

Abstract
Vesicular glutamate transporters (VGLUTs) 1 and 2 show a mutually exclusive distribution in the adult brain that suggests specialization for synapses with different properties of release. Consistent with this distribution, inactivation of the VGLUT1 gene silenced a subset of excitatory neurons in the adult. However, the same cell populations exhibited VGLUT1-independent transmission early in life. Developing hippocampal neurons transiently coexpressed VGLUT2 and VGLUT1 at distinct synaptic sites with different short-term plasticity. The loss of VGLUT1 also reduced the reserve pool of synaptic vesicles. Thus, VGLUT1 plays an unanticipated role in membrane trafficking at the nerve terminal.
Synaptic vesicles actively accumulate the principal excitatory neurotransmitter, glutamate (1, 2), enabling its regulated release by exocytosis. A distinct family of proteins implicated in glutamate release by genetic studies in Caenorhabditis elegans (3) mediates vesicular glutamate transport with the same properties as the transport of native synaptic vesicles (4-8). Heterologous expression of the VGLUTs in inhibitory neurons suffices to confer exocytotic glutamate release (5, 7). However, the VGLUTs were originally isolated as Na+-dependent plasma membrane carriers for inorganic phosphate (9, 10) and mediate a chloride conductance (4), raising questions about the precise nature of their role in excitatory transmission (11).
Despite similar transport activity, VGLUT1 and VGLUT2 exhibit notable complementary patterns of expression in the adult mammalian brain (6, 12). VGLUT1 mRNA is strongly expressed in the cerebral cortex, hippocampus, and cerebellar cortex, and VGLUT2 is expressed in the thalamus, brainstem, and deep cerebellar nuclei. VGLUT1 and VGLUT2 proteins also segregate to distinct synapses with different physiological properties (6, 12). In general, synapses expressing VGLUT1 show a low probability of transmitter release, whereas synapses expressing VGLUT2 show a higher probability of release (6). Thus, VGLUT1 and VGLUT2 may contribute to differences in synaptic transmission that extend beyond their identified role in glutamate uptake. A third VGLUT isoform (VGLUT3) with similar activity is expressed by a small number of cell populations, many of which are not traditionally considered glutamatergic (13-15).
To examine the role of the different VGLUT isoforms in vivo, we inactivated mouse VGLUT1 by deleting the first two transmembrane domains through homologous recombination in embryonic stem cells (fig. S1). Homozygous VGLUT1 knockout mice were born in the expected mendelian ratios from heterozygous parents and exhibited no gross phenotypic abnormalities for about 2 weeks after birth. Examined by Nissl stain, the overall cytoarchitecture of brains from knockout mice was also indistinguishable from that of wild-type mice. Using brain extracts from individual animals at 3 weeks of age, we could not detect VGLUT1 protein in knockout mice (Fig. 1A). The other two isoforms were not up-regulated (Fig. 1A). Synaptic vesicle-enriched membrane fractions from VGLUT1-/- animals also exhibited a reduction in glutamate but not serotonin uptake (Fig. 1B).
Fig. 1.
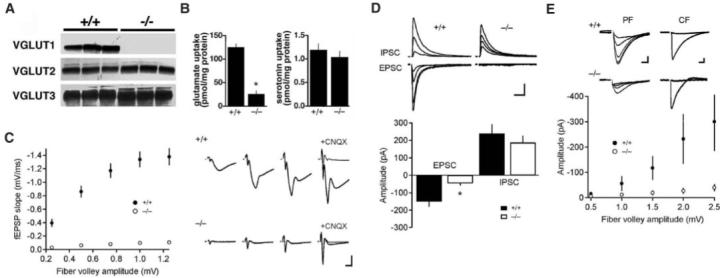
Loss of VGLUT1 affects a subset of excitatory synapses. (A) Western analysis of membranes from the brains of wild-type (+/+) and VGLUT1 knockout (-/-) mice, using antibodies specific for the C termini of VGLUT1 to VGLUT3 (n = 3). (B) Uptake of glutamate and serotonin by LP2 fractions prepared from individual wild-type and VGLUT1 knockout mice. The results indicate the mean ± SEM. (*, P < 0.05; n = 3) (C) Input-output curve for basal synaptic transmission in hippocampal slices from 3-week-old wild-type and VGLUT1-/- mice. fEPSPs were recorded at a range of stimulus intensities from CA1 stratum radiatum. fEPSP slopes in the knockout mice are significantly different from wild-type mice at every fiber volley amplitude (P < 0.0001). However, residual excitatory transmission persists in the knockout, and 100 μM CNQX abolishes the fEPSP in both wild-type and mutant mice. Each point represents the mean ± SEM for each bin (wild type, n = 21; VGLUT1-/-, n = 20). Sample fEPSPs at different stimulus intensities are shown to the right. Scale bar, 0.5 mV (y axis), 5 ms (x axis). (D) EPSCs measured in CA1 pyramidal cells by whole-cell voltage clamp at -70 mV are severely impaired in mutant animals, whereas IPSCs measured at 0 mV (EPSC reversal potential) and the same range of stimulus intensities are not significantly different from those of the wild type. High stimulus intensities reveal residual glutamatergic transmission in VGLUT1-/- mice, which differs significantly from wild-type mice (*, P < 0.05). Error bars show the mean + SEM (wild type, n = 4; VGLUT1-/-, n = 4). Scale bar, 20 pA, 50 ms. (E) In the cerebellum at 3 weeks of age, parallel fiber responses measured in whole-cell recordings from Purkinje cells are impaired but detectable, particularly at high stimulus intensities. Climbing fiber responses are unaffected. Each point represents the mean ± SEM for each bin (wild type, n = 5; VGLUT1-/-, n = 12). Inset shows sample parallel fiber (PF) EPSCs at different stimulus intensities and climbing fiber (CF) responses around the threshold stimulus. PF scale bar, 25 pA, 10 ms. CF scale bar, 400 pA, 10 ms.
