

Abstract
Aims
β-blockers (BB) are strongly recommended after an acute coronary syndrome (ACS), although all patients may not benefit. Causes for variable patient responses to BB are unknown. Given that myocardial ischemia and BB influence metabolic processes regulated by peroxisome proliferator-activated receptor α (PPARα), we hypothesized that interactions between polymorphisms of the PPARα gene (PPARA) and BB treatment would influence clinical outcome following ACS.
Patients & methods
Patients were prospectively enrolled into an ACS registry. A total of 735 ACS patients were genotyped. Mortality and cardiac rehospitalization through 1 year were analyzed in relation to PPARA genotype and BB prescription (597 BB; 138 no BB) at discharge.
Results
Significantly different outcomes associated with BB therapy were observed according to PPARA IVS7 2498 genotype (p = 0.002 for interaction). PPARA IVS7 2498 GG homozygous patients discharged on BB had decreased cardiac rehospitalization (hazard ratio [HR]: 0.52; 95% CI: 0.32–0.86; p = 0.011), while C allele carriers discharged on BB had nearly threefold increased cardiac rehospitalization (HR: 2.92; 95% CI: 1.32–6.92; p = 0.015; genotype interaction p = 0.0005) compared with patients not on BB. PPARA genotype was also associated with differences in PPARα expression, with significantly increased mRNA levels in myocardial samples from normal hearts among GC heterozygotes compared with GG homozygotes (p = 0.04). Transgenic mice with cardiac-specific overexpression of PPARα showed significantly reduced myocardial contractile and chronotropic responses to the β-sympathomimetic dobutamine (p < 0.05) compared with wild-type littermates, supporting the hypothesis that increased PPARα levels result in a blunted β-adrenergic response.
Conclusions
PPARA IVS7 2498 genotype is associated with heterogeneity in 1-year outcome in response to BB among patients following ACS, and may predict which patients benefit from BB therapy, putatively related to the effect of myocardial PPARα expression on β-adrenergic responsiveness.
Keywords: acute coronary syndrome, β-adrenergic receptors, β-blockers, myocardial ischemia, peroxisome proliferator-activated receptor α, pharmacogenetics, PPARα
More than 1.6 million patients are hospitalized with an acute coronary syndrome (ACS) in the USA each year [101]. In accordance with practice guidelines and quality performance measures [1-3], the majority of these patients are discharged on β-blocker (BB) therapy. This practice stems from randomized clinical trials that have demonstrated, on average, an overall benefit of BB therapy for ACS patients [4,5]. However, recent studies suggest that some patients do not benefit from treatment with BB and others may even be harmed [6,7]. While the basis for the observed heterogeneity in response to BB is unknown, one largely unexplored factor is an individual patient’s genetic profile. Recently, we reported that genetic variation in the adrenergic β-2 receptor gene (ADRB2) is strongly associated with mortality after BB therapy among ACS patients [8].
Peroxisome proliferator-activated receptor α (PPARα) is a lipid-activated nuclear receptor transcription factor that controls expression of genes involved in myocardial energy metabolism. Animal studies have shown that PPARα regulates myocardial metabolism under basal conditions and in response to physiologic stressors, including myocardial ischemia [9-15]. BBs can also affect myocardial energy metabolism by decreasing systemic lipolysis and myocardial fatty acid oxidation [16,17], suggesting that the therapeutic response to BBs may be mediated, in part, by cellular lipid metabolic pathways.
The influence of PPARA genotype on clinical outcomes among patients with ACS and specifically in response to BB therapy, has not been reported. We therefore sought to investigate the relationship of PPARA genotype with response to BB therapy following hospitalization for ACS. We examined a SNP in PPARA (PPARA IVS7 2498 G>C) that has previously been reported to be associated with cardiac phenotypes. Specifically, the PPARA IVS7 2498 SNP has been associated with degree of physiologic (in response to intense exercise training) and pathologic (hypertensive) cardiac hypertrophy, progression of coronary artery disease and response to lipid-lowering medications (fibrates) [18-21]. Given that it has been postulated that the associations identified with the PPARA IVS7 2498 G>C SNP could be related to its being in incomplete linkage dis-equilibrium (LD) with PPARA Leu162Val and PPARA IVS1 11394 A>C SNPs [19,22], we also investigated, as a secondary analysis, the individual relationship of these PPARA SNPs and the relationship of PPARA haplotype with response to BB therapy following hospitalization for ACS.
Patients & methods
Subjects
Patients were prospectively enrolled into an ACS registry at two Kansas City, KS, USA, hospitals as previously described [8,23]. Between 1st March 2001 and 31st October 2002, 1199 patients met the criteria for ACS using standard, accepted definitions of myocardial infarction (MI; n = 680) and unstable angina (UA; n = 519) [24,25]. ST-elevation MI (STEMI) patients presented with suggestive cardiac symptoms, diagnostic electrocardiogram (EKG) changes (ST segment elevation or new-onset left bundle branch block [LBBB]), and a positive troponin blood test. Non-STEMI (NSTEMI) patients presented with suggestive cardiac symptoms and/or EKG changes (e.g., ST segment depressions and/or T wave changes), and a positive troponin blood test. UA patients presented with suggestive cardiac symptoms, as defined by at least one of the following: new onset angina (<2 months) of at least Canadian Cardiovascular Society Classification class III, prolonged (>20 min) rest angina, recent (<2 months) worsening of angina, or angina that occurred within 2 weeks of a previous MI [25]. Although EKG changes were not a requirement for diagnosis, nearly half of UA patients had ischemic EKG changes on admission (LBBB 4%, ST-elevations 9%, ST-depressions 12%, T-wave inversions 22%). By definition, all UA patients had a negative troponin blood test. To further increase the specificity of the UA diagnosis, those patients with a diagnostic study that excluded obstructive coronary disease, cardiac perfusion defects or segmental wall motion abnormalities (e.g., coronary angiography, nuclear or echocardiographic stress testing; n = 125) or confirmed an alternative explanation for their presentation (e.g., esophagogastroduodenoscopy) were excluded. Three physicians reviewed the charts of all patients with diagnostic uncertainty (n = 45) and attained consensus on the final diagnosis. Thrombolysis in Myocardial Infarction (TIMI) UA/NSTEMI score was defined using standard, accepted definitions [26].
Each patient was prospectively interviewed during hospitalization to ascertain sociodemo-graphic, economic and health status characteristics. Detailed chart abstractions were performed to obtain patients’ medical history, laboratory results, disease severity and the processes of inpatient care. The study was approved by the Institutional Review Boards of both institutions and written informed consent was provided by each participant with separate consent for the genetic analysis. Race was self-reported. Although there were no differences in gender (93.2% of men vs 92.1% of women), Caucasians (91.5% vs 98.3%; p < 0.001) and older patients (mean age for those consenting = 61 ± 13 years vs 65 ± 13 years for those not consenting; p = 0.004) were less likely to consent to DNA testing. A total of 735 patients enrolled in the genetic substudy had discharge medication status known, constituting the cohort for the current analysis.
