Embryos were harvested between E8-E12.5 and immersion-fixed in 4% paraformaldehyde. Digoxygenin- or Fluorescein-conjugated antisense riboprobes were prepared from cDNAs for Cyclin B2, Cyclin D1, Evx1, Fgf8, Gbx2, Gdf7, Gli1, Gli3, Ihh, Isl1, Lim2, Lfng, Otx2, Pax6, Pax7, Phox2a, Ptc1, Shh, Wnt1, Zic1. One or two color whole-mount in situ hybridizations were conducted using NBT (Roche) or T-NBT (Research Organics, Ohio) histochemistry according to previously established protocols (Agarwala and Ragsdale, 2002).
Cell death assay
Whole-mount cell death assays were carried out on embryos using previously published protocols (Yamamoto and Henderson, 1999; Sanders, 2001; Agarwala et al., 2005).
Bromodeoxyuridine (BrdU) labeling
Pregnant females (E9, E10.5, n=3/age) were injected intraperitoneally with BrdU (Sigma; 50!g BrdU/gram body weight). Embryos were harvested 3 hours later and immersion-fixed in 4% paraformaldehyde. BrdU positive cells were detected using previously established protocols (Agarwala et al., 2005).
Cell counts and midbrain size
Cell death
Cell death at E8.5 and E9 was estimated by counting TUNEL-labeled cells in cross-sections through WT and Shh-/- midbrains. Briefly, all TUNEL-labeled cells were counted from a representative set of sections spanning the anteoposterior axis of Shh-/- and WT midbrains (3/6 total sections/brain, 30!m each). The total numbers of TUNEL-labeled cells for the mutant and wild-type were compared using Student’s t test.
Evx1+ Neurons
The number of spared Evx1+ neurons in the Shh-/- midbrain were compared with the number of Evx1+ neurons in wild-type littermates at E12.5 (n=3 each). All sections (30 μm) containing Evx1+ neurons (average, 9 sections/wildtype brain and 6 sections/mutant brain) were photographed and the total area and volume occupied by the Evx1+ neurons was calculated using ImageJ (NIH). In every third section, all Evx1+ neurons within 3 sampling boxes (subtending 40 μm on each side) placed across the mediolateral axis of the Evx1+ territory, were counted. The total number of Evx1+ neurons in each brain was then adjusted for the total volume occupied by Evx1+ neurons to estimate the total number of Evx1+ neurons/midbrain. Differences in cell numbers between the Shh-/- and WT littermates were evaluated using Student’s t test.
Midbrain Area
Mean cross-sectional area was estimated in Shh-/- and wild-type midbrain flatmounts at E8.5. E9 and E12.5 (n=3/age/genotype) using morphological landmarks and ImageJ software. Midbrain areas in the Shh-/- and WT littermates were compared using Student’s t test.
Acknowledgements
We thank Drs. R. Arkell, R. Barstead, M. Busslinger, P. Chambon, R. Conlon, M. Goulding, P. Gruss, C. Hui, T. Jessell, A. Joyner, W. Klein, A. McMahon, G. Martin, K. Millen, C. Ragsdale, M. Scott, T. Shimogori, C. Tabin, M. Wassef and D. Wolgemuth for DNA reagents and R. Bayly and T. Shimogori for critical reading of the manuscript. This research was supported from University of Texas at Austin start up funds to SA and by grants from NIH to SA and CC.