Schaffer collaterals in stratum radiatum of the CA1 hippocampus are thought to comprise a homogeneous population of excitatory terminals that express VGLUT1. At 3 weeks of age, field recordings from CA1 stratum radiatum of knockout mice showed a substantial reduction in field excitatory postsynaptic potentials (fEPSPs) relative to wild-type littermates (Fig. 1C). Surprisingly, a small amount of residual transmission persisted in the knockouts, particularly at high stimulus intensity. The glutamate receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) abolished this residual response, indicating that it reflected glutamate release. Despite the severe reduction in excitatory postsynaptic currents (EPSCs) (Fig. 1D), whole-cell voltage clamp experiments showed no significant change in inhibitory postsynaptic currents (IPSCs) in knockout CA1 pyramidal neurons. In addition, heterozygous mice expressing 50% of the normal amount of VGLUT1 protein showed fEPSPs indistinguishable from those of wild-type mice (fig. S2), indicating that physiological levels of expression did not limit excitatory transmission.
To assess the specificity for synaptic terminals expressing VGLUT1, we examined slices from the cerebellum. Parallel fiber inputs onto Purkinje cell dendrites of the molecular layer express VGLUT1, whereas climbing fiber terminals onto the same postsynaptic cells express VGLUT2 (6). Consistent with this distribution, whole-cell recordings from Purkinje cells showed a severe reduction in the response to parallel fibers but no change in the climbing fiber response of knockout mice (Fig. 1E). High stimulus intensities nonetheless produced significant parallel fiber EPSCs in VGLUT1-/- animals (Fig. 1E). The unexpected residual activity observed in both the hippocampus and cerebellum may thus reflect a general property of VGLUT1+ synapses rather than a specific feature of one circuit.
Indistinguishable from the wild type at birth, VGLUT1-/- animals older than 2 to 3 weeks fed poorly and did not survive unless maintained separately from littermates. After weaning, they could survive independently for several months, but showed a progressive neurological phenotype that included blindness, uncoordination, and an enhanced startle response. The time course of this decline raised the possibility that high levels of VGLUT1-independent transmission early in development might minimize the effects of VGLUT1 inactivation. Indeed, 3- to 5-day-old mice exhibited VGLUT1-independent transmission in CA1 stratum radiatum that approached 50% of wild-type transmission (Fig. 2, A and B). The residual activity of knockout mice then declined precipitously over the next 2 weeks. Mature animals (older than 2 months) showed essentially no residual excitatory transmission. The magnitude of this defect presumably contributed to the severe adult neurological phenotype (Fig. 2, C to E).
Fig. 2.