Outcomes assessment
All-cause mortality and repeat hospitalization was captured by query of the Social Security Death Masterfile on 1st March 2006, by examination of hospital records and/or telephone follow-up. Hospitalizations for chest pain, heart failure, myocardial infarction or coronary revascularization were defined as cardiac.
Genotyping
Genetic analyses were approved by the Institutional Review Board of Washington University, WA, USA. Specimens were processed in compliance with Health Insurance Portability and Accountability Act (HIPPA) guidelines. DNA isolation, extraction and amplification were performed as previously described [27]. Pyrosequencing was performed using the PSQ™ HS 96A system (Biotage, MA, USA) with MA v2.0 software as previously described [27] using PCR primers (1 pM), 1 ng DNA and conditions listed in Table 1.
Table 1.
Polymorphism primers and genotyping conditions.
| Polymorphism | rs number | Primer sequence | Annealing temperature (°C) |
|---|---|---|---|
| PPARA IVS7 2498 G>C | rs4253778 | Forward 5′-biotinCCTAGTTCTAACACAATCACTCCTT-3′ | 57 |
| Reverse 5′-GGCAATAGTGCCCTAAGTACACCT-3′ | |||
| Internal 5′-GAAATGAAGCTTTTGAATC-3′ | |||
| PPARA L162V G>C | rs1800206 | Forward 5′-biotinATGACAAGTGCGACCGCAGCTGCA-3′ | 66 |
| Reverse 5′-TGTGCAGGGCCACCTTACCTACCGTTGT-3′ | |||
| Internal 5′-TGACATCCCGACAGA-3′ | |||
| PPARA IVS1 11394 A>C | rs135539 | Forward 5′-CCAGGGGGAGGAAAGAGTGAA-3′ | 62 |
| Reverse 5′-biotinGCCACAACTAAGCAGGCAGTG-3′ | |||
| Internal 5′-GCAGAATTTAAATCCTAGGT-3′ |
Human cardiac tissue studies
Cardiac tissue samples were obtained from 34 hearts with no history of myocardial dysfunction and with normal left ventricular ejection fraction determined by echocardiography donated for orthotopic cardiac transplantation but declined for reasons related to size or ABO blood type mismatch. Consent for donation of cardiac tissue for research purposes was obtained from family members by the organ donor organization covering the Colorado—Wyoming—Montana region (Donor Alliance). A total of 0.5 μg of total RNA isolated from human cardiac tissue using an RNeasy® kit (Qiagen Inc., CA, USA) with the addition of DNase was reverse transcribed into cDNA using the SuperScript III™ first strand cDNA synthesis kit (Invitrogen, CA, USA) and random primers. (Typically, 0.1 ng of cDNA, 12.5 nM of each primers and Power Syber Green PCR Master Mix [ABI] with ROX as an internal control were used in the RT-PCR reactions). Data were normalized to 18S expression. eactions were performed in triplicate using the ABI7300 system. The following human-specific primer probe sets were used to detect specific gene expression: hPPARα: Forward: 5′-CGGAGTCCACGCGTGTGAA-3′; hPPARα: Reverse: 5′-GCTGCGGTCGCACTTGTCAT-3′.
Animal studies
All animal experiments and euthanasia protocols were performed in compliance with the National Institutes of Health guidelines for humane treatment of animals and approved by the Animal Care Committee of Washington University. Transgenic mice with the PPARα gene downstream of the myosin heavy chain promoter, resulting in cardiac-specific overexpression of PPARα (MHC-PPARα) have previously been described [28].
In vivo cardiac hemodynamic studies were performed as previously described [29] in intubated, ventilated adult (10–12 weeks) MHC-PPARα mice (n = 5) and age-matched wild-type control littermates (n = 4) anesthetized with thiopental sodium (60 mg/kg). Hemodynamic measurements were recorded, using a 1.4-Fr high-fidelity micromanometer catheter (Millar Instruments, TX, USA) inserted into the left ventricle retrograde across the aortic valve, at baseline and 3 min following the start of each incremental dose of continuously infused intravenous dobutamine (2, 4, 8, 16 and 32 ng/g body weight/min, respectively). Pressure data (continuous aortic pressure, left ventricular systolic and diastolic pressure, heart rate and the derivative of left ventricular pressure over time [dP/dt]) was acquired and digitized with the BioBench computer software data acquisition system (National Instruments).
Statistical analyses
Hardy—Weinberg equilibrium was assessed by χ2 in Caucasians and African—Americans separately. Pair wise linkage (D’) and haplotype analysis was carried out using the Polymorphism and Haplotype Analysis Suite [30]. Patient characteristics were summarized after stratification by discharge BB status and genotype. Continuous variables were reported as mean plus or minus standard deviation and compared using t-tests. Lipid values had skewed distributions, were log-transformed prior to analysis, and were summarized by median and interquartile range. Categorical variables were summarized by frequency and compared using χ2 or Fisher’s exact tests, when the expected cell size was less than 5.
The primary outcome was time to rehospitalization for cardiac events through 12 months. Time to all-cause mortality, to the combined end point of all-cause mortality or cardiac rehospitalization, and to all-cause rehospitalization were also examined. Event rates by PPARA IVS7 2498 genotype and by BB status within PPARA IVS7 2498 genotypes were calculated using Kaplan—Meier analysis and compared using log-rank tests. (GC and CC genotypes were combined into one group due to the low minor allele frequency of C allele).
The relative hazard associated with BB use within genotype was estimated using Cox proportional hazard models, including BB, genotype and BB by genotype interaction, both in crude analysis and adjusting for other patient and process of care characteristics (receipt of aspirin within 24 h of admission, angiotensinconverting enzyme inhibitor at discharge for patients with left ventricular systolic dysfunction, aspirin at discharge, tobacco education for current smokers, and whether reperfusion was done in a timely fashion) [101].
To account for the nonrandom allocation of BB therapy to patients, propensity scores for BB use were calculated using nonparsimonious logistic regression models that included all variables in Table 2 as well as second-order interactions with p-values less than 0.2. The resulting model had good discrimination (c = 0.74) and sufficient overlap between BB groups to permit adjustment. Propensity for BB use (on the logit scale) was then included as a covariate in the proportional hazards model, along with site and Table 3 variables that were significantly associated with PPARA IVS7 2498 genotype. All continuous variables were modeled using restricted cubic splines [31]. Proportional hazard assumptions were verified using Schoenfeld residuals. p-values less than 0.05 were considered statistically significant. Analyses were performed using SAS version 9.1 (SAS Institute, Inc., NC, USA) and R version 2.2.0 [32]. In addition to our primary analysis of the association of PPARA IVS7 248 genotype with cardiac rehospitalization through 12 months, we performed a secondary analysis where PPARA IVS7 2498 SNP and PPARA haplotype were added to the proportional hazards model, individually and together, to assess which provided the most prognostic information on cardiac outcomes or on BB response. These analyses were performed among African—Americans and Caucasians separately, to minimize confounding by racial admixture. Given that three SNPs contributed to this analysis, a conservative Bonferroni correction was applied and a p-value of less than 0.016 was considered statistically significant.