References
- Agarwala S, Aglyamova GV, Marma AK, Fallon JF, Ragsdale CW. Differential susceptibility of midbrain and spinal cord patterning to floor plate defects in the talpid2 mutant. Dev Biol. 2005;288:206–220. doi: 10.1016/j.ydbio.2005.09.034. [DOI] [PubMed] [Google Scholar]
- Agarwala S, Ragsdale CW. A role for midbrain arcs in nucleogenesis. Development. 2002;129:5779–5788. doi: 10.1242/dev.00179. [DOI] [PubMed] [Google Scholar]
- Agarwala S, Sanders TA, Ragsdale CW. Sonic Hedgehog Control of Size and Shape in Midbrain Pattern Formation. Science. 2001;291:2147–2150. doi: 10.1126/science.1058624. [DOI] [PubMed] [Google Scholar]
- Aglyamova GV, Agarwala S. Gene expression analysis of the Hedgehog signaling cascade in the chick midbrain and spinal cord. Dev Dyn. 2007;236:1363–1373. doi: 10.1002/dvdy.21146. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Shigeno C, Hiraki Y, Shukunami C, Kohno H, Akagi M, Konishi J, Nakamura T. Cloning of a mouse smoothened cDNA and expression patterns of hedgehog signalling molecules during chondrogenesis and cartilage differentiation in clonal mouse EC cells, ATDC5. Biochem Biophys Res Commun. 1997;235:142–147. doi: 10.1006/bbrc.1997.6750. [DOI] [PubMed] [Google Scholar]
- Alcedo J, Ayzenzon M, Von Ohlen T, Noll M, Hooper JE. The Drosophila smoothened Gene Encodes a Seven-Pass Membrane Protein, a Putative Receptor for the Hedgehog Signal. Cell. 1996;86:221–232. doi: 10.1016/s0092-8674(00)80094-x. [DOI] [PubMed] [Google Scholar]
- Aoto K, Nishimura T, Eto K, Motoyama J. Mouse GLI3 regulates Fgf8 expression and apoptosis in the developing neural tube, face, and limb bud. Dev Biol. 2002;251:320–332. doi: 10.1006/dbio.2002.0811. [DOI] [PubMed] [Google Scholar]
- Bai CB, Stephen D, Joyner AL. All Mouse Ventral Spinal Cord Patterning by Hedgehog Is Gli Dependent and Involves an Activator Function of Gli3. Developmental Cell. 2004;6:103–115. doi: 10.1016/s1534-5807(03)00394-0. [DOI] [PubMed] [Google Scholar]
- Bally-Cuif L, Cholley B, Wassef M. Involvement of Wnt-1 in the formation of the mes/metencephalic boundary. Mechanisms of Development. 1995;53:23–34. doi: 10.1016/0925-4773(95)00421-1. [DOI] [PubMed] [Google Scholar]
- Bayly RD, Ngo M, Aglyamova GV, Agarwala S. Regulation of ventral midbrain patterning by Hedgehog signaling. Development. 2007;134:2115–2124. doi: 10.1242/dev.02850. [DOI] [PubMed] [Google Scholar]
- Blaess S, Corrales JD, Joyner AL. Sonic hedgehog regulates Gli activator and repressor functions with spatial and temporal precision in the mid/hindbrain region. Development. 2006;133:1799–1809. doi: 10.1242/dev.02339. [DOI] [PubMed] [Google Scholar]
- Bourikas D, Pekarik V, Baeriswyl T, Grunditz A, Sadhu R, Nardo M, Stoeckli ET. Sonic hedgehog guides commissural axons along the longitudinal axis of the spinal cord. Nat Neurosci. 2005;8:297–304. doi: 10.1038/nn1396. [DOI] [PubMed] [Google Scholar]
- Chi CL, Martinez S, Wurst W, Martin GR. The isthmic organizer signal FGF8 is required for cell survival in the prospective midbrain and cerebellum. Development. 2003;130:2633–2644. doi: 10.1242/dev.00487. [DOI] [PubMed] [Google Scholar]
- Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- Dahmane N, Lee J, Robins P, Heller P, Ruiz i Altaba A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature. 1997;389:876–881. doi: 10.1038/39918. [DOI] [PubMed] [Google Scholar]
- Dahmann C, Basler K. Compartment boundaries: at the edge of development. Trends Genet. 1999;15:320–326. doi: 10.1016/s0168-9525(99)01774-6. [DOI] [PubMed] [Google Scholar]
- Downes KM, Davies T. Staging of gastrulating mouse embryos by morphological landmarks in the dissecting microscope. Development. 1993;118:1255–1266. doi: 10.1242/dev.118.4.1255. [DOI] [PubMed] [Google Scholar]
- Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- Ericson J, Morton S, Kawakami A, Roelink H, Jessell TM. Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell. 1996;87:661–673. doi: 10.1016/s0092-8674(00)81386-0. [DOI] [PubMed] [Google Scholar]
- Fedtsova N, Turner EE. Signals from the ventral midline and isthmus regulate the development of Brn3.0-expressing neurons in the midbrain. Mechanisms of Development. 2001;105:129–144. doi: 10.1016/s0925-4773(01)00399-9. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Johnson RL, Milenkovic L, McMahon JA, Scott MP. Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev. 1996;10:301–312. doi: 10.1101/gad.10.3.301. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Goulding MD, Chalepakis G, Deutsch U, Erselius JR, Gruss P. Pax-3, a novel murine DNA binding protein expressed during early neurogenesis. EMBO J. 1991;10:1135–1147. doi: 10.1002/j.1460-2075.1991.tb08054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulding MD, Lumsden A, Gruss P. Signals from the notochord and floor plate regulate the region-specific expression of two Pax genes in the developing spinal cord. Development. 1993;117:1001–1016. doi: 10.1242/dev.117.3.1001. [DOI] [PubMed] [Google Scholar]
- Guo C, Qiu H-Y, Huang Y, Chen H, Yang R-Q, Chen S-D, Johnson RL, Chen Z-F, Ding Y-Q. Lmx1b is essential for Fgf8 and Wnt1 expression in the isthmic organizer during tectum and cerebellum development in mice. Development. 2007;134:317–325. doi: 10.1242/dev.02745. [DOI] [PubMed] [Google Scholar]
- Gutin G, Fernandes M, Palazzolo L, Paek H, Yu K, Ornitz DM, McConnell SK, Hebert JM. FGF signalling generates ventral telencephalic cells independently of SHH. Development. 2006;133:2937–2946. doi: 10.1242/dev.02465. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J. Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- Ishibashi M, McMahon AP. A sonic hedgehog-dependent signaling relay regulates growth of diencephalic and mesencephalic primordia in the early mouse embryo. Development. 2002;129:4807–4819. doi: 10.1242/dev.129.20.4807. [DOI] [PubMed] [Google Scholar]
- Jacob J, Briscoe J. Gli proteins and the control of spinal-cord patterning. EMBO Rep. 2003;4:761–765. doi: 10.1038/sj.embor.embor896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson A, Tam P. Cephalic neurulation in the mouse embryo analyzed by SEM and morphometry. Anat. Rec. 1982;203:375–396. doi: 10.1002/ar.1092030308. [DOI] [PubMed] [Google Scholar]
- Jarov A, Williams KP, Ling LE, Koteliansky VE, Duband J-L, Fournier-Thibault C. A dual role for Sonic hedgehog in regulating adhesion and differentiation of neuroepithelial cells. Developmental Biology. 2003;261:520–536. doi: 10.1016/s0012-1606(03)00351-8. [DOI] [PubMed] [Google Scholar]
- Jessell TM. Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat Rev Genet. 2000;1:20–29. doi: 10.1038/35049541. [DOI] [PubMed] [Google Scholar]
- Khokha MK, Hsu D, Brunet LJ, Dionne MS, Harland RM. Gremlin is the BMP antagonist required for maintenance of Shh and Fgf signals during limb patterning. Nat Genet. 2003;34:303–307. doi: 10.1038/ng1178. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Pereira FA, Qiu Y, Chen C-H, Beachy PA, Tsai SY, Tsai M-J. Mediation of Sonic Hedgehog-Induced Expression of COUP-TFII by a Protein Phosphatase. Science. 1997;278:1947–1950. doi: 10.1126/science.278.5345.1947. [DOI] [PubMed] [Google Scholar]
- Lawrence PA, Casal J, Struhl G. The hedgehog morphogen and gradients of cell affinity in the abdomen of Drosophila. Development. 1999;126:2441–2449. doi: 10.1242/dev.126.11.2441. [DOI] [PubMed] [Google Scholar]
- Litingtung Y, Chiang C. Specification of ventral neuron types is mediated by an antagonistic interaction between Shh and Gli3. Nat Neurosci. 2000;3:979–985. doi: 10.1038/79916. [DOI] [PubMed] [Google Scholar]