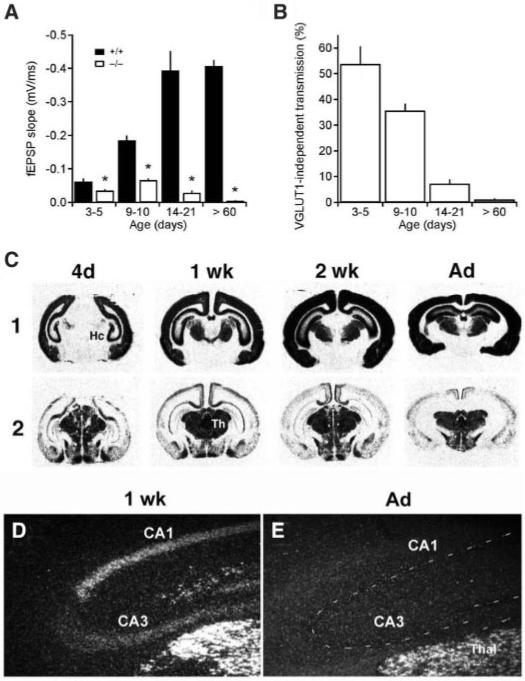
Developmental changes in VGLUT1-independent transmission correlate with the transient expression of VGLUT2 in hippocampal pyramidal neurons. (A) fEPSP slopes measured at a fiber volley amplitude of 0.25 mV increase with age in wild-type mice, but decline in VGLUT1-/- mice after postnatal day 9 to 10. Differences between wild-type and VGLUT1-/- mice are significant (*, P < 0.001). Error bars show mean + SEM. (wild type, n = 15 to 40; VGLUT1-/-, n = 18 to 43). (B) VGLUT1-independent synaptic transmission expressed as a percentage of the wild type (at a fiber volley amplitude of 0.25 mV) constitutes a larger percentage of excitatory synaptic transmission in hippocampi from younger mice and declines through development. Each bar represents the data collected from 15 to 43 slices; error bars show mean + SEM. (C) In situ hybridization from the developing mouse brain with 35S-labeled VGLUT1 and two antisense RNA probes. VGLUT1 expression increases progressively from 4 days (4d) to adulthood (Ad). In contrast, VGLUT2 expression does not change in the thalamus but reaches a peak at 1 week (1 wk) in the hippocampus, followed by a rapid decline and disappearance by adulthood. Hc, hippocampus; Th, thalamus. (D and E) Darkfield microscopy of sections hybridized with 35S-labeled antisense VGLUT2 RNA and dipped in autoradiographic emulsion shows substantial labeling of the pyramidal cell layer in CA1 to CA3 from 1-week-old (D) but not adult (E) wild-type mice. In the adult, the pyramidal cell layer is indicated by a dashed line, and the thalamus (Thal) serves as a control for the labeling.
To identify the VGLUT isoform transiently coexpressed with VGLUT1, we performed in situ hybridization through development. Hippocampal pyramidal cells in CA1 to CA3 and dentate gyrus granule cells expressed substantial amounts of VGLUT2 at 1 week after birth, followed by a rapid reduction over the next 2 weeks (Fig. 2, C to E), a pattern correlating with VGLUT1-independent transmission and the temporal profile of neurological decline. The expression of both VGLUT1 and VGLUT2 by developing hippocampal neurons represents a significant divergence from the mutually exclusive expression of these isoforms in adulthood.
When coexpressed in hippocampal pyramidal cells, VGLUT1 and VGLUT2 might (i) colocalize on the same synaptic vesicles, (ii) segregate to different vesicles located at the same release site, or (iii) target to distinct release sites. Each possibility predicts distinct physiological deficits to account for the large overall reduction in excitatory transmission observed in VGLUT1-/- mice. In light of its role in vesicle filling, the deletion of VGLUT1 might be expected to reduce quantal size. However, miniature EPSC (mEPSC) amplitude, reflecting the amount of glutamate released per vesicle (as well as the postsynaptic response), did not differ in knockout mice relative to wild-type littermates (Fig. 3, A and B). Because wild-type transmission is mediated predominantly by VGLUT1 and the VGLUT1-independent activity by VGLUT2, we conclude that the two isoforms localize to different synaptic vesicles (16).
Fig. 3.