Table 2.
Patient characteristics by discharge β-blocker status.
| Characteristic | Discharged on β-blocker (n= 597) |
Not discharged on β-blocker (n= 138) |
p-value |
|---|---|---|---|
| Age, mean (SD) | 60.7 (12.3) | 58.7 (13.0) | 0.10 |
| Sex, n (%) | 0.74 | ||
| Male | 381 (63.8) | 86 (62.3) | |
| Female | 216 (36.2) | 52 (37.7) | |
| Race, n (%) | 0.385 | ||
| Caucasian | 467 (78.4) | 100 (72.5) | |
| African—American | 105 (17.6) | 35 (25.4) | |
| Hispanic | 16 (2.7) | 3 (2.2) | |
| Asian | 1 (0.2) | 0 (0.0) | |
| Native American | 3 (0.5) | 0 (0.0) | |
| East Indian | 4 (0.7) | 0 (0.0) | |
| Prior MI, n (%) | 199 (33.3) | 47 (34.1) | 0.87 |
| Prior PCI, n (%) | 191 (32.0) | 39 (28.3) | 0.39 |
| Prior CABG, n (%) | 112 (18.8) | 19 (13.8) | 0.17 |
| Chronic heart failure, n (%) | 44 (7.4) | 12 (8.7) | 0.60 |
| Hypertension, n (%) | 398 (66.7) | 89 (64.5) | 0.63 |
| Hyperlipidemia, n (%) | 370 (62.0) | 71 (51.4) | 0.02 |
| Diabetes, n (%) | 162 (27.1) | 43 (31.2) | 0.34 |
| Renal failure, n (%) | 15 (2.5) | 4 (2.9) | 0.77 |
| Chronic lung disease, n (%) | 62 (10.4) | 26 (18.8) | 0.006 |
| Smoking status, n (%) | 0.86 | ||
| Current (≤ 30 days) | 214 (35.9) | 49 (35.5) | |
| Former (>30 days) | 218 (36.6) | 48 (34.8) | |
| Never or <100 cigarettes total | 164 (27.5) | 41 (29.7) | |
| Admission BMI, mean (SD) | 29.5 (6.2) | 30.0 (6.8) | 0.46 |
| Systolic blood pressure, mean (SD) | 138 (27) | 134 (27) | 0.19 |
| Ejection fraction <40%, n (%) | 134 (23.5) | 20 (15.6) | 0.05 |
| Total cholesterol (mg/dl), median (IQR) | 178 (151–210) | 167 (147–198) | 0.29 |
| HDL (mg/dl), median (IQR) | 39 (32–49) | 42 (33–51) | 0.13 |
| LDL (mg/dl), median (IQR) | 102 (77–130) | 92 (71–118) | 0.08 |
| Triglycerides (mg/dl), median (IQR) | 143 (107–214) | 142 (90–229) | 0.57 |
| ACS type, n (%) | <0.001 | ||
| STEMI | 184 (30.8) | 25 (18.1) | |
| NSTEMI | 189 (31.7) | 36 (26.1) | |
| Unstable angina | 224 (37.5) | 77 (55.8) | |
| TIMI STEMI risk score, mean (SD) | 3.1 (2.2) | 3.6 (2.0) | 0.29 |
| TIMI UA/NSTEMI risk score, mean (SD) | 3.0 (1.4) | 2.6 (1.3) | 0.005 |
| Primary reperfusion for STEMI, n (%) | 136 (73.9) | 13 (52.0) | 0.02 |
| Coronary angiography, n (%) | 499 (83.6) | 102 (73.9) | 0.008 |
| Treatment strategy, n (%) | <0.001 | ||
| Medical | 201 (33.7) | 74 (53.6) | |
| PCI | 364 (61.0) | 63 (45.7) | |
| CABG | 32 (5.4) | 1 (0.7) | |
| PPARA IVS7 2498 G>C, n (%) | 0.19 | ||
| GG | 328 (57.6) | 63 (48.8) | |
| GC | 171 (30.1) | 47 (36.4) | |
| CC | 70 (12.3) | 19 (14.7) | |
| PPARA L162 V C>G, n (%) | 0.54 | ||
| CC | 507 (89.4) | 119 (92.2) | |
| CG | 56 (9.9) | 9 (7.0) | |
| GG | 4 (0.7) | 1 (0.8) | |
| PPARA IVS1 11394 A>C, n (%) | 0.05 | ||
| AA | 144 (25.2) | 40 (30.5) | |
| AC | 299 (52.3) | 53 (40.5) | |
| CC | 129 (22.6) | 38 (29.0) |
To convert values for cholesterol to millimoles per liter, multiply by 0.02586. To convert values for triglycerides to millimoles per liter, multiply by 0.01129.
ACS: Acute coronary syndrome; BMI: Body-mass index; CABG: Coronary artery bypass graft; HDL: High-density lipoprotein;IQR: Interquartile range; LDL: Low-density lipoprotein; MI: Myocardial infarction; NSTEMI: Non-ST-elevation myocardial infarction;PCI: Percutaneous coronary intervention; SD: Standard deviation; STEMI: ST-elevation myocardial infarction; UA: Unstable angina.
Table 3.
Patient characteristics by genotype .