- Marigo V, Davey RA, Zuo Y, Cunningham JM, Tabin CJ. Biochemical evidence that patched is the Hedgehog receptor. Nature. 1996;384:176–179. doi: 10.1038/384176a0. [DOI] [PubMed] [Google Scholar]
- Marti E, Takada R, Bumcrot DA, Sasaki H, McMahon AP. Distribution of Sonic hedgehog peptides in the developing chick and mouse embryo. Development. 1995;121:2537–2547. doi: 10.1242/dev.121.8.2537. [DOI] [PubMed] [Google Scholar]
- Martin GR. Why thumbs are up. Nature. 1995;374:410–411. doi: 10.1038/374410a0. [DOI] [PubMed] [Google Scholar]
- McMahon A, Bradley A. The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell. 1990;62:1073–1085. doi: 10.1016/0092-8674(90)90385-r. [DOI] [PubMed] [Google Scholar]
- Millet S, Bloch-Gallego E, Simeone A, Alvarado-Mallart RM. The caudal limit of Otx2 gene expression as a marker of the midbrain/hindbrain boundary: a study using in situ hybridisation and chick/quail homotopic grafts. Development. 1996;122:3785–3797. doi: 10.1242/dev.122.12.3785. [DOI] [PubMed] [Google Scholar]
- Patten I, Kulesa P, Shen MM, Fraser S, Placzek M. Distinct modes of floor plate induction in the chick embryo. Development. 2003;130:4809–4821. doi: 10.1242/dev.00694. [DOI] [PubMed] [Google Scholar]
- Pierani A, Brenner-Morton S, Chiang C, Jessell TM. A sonic hedgehog- independent, retinoid-activated pathway of neurogenesis in the ventral spinal cord. Cell. 1999;97:903–915. doi: 10.1016/s0092-8674(00)80802-8. [DOI] [PubMed] [Google Scholar]
- Rodriguez I, Basler K. Control of compartmental affinity boundaries by hedgehog. Nature. 1997;389:614–618. doi: 10.1038/39343. [DOI] [PubMed] [Google Scholar]
- Roelink H, Porter JA, Chiang C, Tanabe Y, Chang DT, Beachy PA, Jessell TM. Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of sonic hedgehog autoproteolysis. Cell. 1995;81:445–455. doi: 10.1016/0092-8674(95)90397-6. [DOI] [PubMed] [Google Scholar]
- Sanders TA. Committee on Neurobiology. University of Chicago; Chicago: 2001. Arcuate Patterning as a central feature of ventral midbbrain development during early embryogenesis. [Google Scholar]
- Sanders TA, Lumsden A, Ragsdale CW. Arcuate plan of chick midbrain development. J Neurosci. 2002;22:10742–10750. doi: 10.1523/JNEUROSCI.22-24-10742.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlichting K, Demontis F, Dahmann C. Cadherin Cad99C is regulated by Hedgehog signaling in Drosophila. Developmental Biology. 2005;279:142–154. doi: 10.1016/j.ydbio.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, Scott MP, Pennica D, Goddard A, Phillips H, Noll M, Hooper JE, de Sauvage F, Rosenthal A. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384:129–134. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- Theiler K. Atlas of Embryonic Development. Springer-Verlag; New York: 1989. The House Mouse. [Google Scholar]
- Wijgerde M, McMahon JA, Rule M, McMahon AP. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev. 2002;16:2849–2864. doi: 10.1101/gad.1025702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Henderson CE. Patterns of programmed cell death in populations of developing spinal motoneurons in chicken, mouse, and rat. Dev Biol. 1999;214:60–71. doi: 10.1006/dbio.1999.9413. [DOI] [PubMed] [Google Scholar]
- Ye W, Shimamura K, Rubenstein JLR, Hynes MA, Rosenthal A. FGF and Shh Signals Control Dopaminergic and Serotonergic Cell Fate in the Anterior Neural Plate. Cell. 1998;93:755–766. doi: 10.1016/s0092-8674(00)81437-3. [DOI] [PubMed] [Google Scholar]
- Zervas M, Millet S, Ahn S, Joyner AL. Cell Behaviors and Genetic Lineages of the Mesencephalon and Rhombomere 1. Neuron. 2004;43:345–357. doi: 10.1016/j.neuron.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Zhang XM, Ramalho-Santos M, McMahon AP. Smoothened Mutants Reveal Redundant Roles for Shh and Ihh Signaling Including Regulation of L/R Asymmetry by the Mouse Node. Cell. 2001;105:781–792. [PubMed] [Google Scholar]