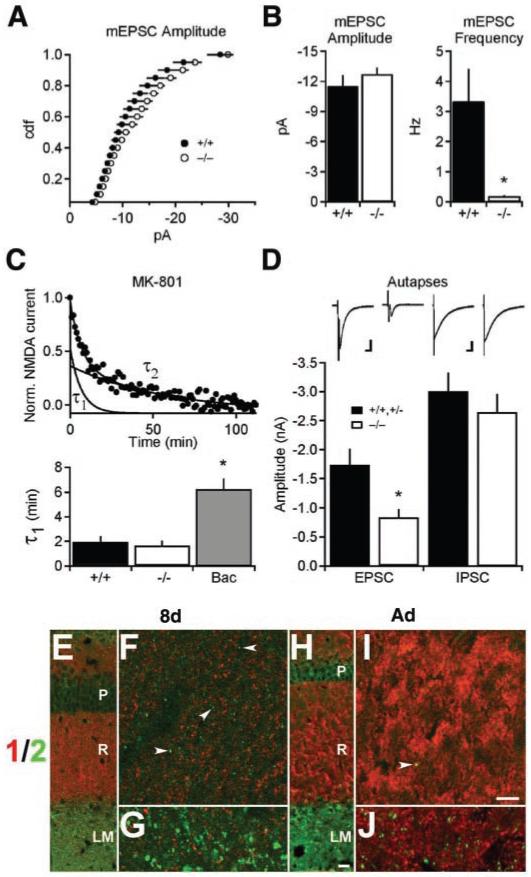
VGLUT1-independent transmission arises from a distinct subpopulation of release sites made by a single cell. (A) The distribution of miniature EPSC (mEPSC) amplitudes recorded from stratum radiatum does not differ between mutant and wild-type mice (P > 0.1, Kolmogorov-Smirnov; wild type, n = 6; VGLUT1-/-, n = 5). (B) Mean mEPSC amplitude does not differ between knockout and wild-type animals. In contrast, the frequency of mEPSCs is severely reduced (*, P < 0.05; wild type, n = 6; VGLUT1-/-, n = 5). (C) In the presence of the use-dependent NMDA receptor antagonist MK-801, the decay rate of NMDA EPSCs from 9- to 10-day-old VGLUT1-/- slices does not differ from that of the wild type (P > 0.1). Decay curves could be fit by two exponentials, τ1 and τ2. The GABAB agonist baclofen (Bac) (60 μM) increases τ1, indicating its sensitivity to changes in probability of release (*, P < 0.01), whereas τ2 does not change under all conditions (P > 0.1; wild type, n = 7; VGLUT1-/-, n = 11; baclofen, n = 8). (D) Autaptic cultures prepared from newborn VGLUT1-/- hippocampi (10 to 14 days in culture) show reduced EPSC amplitudes relative to combined cultures of wild-type and VGLUT1+/- heterozygotes (P < 0.01; wild type/heterozygote, n = 26; VGLUT1-/-, n = 33). Autaptic IPSCs from knockout cultures were not significantly different from those of the wild type (P > 0.1; wild type/heterozygote, n = 31; VGLUT1-/-, n = 38). EPSC scale bar, 500 pA, 10 ms. IPSC scale bar, 1 nA, 50 ms. (E to J) Immunofluorescence for VGLUT1 (red) and VGLUT2 (green) in 8-day-old (8d) or adult (Ad) wild-type mouse CA1 hippocampus. (F) and (I) show high-magnification fields from the stratum radiatum (marked by the R) of (E) and (H), respectively. (G) and (J) show high-magnification fields of the corresponding stratum lacunosum moleculare (LM). VGLUT1 does not colocalize with VGLUT2 at either age. Fine puncta in stratum radiatum label for VGLUT2 (arrowheads) at 8 days (C) but not adulthood (I), whereas larger puncta in stratum lacunosum moleculare stain for VGLUT2 at both ages [(D) and (J)]. P, stratum pyramidale. Scale bar in (H), 60 μm [applies to (E) and (H)]; scale bar in (I), 4 μm [applies to (F), (G), (I), and (J)].
The loss of VGLUT1 substantially reduced the frequency of mEPSCs (Fig. 3B). To determine whether this reflected a reduction in the probability of releasing a vesicle filled with transmitter from a synaptic site containing both filled and empty vesicles, we took advantage of the use-dependent N-methyl-D-aspartate (NMDA) receptor antagonist MK-801 (17, 18). If filled vesicles co-mingle with empty vesicles, then exocytosis of empty vesicles should reduce the apparent probability of release and increase the time required for inhibition by MK-801. However, the decay in NMDA response resulting from MK-801 was not affected by the loss of VGLUT1 (Fig. 3C). The γ-aminobutyric acid type B (GABAB) agonist baclofen, which reduces the probability of transmitter release (18), caused a clear prolongation in the first component of MK-801-dependent decay (Fig. 3C), even though this concentration (60 μM) reduces fEPSPs to a smaller extent than the loss of VGLUT1, confirming the sensitivity of this assay to changes in release probability. The residual responses present in VGLUT1 knockout mice thus resembled those of the wild type in terms of quantal size and the probability of release. Because the only parameter severely affected was the frequency of spontaneous release (Fig. 3B), the loss of VGLUT1 appears to have silenced the population of release sites normally expressing this isoform, presumably resulting from the fusion of empty vesicles. Thus, VGLUT1 and VGLUT2 must be expressed at distinct synaptic sites.
Immunofluorescence in stratum radiatum confirmed the localization of VGLUT1 and VGLUT2 to distinct synapses. At 8 days after birth, VGLUT2 localized to puncta that did not double label for VGLUT1. VGLUT2+ terminals in stratum radiatum were also substantially smaller than those positive for VGLUT1 (Fig. 3F). In the adult, the labeling for VGLUT2 in stratum radiatum essentially disappeared (Fig. 3, H and J), consistent with the developmental regulation of VGLUT2 mRNA. The persistent expression of VGLUT2 by terminals in adult stratum lacunosum moleculare reflects distinct inputs from the nucleus reuniens of the thalamus (6).