| Characteristic |
PPARA IVS7 2498 |
PPARA L162 V |
PPARA IVS1 11394 |
|||
|---|---|---|---|---|---|---|
| GG (n=391) | GC/CC (n=307) | CC (n=626) | CG/GG (n=70) | AA (n=184) | AC/CC (n=519) | |
| Age, mean (SD) | 61.4 (12.3)* | 59.4 (12.6)* | 60.4 (12.6) | 60.5 (12.2) | 60.7 (11.8) | 60.2 (12.7) |
| Sex, n (%) | ||||||
| Male | 258 (66.0) | 188 (61.2) | 398 (63.6) | 47 (67.1) | 113 (61.4) | 340 (65.5) |
| Female | 133 (34.0) | 119 (38.8) | 228 (36.4) | 23 (32.9) | 71 (38.6) | 179 (34.5) |
| Race, n (%) | ||||||
| Caucasian | 358 (91.8)*** | 188 (61.2)*** | 485 (77.5)* | 63 (91.3)* | 158 (85.9)*** | 391 (75.5)*** |
| African— American |
15 (3.8)*** | 109 (35.5)*** | 116 (18.5)* | 4 (5.8)* | 15 (8.2)*** | 111 (21.4)*** |
| Other | 17 (4.4)*** | 10 (3.3)*** | 25 (4.0)* | 2 (2.9)* | 11 (6.0)*** | 16 (3.1)*** |
| Prior MI, n (%) | 125 (32.0) | 109 (35.5) | 207 (33.1) | 23 (32.9) | 65 (35.3) | 171 (32.9) |
| Prior PCI, n (%) | 131 (33.5) | 87 (28.3) | 196 (31.3) | 26 (37.1) | 63 (34.2) | 157 (30.3) |
| Prior CABG, n (%) | 74 (18.9) | 55 (17.9) | 109 (17.4) | 18 (25.7) | 35 (19.0) | 95 (18.3) |
| Chronic heart failure, n (%) |
22 (5.6)* | 33 (10.7)* | 45 (7.2) | 8 (11.4) | 19 (10.3) | 34 (6.6) |
| Hypertension, n (%) |
251 (64.2) | 208 (67.8) | 405 (64.7) | 50 (71.4) | 125 (67.9) | 337 (64.9) |
| Hyperlipidemia, n (%) |
248 (63.4)* | 172 (56.0)* | 379 (60.5) | 44 (62.9) | 120 (65.2) | 304 (58.6) |
| Diabetes, n (%) | 103 (26.3) | 89 (29.0) | 169 (27.0) | 22 (31.4) | 47 (25.5) | 147 (28.3) |
| Renal failure, n (%) |
6 (1.5)* | 12 (3.9)* | 16 (2.6) | 3 (4.3) | 4 (2.2) | 15 (2.9) |
| Chronic lung disease, n (%) |
41 (10.5) | 43 (14.0) | 71 (11.3) | 10 (14.3) | 31 (16.8)* | 51 (9.8)* |
| Smoking status, n (%) | ||||||
| Current (≤ 30 days) |
132 (33.8) | 117 (38.2) | 226 (36.2) | 21 (30.0) | 65 (35.3) | 188 (36.3) |
| Former (>30 days) | 148 (37.9) | 108 (35.3) | 234 (37.4) | 21 (30.0) | 76 (41.3) | 181 (34.9) |
| Never or <100 cigarettes total |
111 (28.4) | 81 (26.5) | 165 (26.4) | 28 (40.0) | 43 (23.4) | 149 (28.8) |
| Admission BMI, mean (SD) |
29.5 (5.8) | 29.8 (7.0) | 29.6 (6.3) | 29.5 (7.0) | 30.1 (6.0) | 29.4 (6.4) |
| Systolic blood pressure, mean (SD) |
136 (26) | 139 (28) | 137 (27)* | 129 (21)* | 133 (23)* | 138 (28)* |
| Ejection fraction <40%, n (%) |
90 (24.2) | 56 (19.2) | 136 (22.9) | 11 (16.9) | 35 (20.6) | 114 (22.9) |
| Total cholesterol (mg/dl), median (IQR) |
177 (151–210) | 175 (147–205) | 176 (149–209) |
173 (144–192) | 178 (147–210) | 177 (150–207) |
| HDL (mg/dl), median (IQR) |
39 (32–48) | 41 (33–51) | 40 (33–49)* | 36 (29–46)* | 38 (32–46)* | 40 (33–50)* |
| LDL (mg/dl), median (IQR) |
100 (74–125) | 98 (76–132) | 99 (76–129)* | 94 (66–113)* | 102 (76–130) | 99 (76–127) |
| Triglycerides (mg/dl), median (IQR) |
146 (107–230)* | 140 (98–202)* | 143 (103–214) |
165 (114–265) | 147 (107–235) | 142 (100–214) |
| ACS type, n (%) | ||||||
| STEMI | 116 (29.7)** | 83 (27.0)** | 181 (28.9) | 21 (30.0) | 53 (28.8) | 146 (28.1) |
| NSTEMI | 136 (34.8)** | 80 (26.1)** | 201 (32.1) | 18 (25.7) | 50 (27.2) | 167 (32.2) |
| Unstable angina | 139 (35.5)** | 144 (46.9)** | 244 (39.0) | 31 (44.3) | 81 (44.0) | 206 (39.7) |
| TIMI STEMI risk score, mean (SD) |
3.0 (2.1) | 3.3 (2.3) | 3.1 (2.2) | 3.2 (2.0) | 3.0 (1.8) | 3.1 (2.2) |
| TIMI UA/NSTEMI risk score, mean (SD) |
3.1 (1.4)* | 2.8 (1.3)* | 2.9 (1.4) | 2.9 (1.3) | 2.9 (1.3) | 2.9 (1.4) |
| Primary reperfusion for STEMI, n (%) |
86 (74.1) | 55 (66.3) | 126 (69.6) | 17 (81.0) | 38 (71.7) | 104 (71.2) |
| Coronary angiography, n (%) |
345 (88.2)*** | 227 (73.9)*** | 514 (82.1) | 59 (84.3) | 150 (81.5) | 425 (81.9) |
| Treatment strategy, n (%) | ||||||
| Medical | 110 (28.1)*** | 146 (47.6)*** | 227 (36.3) | 25 (35.7) | 73 (39.7) | 189 (36.4) |
| PCI | 261 (66.8)*** | 150 (48.9)*** | 371 (59.3) | 42 (60.0) | 102 (55.4) | 307 (59.2) |
| CABG | 20 (5.1)*** | 11 (3.6)*** | 28 (4.5) | 3 (4.3) | 9 (4.9) | 23 (4.4) |
| Discharge β-blocker, n (%) |
329 (83.9) | 241 (78.5) | 507 (81.0) | 60 (85.7) | 144 (78.3) | 428 (82.5) |
To convert values for cholesterol to millimoles per liter, multiply by 0.02586. To convert values for triglycerides to millimoles per liter, multiply by 0.01129.
p< 0.001
p < 0.01
p < 0.05.
ACS: Acute coronary syndrome; BMI: Body-mass index; CABG: Coronary artery bypass graft; HDL: High-density lipoprotein; IQR: Interquartile range; LDL: Low-density lipoprotein; MI: Myocardial infarction; NSTEMI: Non-ST-elevation myocardial infarction; PCI: Percutaneous coronary intervention; SD: Standard deviation; STEMI: ST-elevation myocardial infarction; UA: Unstable angina.
Hemodynamic measurements (heart rate and dP/dt) and mRNA expression level values were summarized by mean and standard deviation and compared using t-tests. p-values less than 0.05 were considered statistically significant.
Results
Baseline characteristics & PPARA genotype frequencies
Of the 735 samples from consecutive ACS patients in the genetic substudy, genotypes for PPARA IVS7 2498, PPARA IVS1 11394 and PPARA Leu162Val SNPs were successfully obtained in 698 (95%), 703 (96%) and 696 (95%), respectively. None of the variants deviated significantly from Hardy—Weinberg equilibrium. PPARA IVS7 2498 C and PPARA IVS1 11394 C alleles exhibited significantly different frequency in Caucasian and African—American ACS patients (PPARA IVS7 2498: 0.20 vs 0.67; PPARA IVS1 11394: 0.43 vs 0.30, respectively; p < 0.001). Baseline clinical and demographic data are listed in Tables 2 & 3 for the patients in whom genetic specimens were acquired. When the patients were stratified according to PPARA IVS7 2498 genotype (Table 3), univariate analyses demonstrated significant differences in age, race, chronic heart failure, hyperlipidemia, renal failure, triglycerides, ACS type, TIMI UA/NSTEMI risk score, receipt of cardiac catheterization and revascularization. Compared with patients with the PPARA IVS7 2498 GG genotype, carriers of the PPARA IVS7 2498 C allele (GC or CC genotype) were younger (59.4 ± 12.6 years vs 61.4 ± 12.3 years), had less hyperlipidemia (56.0 vs 63.4%) and had lower fasting serum triglycerides (140 vs 146 mg/dl). In addition, they had higher prevalence of chronic heart failure (10.7 vs 5.6%), and renal failure (3.9 vs 1.5%) and were less likely to receive cardiac catheterization and revascularization. Most of the patients who were discharged on BB were discharged on metoprolol (∼66%), atenolol (∼8%) or carvedilol (∼6%). There was no significant difference between BB when PPARA IVS7 2498 GG homozygotes were compared with PPARA IVS7 2498 C allele carriers (Table 4).
Table 4.
Discharge BB for the ACS genetic substudy stratified by PPARA IVS7 2498 genotype.
|
PPARA IVS7 2498 C allele |
|||
|---|---|---|---|
|
Allele present (n = 307) |
Allele absent (n = 391) |
p-value | |
| Metoprolol | 196 (63.8%) | 270 (69.1%) | 0.147 |
| Atenolol | 25 (8.1%) | 28 (7.2%) | 0.627 |
| Carvedilol | 17 (5.5%) | 21 (5.4%) | 0.923 |
| Nadolol | 1 (0.3%) | 1 (0.3%) | 1.000 |
| Propanolol | 0 (0.0%) | 2 (0.5%) | 0.507 |
| Sotalol | 2 (0.7%) | 3 (0.8%) | 1.000 |
| Labetalol | 1 (0.3%) | 2 (0.5%) | 1.000 |
| Other β-blockers | 8 (2.6%) | 6 (1.5%) | 0.316 |
ACS: Acute coronary syndrome; BB: β-blocker.
Interaction between PPARA IVS7 2498 genotype & BB treatment influences clinical outcome following ACS
In unadjusted analyses, PPARA IVS7 2498 genotype showed no direct association with all-cause mortality (p = 0.46), all-cause rehospitalization (p = 0.12), cardiac rehospitalization (p = 0.45), or all-cause mortality and cardiac rehospitalization (p = 0.44; data not shown) through 1 year (Figure 1). However, a significant PPARA IVS7 2498 genotype by BB therapy interaction effect on cardiac rehospitalization was observed (p = 0.002 for interaction; Figure 2). Compared with patients not discharged on BB, at 1 year, patients with the PPARA IVS7 2498 GG genotype who were discharged on BB showed significantly decreased cardiac rehospitalization (p = 0.002). By contrast, patients with the GC or CC genotype receiving BB therapy showed a trend toward increased cardiac rehospitalization (p = 0.058) compared with patients not receiving BB therapy. A significant genotype by BB interaction was also found for all-cause rehospitalization (interaction p-value = 0.003) and the combined end point of all-cause mortality or cardiac rehospitalization (interaction p-value = 0.008) but not for all-cause mortality alone (interaction p-value = 0.44). When genotypes were compared within treatment groups, there was significantly increased cardiac rehospitalization in the non-BB-treated patients with the PPARA IVS7 2498 GG genotype compared with the non-BB treated patients with the PPARA IVS7 2498 GC or CC genotypes (Figure 2; p < 0.001). There was no significant difference in cardiac rehospitalization between genotype groups if only the BB-treated group was examined.
Figure 1.
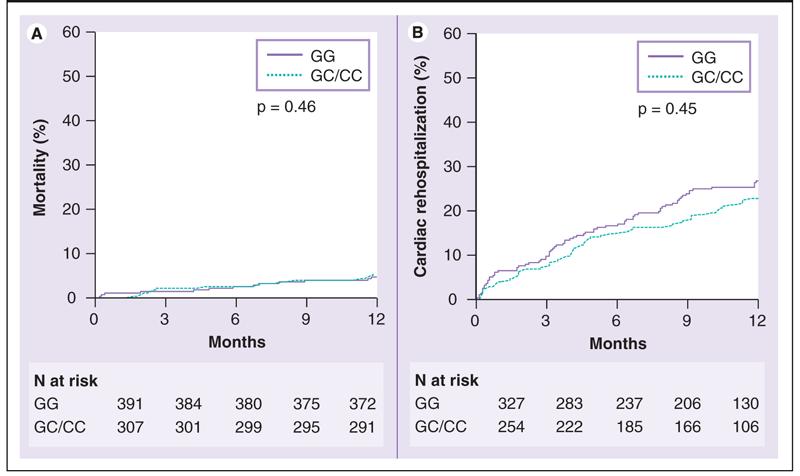
Kaplan-Meier estimates of mortality (left) and cardiac rehospitalization (right) in acute coronary syndrome genetic substudy stratified by PPARA IVS7 2498 genotype.
Figure 2.
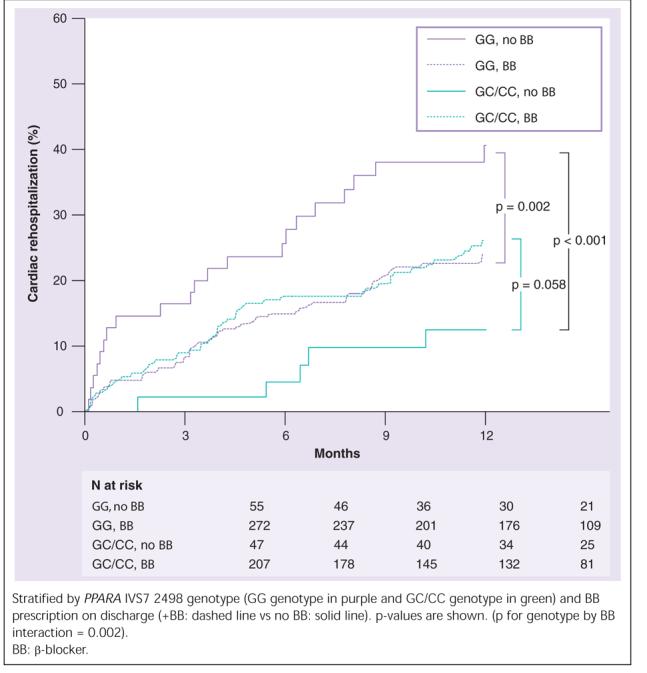
Kaplan-Meier estimates of cardiac rehospitalization in acute coronary syndrome genetic substudy.
In multivariable analysis adjusting for propensity of BB therapy and all factors that differed by PPARA IVS7 2498 genotype, genotype remained an independent predictor of response to BB therapy. Among ACS patients homozygous for the PPARA IVS7 2498 GG genotype, discharge on BB therapy was associated with a 48% relative risk reduction in cardiac rehospitalization (hazard ratio [HR]: 0.52; 95% CI: 0.32–0.86; p = 0.011). Discharge on BB in carriers of the PPARA IVS7 2498 C allele was associated with a nearly threefold relative increase in the risk of cardiac rehospitalization (HR: 2.92; 95% CI: 1.32–6.92; p = 0.015; genotype interaction p = 0.0005). Similar results were found for the combined end point of all-cause mortality plus cardiac rehospitalization, although the reduction in all-cause mortality and cardiac rehospitalization observed in the GG homozygous group was not statistically significant (HR: 0.73; 95% CI: 0.44–1.20; p = 0.21 for GG homozygous; HR: 2.73; 95% CI: 1.23–6.06; p = 0.014 for C allele carries; p-value for interaction = 0.005). HRs and CIs are shown in Figure 3.