To exclude the possibility that synapses formed by a small, distinct population of cells might contribute to the residual activity observed in VGLUT1 knockouts, we prepared cultures from the newborn hippocampus in which single neurons form synapses exclusively onto themselves (autapses). Neurons from VGLUT1-/- animals showed a clear reduction in EPSC but not IPSC amplitude relative to wild-type littermates (Fig. 3D). In addition, we detected EPSCs in essentially all the neurons that did not exhibit IPSCs. Because all excitatory neurons in the hippocampus appear to express VGLUT1 (6, 11), the residual activity in knockouts must have derived from the same cells normally expressing this isoform. Indeed, many pyramidal neurons expressed VGLUT2 transiently during development in vivo (Fig. 2, C and D).
The residual excitatory transmission observed in VGLUT1 knockout mice provided an opportunity to explore the differences in function between VGLUT1 and VGLUT2. The VGLUT isoforms exhibit very similar transport activity in standard radiotracer flux assays (6, 7, 12-15, 19). However, VGLUT1 and VGLUT2 differ in their steady-state subcellular location when stably expressed in PC12 cells (6), indicating differences in membrane trafficking. To determine whether the two isoforms might recycle differently at the nerve terminal, we examined the response of knockouts to repetitive stimulation, taking advantage of the larger VGLUT1-independent (VGLUT2) response 9 to 10 days after birth and comparing it with the wild-type response, reflecting predominantly VGLUT1. In stratum radiatum, excitatory transmission in VGLUT1-/- animals depressed more rapidly than that in wild-type animals in response to stimulation at 10 Hz, and it recovered more slowly (Fig. 4). Thus, VGLUT1+ and VGLUT2+ synapses made by the same cell exhibited different short-term plasticity, indicating that the distinct vesicles containing these isoforms recycle differently. The altered response in knockout mice may reflect intrinsic differences between VGLUT1 and VGLUT2 proteins or variation in the properties of release at the distinct sites where they are located. Munc13 isoforms also appear to segregate to different release sites formed by individual hippocampal neurons, with important consequences for short-term plasticity (20).
Fig. 4.
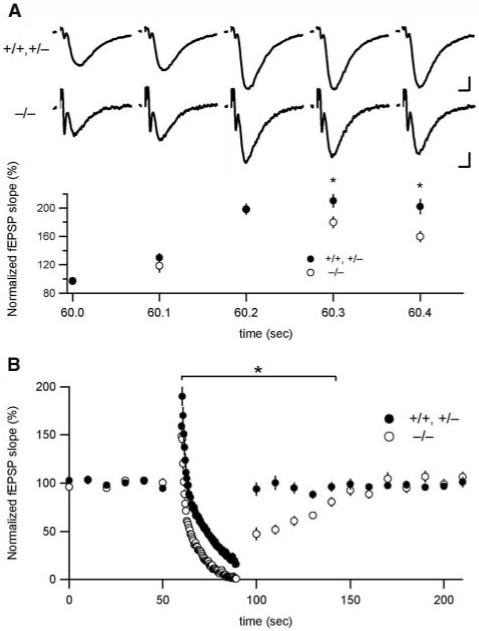
VGLUT1-independent transmission responds differently to repetitive stimulation. (A) Initial responses to 10-Hz repetitive stimulation overlap in postnatal day 9 to 10 in hippocampal slices from wild-type and knockout mice but begin to separate after three stimuli (*, P < 0.05; wild type/heterozygote, n = 31; VGLUT1-/-, n = 28). fEPSP slopes were measured at 0.1 Hz before the onset of stimulation and amplitudes were normalized to baseline fEPSPs. Some data points overlap and are masked. Sample traces above the graph correspond to the first five stimuli of the stimulation train in wild type/heterozygote (top) and knockout (bottom). Error bars show mean ± SEM. +/+,+/- scale bar, 0.2 mV, 5 ms. -/- scale bar, 0.1 mV, 5 ms. (B) The response to prolonged repetitive stimulation (300 pulses at 10 Hz) declines more rapidly and recovers more slowly in knock- out mice relative to that of the wild type (*, P < 0.05; wild type/heterozygote, n = 31; VGLUT1-/-, n = 28). Responses to repetitive stimulation were binned (bin size = 0.4 s) and normalized to fEPSP slopes measured before the onset of stimulation.