Figure 3.
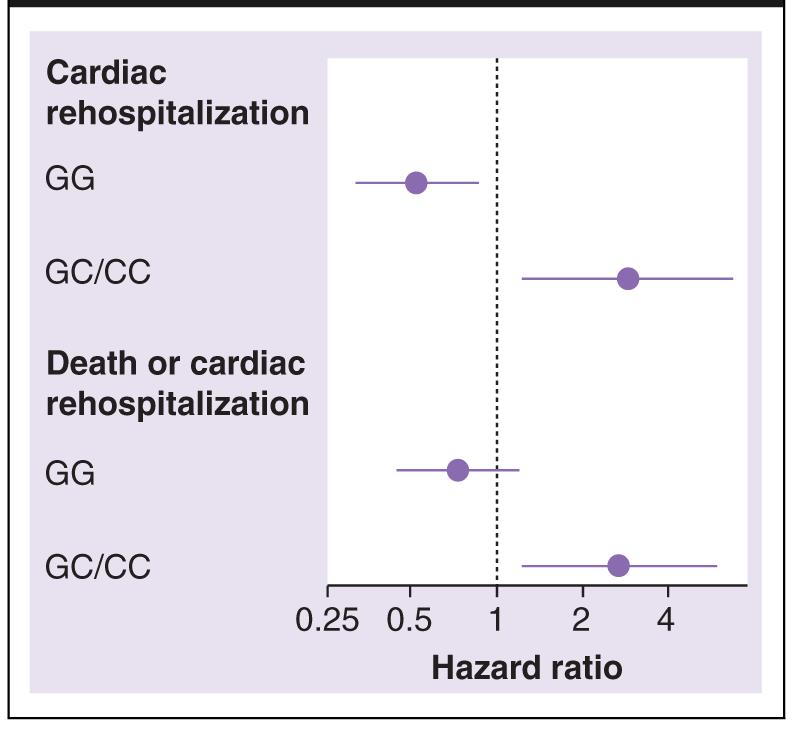
Adjusted hazard ratios for β-blocker use in acute coronary syndrome genetic substudy, by end point and PPARA IVS7 2498 genotype.
Population stratification
Given the racial differences in allele frequency for PPARA IVS7 2498, the effect of PPARA genotype on response to BB therapy was examined separately in Caucasian subjects, our largest racial group. The results were consistent with the findings in the entire group, showing a significant genotype by BB interaction (p = 0.006 for interaction; Figure 4). Additional analyses demonstrated that there was no difference in cardiac rehospitalization when stratified by race and there was no significant race by BB therapy interaction (p = 0.66). Collectively, these data suggest that the observed effects of BB on outcome as a function of PPARA IVS7 2498 genotype are independent of race.
Figure 4.
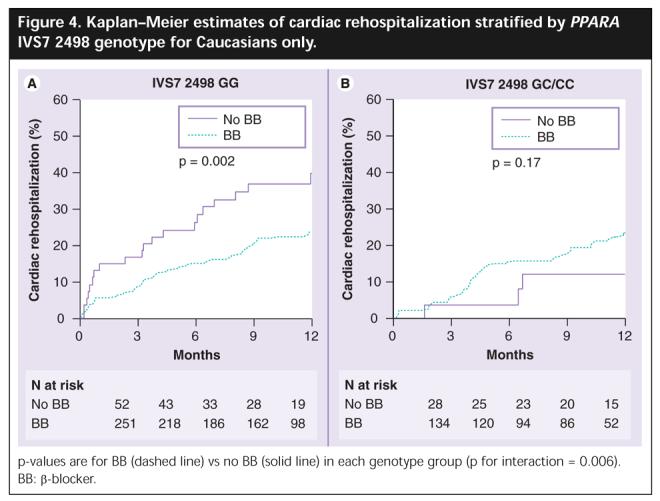
Kaplan-Meier estimates of cardiac rehospitalization stratified by PPARA IVS7 2498 genotype for Caucasians only.
The PPARA IVS7 2498 genotype by BB interaction is not affected by PPARA Leu162Val or PPARA IVS1 11394 genotype
Since haplotype analysis may contribute additional information on the association of genetic variation with phenotypes, we assessed if additional PPARA variants provided improved association with cardiac outcome or affected genotype by BB interaction. SNP and haplotype association analyses were performed among African—Americans and Caucasians separately. No statistically significant association was observed between the rare variants of PPARA IVS7 2498 and PPARA Leu162Val or PPARA IVS1 11394 in the African—American patients (Table 5). Incomplete LD was observed between the rare variants of each of the PPARA SNPs in Caucasians (Table 5).
Table 5.
Pairwise linkage (D’) and haplotype analysis determined by Phase analysis for PPARA IVS7 2498—Leu162Val—IVS1 11394 haplotype.
| D’ |
||
|---|---|---|
| Caucasians | African—Americans | |
| PPARA IVS7 2498 — PPARA L162V | 0.5124** | -1 |
| PPARA IVS7 2498 — PPARA IVS1 11394 | -0.4444** | 0.1464 |
| PPARA L162V — PPARA IVS1 11394 | -0.5454* | 0.4155 |
p < 0.01
p < 0.001
A multivariate proportional hazards model predicting outcome was used to assess the independent contribution of the main effect for PPARA IVS7 2498 SNP and the main effect of the haplotype, in Caucasian subjects, for this region as well as their interactions with BB use. The PPARA IVS7 2498 SNP main effect was significant (p = 0.012) and its interaction with BB use was also significant (p = 0.0156). Neither the main effect for the haplotype nor its interaction with BB use were significant, suggesting that the haplotype did not provide significant predictive power for outcome above that provided by PPARA IVS7 2498 alone. Therefore, once the genotype at PPARA IVS7 2498 was known, haplotype did not provide any additional prognostic information on cardiac outcomes or differential response to BB versus non-BB treatment strategies.
PPARA IVS7 2498 GC heterozygote individuals have increased cardiac PPARα mRNA expression compared with GG homozygote individuals
To investigate the potential mechanism of the clinical association, we determined levels of PPARα mRNA expression in cardiac tissue from 34 hearts (17 GG and 17 GC; none of the individuals were CC homozygotes) with no history of myocardial dysfunction and with normal left ventricular ejection fraction determined by echocardiography donated for orthotopic cardiac transplantation but declined for reasons related to size or ABO blood type mismatch. Mean PPARα expression in cardiac tissue from PPARA IVS7 2498 GC heterozygote individuals was significantly greater than expression in PPARA IVS7 2498 GG homozygote individuals (Figure 5; 1.20 ± 0.30 vs 0.94 ± 0.40; p = 0.04).
Figure 5.
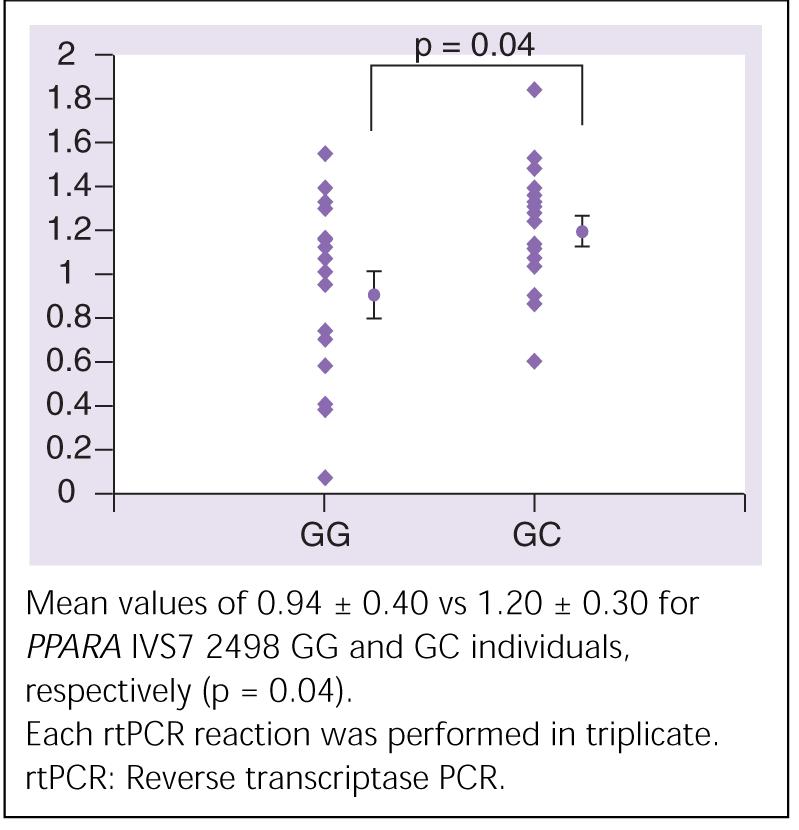
Mean (± standard error) mRNA expression (determined by rtPCR) from normal cardiac tissue from PPARA IVS7 2498 GG (n = 17) and GC (n = 17) individuals.
Hemodynamic response to β-adrenergic stimulation is influenced by activation of the cardiac PPARα pathway
To determine the effect of increased PPARα expression on β-adrenergic responsiveness, we studied changes in heart rate and contractility in response to incremental doses of the β-sympathomimetic dobutamine in an animal model that mimics the relative differences in cardiac PPARα expression demonstrated by our mRNA expression experiments in humans. Transgenic mice with cardiac-specific overexpression of PPARα (MHC-PPARα) [28], compared with nontransgene littermates, had significantly blunted response, as determined by myocardial dP/dt and heart rate, to incremental doses of the β-sympathomimetic dobutamine (Figure 6).
Figure 6.
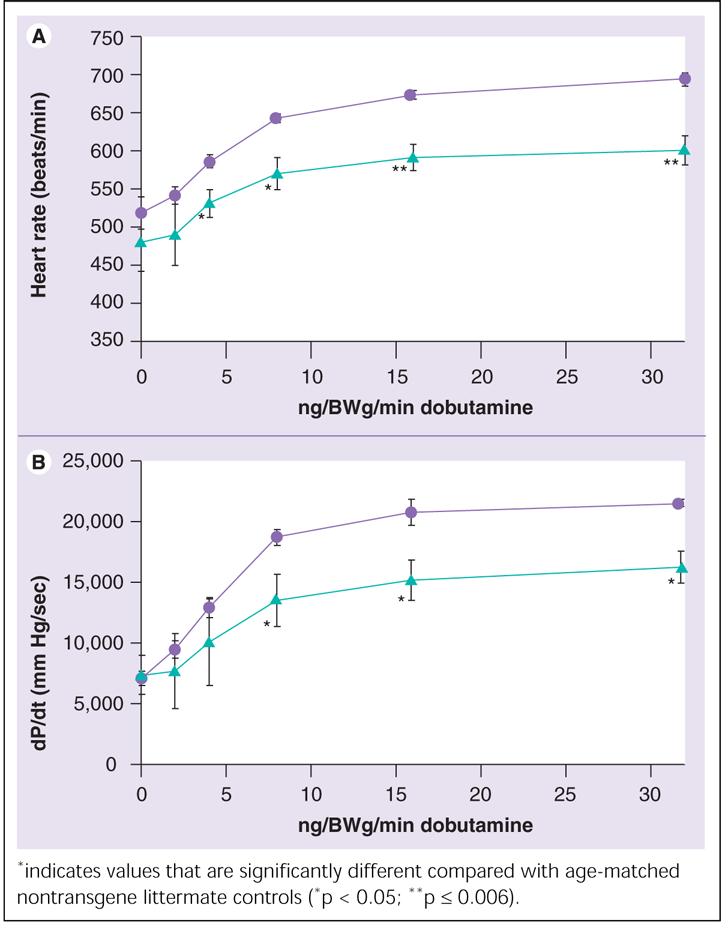
Mean (± standard deviation) heart rate (top) and dP/dt (bottom) response to incremental doses of dobutamine in the MHC-PPARα mice (n = 5, triangles) and age-matched wild-type control littermates (n = 4, circles).
Discussion
Our results demonstrate a significant association between PPARA IVS7 2498 genotype and 1-year outcome in response to BB versus non-BB treatment strategies among ACS patients. This association was observed after multivariable adjustment, including adjustment for the propensity to be prescribed a BB, and suggests there may be both benefit and potential harm associated with BB therapy among ACS patients as a function of patients’ PPARA genotype. While patients with the PPARA IVS7 2498 GG genotype (56% of our population) who were discharged on BB showed significantly decreased cardiac rehospitalization rates compared with similar patients not receiving BB therapy (p = 0.002), patients with the GC or CC genotype receiving BB therapy showed a trend toward increased cardiac rehospitalization rates (p = 0.058). Although this study was not a randomized trial of BB treatment, and the observed associations cannot prove causality, these data support a potentially important pharmacogenetic association between patients’ genotype, treatment and outcomes.
Moreover, we observed marked differences in cardiac rehospitalization rates between genotype groups in the non-BB treated patients, such that the patients with the PPARA IVS7 2498 GG genotype demonstrate significantly increased cardiac rehospitalization in the absence of BB than untreated PPARA IVS7 2498 C allele carriers. Whilst a marked reduction in cardiac rehospitalization is seen with BB in patients with the PPARA IVS7 2498 GG genotype, it still does not attain the favorable outcomes of untreated C-carriers, although their cardiac rehospitalization rates are virtually identical to those of C-carriers treated with BB (Figure 2). These data suggest that an underlying physiologic difference in the myocardium related to PPARA genotype may result in patients with the PPARA IVS7 2498 GC/CC genotypes gaining at best, no benefit, and at worst, harm, from BB therapy. By contrast, patients with the PPARA IVS7 2498 GG genotype derive a significant benefit from BB treatment post-ACS.