Our findings indicate that the vast majority of excitatory synapses in CA1 stratum radiatum of VGLUT1 knockout mice are silent, presumably a result of the fusion of empty synaptic vesicles. To determine whether the loss of VGLUT1 also influenced synapse morphology, we examined the ultrastructure of excitatory synapses in the hippocampus and cerebellum. Wild-type and VGLUT1 knockout mice did not differ in the number of asymmetric synapses (Fig. 5G) or the size of the postsynaptic density (Fig. 5H). The trophic interactions required for synapse maintenance may therefore depend on the release of factors other than glutamate. However, excitatory terminals in stratum radiatum of hippocampal CA1 from VGLUT1-/- animals showed a severe reduction in the number of synaptic vesicles at a time (3 weeks) when there was little residual transmission (Fig. 5, A and B) or when there is none (>7 weeks). In the granule cell layer of the cerebellum, a subset of mossy fibers in knockout mice also showed a substantial reduction in synaptic vesicles (Fig. 5, C and D). Similarly, parallel fiber terminals in the cerebellum of knockout mice contained many fewer synaptic vesicles than wild-type mice (Fig. 5, E and F). In contrast, adjacent climbing fiber terminals showed no morphological defect in the knockout (Fig. 5F), indicating specificity for terminals expressing VGLUT1. Quantitation confirmed the loss of synaptic vesicles in excitatory terminals of both the hippocampus and cerebellum (Fig. 5I). Morphometric analysis of the VGLUT1 knockouts further indicated a severe reduction in the number of vesicles >100 nm away from the active zone in both brain regions (Fig. 5J). The number of vesicles less than 100 nm from the plasma membrane, including morphologically docked vesicles, did not significantly differ between the knockout and wild type, indicating the selectivity of the defect for vesicles at a distance from the active zone (21). The hippocampal and cerebellar terminals of VGLUT1 knockout animals showed irregular, tubulovesicular membranes not observed in wild-type mice (Fig. 5, A to F).
Fig. 5.
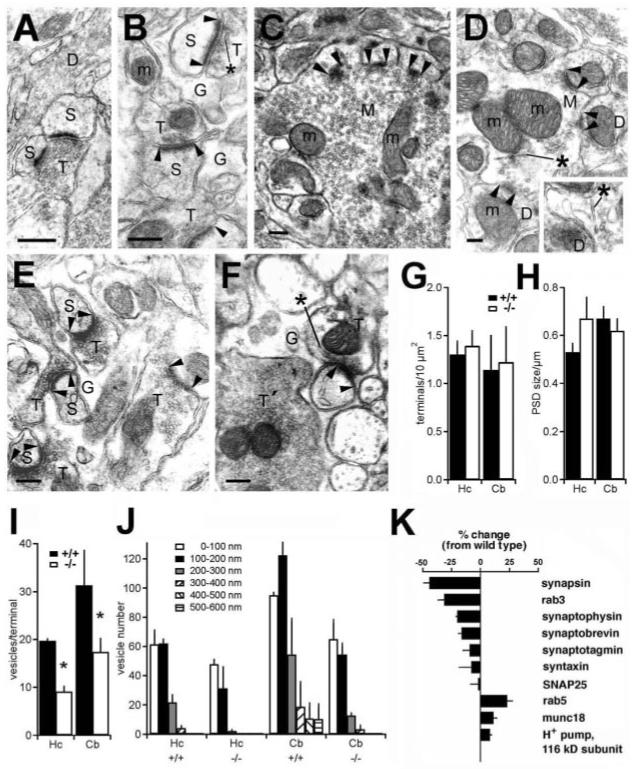
Loss of VGLUT1 specifically reduces the reserve pool of synaptic vesicles. (A) In CA1 stratum radiatum of wildtype mice, a terminal packed with round and homogenous synaptic vesicles makes an asymmetric synapse. (B) An excitatory terminal in stratum radiatum of knockout mice contains many fewer vesicles. The vesicles also have unusual, often elongated shapes (*) and cluster close to the active zone. In the granule cell layer of the cerebellum, a subpopulation of mossy fibers in knockout mice (D) shows a substantial reduction in the number of synaptic vesicles relative to the number in the wild type (C). In the molecular layer of wild-type cerebellum (E), parallel fiber terminals are packed with synaptic vesicles, whereas in knockout mice (F), parallel fiber terminals (T), but not climbing fiber terminals (T′), show a marked reduction in vesicle number. Directly adjacent to the affected parallel fiber terminals, climbing fibers, which express VGLUT2, contain normal numbers of vesicles. In knockout cerebellum, mossy and parallel fiber terminals [(D) and (F)] often exhibit irregular, cylindrical membranes (*) similar to those observed in the hippocampus (B). M, cerebellar mossy fiber terminal; D, dendrite; S, dendritic spine; G, astroglial process; m, mitochondrion; arrowheads, synaptic complexes. Scale bars, 250 nm. Quantitation of excitatory terminal density (G) and postsynaptic density (PSD) size (H) in the hippocampus (Hc) and cerebellum (Cb) shows no significant difference between wild-type (+/+) and VGLUT1 knockout (-/-) mice at 3 weeks or as adults (>7 weeks). (I) A substantial reduction in the number of synaptic vesicles is observed in both the hippocampus (*, P < 0.01) and the cerebellum (*, P < 0.01) of knockout mice. (J) Quantitation of synaptic vesicles as a function of distance from the active zone shows no significant difference between +/+ and -/- mice in the first 100-nm zone. However, the knockout mice in all cases show a substantial reduction in the number of vesicles located >100 nm away from the synapse (P = 0.03 for each condition). Hc, hippocampus; Cb, cerebellum. The data represent the mean + SEM of 3-week-old mice (n = 3). (K) Quantitative Western analysis shows that synaptic vesicle-associated proteins synapsin 1 and rab3 decrease substantially in the absence of VGLUT1. Synaptophysin and synaptobrevin show significant but smaller (∼20%) reductions. The endosomal guanosine triphos phatase rab5 increases. Error bars represent the mean + SEM (n = 3).
To determine whether the loss of synaptic vesicles correlated with biochemical changes, we immunoblotted hippocampal extracts from 3-week-old animals for presynaptic proteins. By quantitative Western analysis, synaptic vesicle proteins such as synaptophysin and the v-soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor (SNARE) synaptobrevin showed modest but significant reductions (15 to 25%) in knockout mice relative to wild-type mice, whereas other vesicle proteins were little affected (Fig. 5K). However, rab3 and synapsin in particular showed large reductions approaching 50% of wild-type levels. Synapsin associates with the reserve pool of synaptic vesicles to promote recycling under conditions of high-frequency stimulation (22, 23), suggesting a correlation with the morphological loss of synaptic vesicles distant from the active zone. The early endosomal protein rab5 showed an increase in the absence of VGLUT1, possibly reflecting the pleomorphic tubulovesicular structures observed in excitatory terminals of the knockout by electron microscopy and consistent with a defect in endocytosis.
In contrast to these results, previous studies that interfered with vesicle filling by inhibiting either the vacuolar H+ pump that provides the driving force for transport (24), or the vesicular acetylcholine transporter (25), did not reveal effects on vesicle recycling. Furthermore, knockout mice with severely impaired or no exocytotic glutamate release have no defect in the reserve (or readily releasable) pool of synaptic vesicles (26-28). The loss of reserve pool vesicles in VGLUT1 knockout mice must therefore reflect a specific role for the protein in synaptic vesicle recycling (29).
VGLUT1 and VGLUT2 target to distinct synaptic release sites formed by a single hippocampal neuron during development. Developmental coexpression of VGLUT2 with VGLUT1 may occur at multiple synapses. In the cerebellum, a wave of VGLUT1 starting at the bottom of the molecular layer replaces the VGLUT2 in parallel fibers 2 to 3 weeks after birth (30). Consistent with this, we have found that parallel fiber synapses also exhibit VGLUT1-independent transmission, presumably due to VGLUT2. Thus, the developmental maturation of VGLUT1+ projections in multiple brain regions involves the transient expression of distinct, VGLUT2+ release sites that may either undergo elimination or simply switch isoforms.
Supplementary Material
Acknowledgments
We thank N. Killeen, N. Calakos, D. House, V. Stein, C. Zhou, and G. Li for assistance, and M. Scanziani, H. Li, and members of the Edwards, Nicoll, and Pleasure laboratories for suggestions. T.Q., F.A.C., and J.S.-M. were supported by the Norwegian Research Council and the European Union. K.K. is supported by NSF, J.J. and D.R.C. by NIH, R.T.F. by NINDS, and R.H.E. by the National Institute of Mental Health, National Institute on Drug Abuse, National Institute of Neurological Disorders and Stroke, and the Mathers Foundation. R.A.N. is a member of the Keck Center for Integrative Neuroscience and the Silvio Conte Center for Neuroscience Research and is supported by the NIH and the Bristol-Myers Squibb Co.
References and Notes
- 1.Maycox PR, Deckwerth T, Hell JW, Jahn R. J. Biol. Chem. 1988;263:15423. [PubMed] [Google Scholar]
- 2.Carlson MD, Kish PE, Ueda T. J. Biol. Chem. 1989;264:7369. [PubMed] [Google Scholar]
- 3.Lee RY, Sawin ER, Chalfie M, Horvitz HR, Avery L. J. Neurosci. 1999;19:159. doi: 10.1523/JNEUROSCI.19-01-00159.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bellocchio EE, Reimer RJ, Fremeau RTJ, Edwards RH. Science. 2000;289:957. doi: 10.1126/science.289.5481.957. [DOI] [PubMed] [Google Scholar]
- 5.Takamori S, Rhee JS, Rosenmund C, Jahn R. Nature. 2000;407:189. doi: 10.1038/35025070. [DOI] [PubMed] [Google Scholar]
- 6.Fremeau RT, Jr., et al. Neuron. 2001;31:247. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- 7.Takamori S, Rhee JS, Rosenmund C, Jahn R. J. Neurosci. 2001;21:RC182. doi: 10.1523/JNEUROSCI.21-22-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herzog E, et al. J. Neurosci. 2001;21:RC181. doi: 10.1523/JNEUROSCI.21-22-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ni B, Rosteck PR, Nadi NS, Paul SM. Proc. Natl. Acad. Sci. U.S.A. 1994;91:5607. doi: 10.1073/pnas.91.12.5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aihara Y, et al. J. Neurochem. 2000;74:2622. doi: 10.1046/j.1471-4159.2000.0742622.x. [DOI] [PubMed] [Google Scholar]
- 11.Bellocchio EE, et al. J. Neurosci. 1998;18:8648. doi: 10.1523/JNEUROSCI.18-21-08648.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Varoqui H, Schafer MK, Zhu H, Weihe E, Erickson JD. J. Neurosci. 2002;22:142. doi: 10.1523/JNEUROSCI.22-01-00142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gras C, et al. J. Neurosci. 2002;22:5442. doi: 10.1523/JNEUROSCI.22-13-05442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fremeau RT, Jr., et al. Proc. Natl. Acad. Sci. U.S.A. 2002;99:14488. doi: 10.1073/pnas.222546799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schafer MK, Varoqui H, Defamie N, Weihe E, Erickson JD. J. Biol. Chem. 2002;277:50734. doi: 10.1074/jbc.M206738200. [DOI] [PubMed] [Google Scholar]
- 16.If both isoforms were present on the same vesicle, the loss of VGLUT1 should have reduced quantal size, assuming that the amount of transporter expressed limits the rate of vesicle filling. If, however, a single VGLUT protein suffices to fill a vesicle to thermodynamic equilibrium, and if many vesicles contain VGLUT2 as well as VGLUT1, the knockout could not have substantially reduced excitatory transmission. A large number of synaptic vesicles must therefore contain only one isoform.
- 17.Hessler NA, Shirke AM, Malinow R. Nature. 1993;366:569. doi: 10.1038/366569a0. [DOI] [PubMed] [Google Scholar]
- 18.Rosenmund C, Clements JD, Westbrook GL. Science. 1993;262:754. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- 19.Takamori S, Malherbe P, Broger C, Jahn R. EMBO Rep. 2002;3:798. doi: 10.1093/embo-reports/kvf159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenmund C, et al. Neuron. 2002;33:411. doi: 10.1016/s0896-6273(02)00568-8. [DOI] [PubMed] [Google Scholar]
- 21.The ratio of vesicles >100 nm from the cell membrane/vesicles. <100 nm from the membrane was 1.73 ± 0.21 (mean ± SEM) in the wild type and 0.88 ± 0.14 in the knockout based on all the data. P = 0.004 by analysis of variance (General Linear Model, MINITAB).
- 22.Rosahl TW, et al. Nature. 1995;375:488. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- 23.Ryan TA, Li L, Chin LS, Greengard P, Smith SJ. J. Cell Biol. 1996;134:1219. doi: 10.1083/jcb.134.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Q, Petersen CCH, Nicoll RA. J. Physiol. 2000;525:195. doi: 10.1111/j.1469-7793.2000.t01-1-00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parsons RL, Calupca MA, Merriam LA, Prior C. J. Neurophysiol. 1999;81:2696. doi: 10.1152/jn.1999.81.6.2696. [DOI] [PubMed] [Google Scholar]
- 26.Verhage M, et al. Science. 2000;287:864. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- 27.Augustin I, Rosenmund C, Sudhof TC, Brose N. Nature. 1999;400:457. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- 28.Altrock WD, et al. Neuron. 2003;37:787. doi: 10.1016/s0896-6273(03)00088-6. [DOI] [PubMed] [Google Scholar]
- 29.Although the altered morphology of excitatory terminals might be thought to contribute to the enhanced short-term depression observed in VGLUT1 knockout mice, the changes presumably occur at silent synapses that would otherwise have expressed VGLUT1, rather than the functional synapses expressing VGLUT2.
- 30.Miyazaki T, Fukaya M, Shimizu H, Watanabe M. Eur. J. Neurosci. 2003;17:2563. doi: 10.1046/j.1460-9568.2003.02698.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.