The mechanism whereby genetic variation in PPARA IVS7 2498 confers an underlying physiologic difference in the myocardium and, consequently, a variable response to BB has never been reported. We propose, based on our cardiac PPARα mRNA expression data, that the effect may be related to differential PPARα activity that modulates myocardial responses to β-adrenergic stimulation via direct or indirect metabolic effects. In our experiments comparing quantitative responses to β-adrenergic stimulation, we observed blunted adrenergic response in transgenic mice with cardiac-specific overexpression of PPARα compared with nontransgene littermates. Taken together, these data suggest that PPARA IVS7 2498 C allele carriers at baseline are already less responsive to β-adrenergic stimulation compared with GG homozygotes, reducing or eliminating the mechanism of benefit derived from BB administration after ACS and potentially unmasking hazardous effects.
Although these data do not follow the classic pharmacogenetic paradigm where the difference in genotype groups is exaggerated when stratified by treatment, this association represents an important pharmacogenetic interaction. If validated and confirmed by additional prospective studies, these data provide evidence that ACS patients may benefit from being genotyped for the PPARA IVS7 2498 genotype prior to initiating BB therapy. Putatively only those ACS patients with the PPARA IVS7 2498 GG genotype should receive BB while patients with PPARA IVS7 2498 GC/CC genotypes should be considered for alternative therapy.
Although the PPARA IVS7 2498 SNP is an intronic SNP with unknown molecular effects, as discussed above, several strong phenotypic associations of this SNP have been previously described [19,33-35]. The PPARA IVS7 2498 SNP has been associated with degree of physiologic and pathologic cardiac hypertrophy, progression of coronary artery disease and response to lipid-lowering medications [18-21]. It has also been postulated that the associations identified with the PPARA IVS7 2498 SNP could be related to its being in LD with PPARA Leu162Val and PPARA IVS1 11394 SNPs. The results of our haplotype and statistical remodeling analyses, however, do not provide evidence to support this speculation. It is quite possible, however, that the PPARA IVS7 2498 SNP is in LD with another, as yet undiscovered, coding or promoter SNP. Alternatively, given its location and its association with different levels of PPARα mRNA expression, this SNP may disrupt a microRNA site or be in LD with a SNP that disrupts microRNA site. These possibilities clearly warrant further investigation.
Some potential limitations of this study should be considered when interpreting these results. First, the blood samples were obtained from a subset of participants in a registry conducted at two hospitals in a single city. The lack of more diversity in the population and the relatively modest number of patients may have limited the ability to examine the effect of genotype on certain outcomes. We did not find a significant association between PPARA IVS7 2498 genotypes and 1-year survival following ACS or a genotype-by-BB therapy interaction for mortality, although the study had limited power to detect such an association. An additional concern is the potential introduction of selection bias in either patients who participated in the genetic portion of our study or in whom BB therapy was recommended. However, it is unlikely that either patients refusing to participate or physicians initiating BB therapy were aware of patients’ genotype. In addition, we did not have access to pharmacy records and were not able to independently confirm that patients remained on BB for the entire year. However, a previous investigation suggests that approximately 80% of ACS patients discharged on BB will continue taking them for the next year [36]. Finally, this was not a randomized clinical trial of BB stratified by genotype. Although we used propensity models to adjust for observable bias associated with the nonrandom allocation of BB therapy, residual confounding cannot be definitely excluded. We propose that small, genotype-directed prospective studies could be considered to validate and extend these findings.
Conclusion
In conclusion, our data demonstrate a significant association between the PPARA IVS7 2498 genotype and heterogeneity in 1-year outcome in response to BB versus non-BB treatment strategies among ACS patients. These observations are the first to link specific sequence variations in PPARA and response to BB therapy with variable outcomes among patients with cardiovascular disease; the first to demonstrate differential PPARα mRNA expression in cardiac tissue resulting from PPARA IVS7 2498 genotype, providing a molecular mechanism for the clinical associations; and have important practical implications for the treatment of ACS patients. If confirmed by additional prospective studies, these data provide evidence suggesting that ACS patients may benefit from genotyping for the PPARA IVS7 2498 genotype prior to initiating BB therapy.
Future perspective
The PPARs represent promising targets for further advances in pharmacogenetic investigation and intervention. Our current data support that ACS patients may benefit from genotyping for the PPARA IVS7 2498 genotype prior to initiating BB therapy. Future studies are anticipated to confirm these findings in other patient populations and, potentially, to identify other PPARA genotypes that will predict BB responsiveness. Additional investigation into the mechanism by which PPARA genotype affects BB responsiveness may provide new insights into PPAR and β-adrenergic receptor biology. Ultimately, knowledge of relevant PPARA genotypes, as well as a better understanding of the underlying functional mechanism, may help guide individualized therapy for patients with ACS.
Executive summary.
PPARA IVS7 2498 genotype showed no direct association with all-cause mortality (p = 0.46), all-cause rehospitalization (p = 0.12), cardiac rehospitalization (p = 0.45), or all-cause mortality and cardiac rehospitalization (p = 0.44).
A significant PPARA IVS7 2498 genotype by β-blocker (BB) therapy interaction effect on cardiac rehospitalization was observed (p = 0.002 for interaction).
Among acute coronary syndrome patients homozygous for the PPARA IVS7 2498 GG genotype, receiving BB therapy was associated with a 48% relative risk reduction in cardiac rehospitalization (HR: 0.52; 95% CI: 0.32–0.86; p = 0.011).
In carriers of the PPARA IVS7 2498 C allele, receiving BB therapy was associated with a nearly threefold relative increase in the risk of cardiac rehospitalization (HR: 2.92; 95% CI: 1.32–6.92; p = 0.015; genotype interaction p = 0.0005).
Carriers of the PPARA IVS7 2498 C allele have increased cardiac PPARα mRNA expression compared with GG homozygote individuals (1.20 ± 0.30 vs 0.94 ± 0.40; p = 0.04).
Transgenic mice with cardiac-specific overexpression of PPARα, compared with nontransgene littermates, had blunted response, as determined by myocardial dP/dt and heart rate, to incremental doses of the β-sympathomimetic dobutamine.
Footnotes
Financial & competing interests disclosure The authors would like to thank Derek Van Booven for invaluable informatics assistance. This work has been funded by NIH Specialized Center for Clinically-Oriented Research (SCCOR) in Cardiac Dysfunction and Disease #1 P50 HL077113 and Functional Polymorphism Analysis in Drug Pathways UO1 GM063340. DPK is a scientific consultant to Bristol-Myers-Squibb, Pfizer, and Phrixus. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research The author states that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Bibliography
- 1.Krumholz HM, Anderson JL, Brooks NH, et al. ACC/AHA clinical performance measures for adults with ST-elevation and non-ST-elevation myocardial infarction. A report of the American College of Cardiology/American Heart Association Task Force on Performance Measures (writing committee to develop performance measures on st-elevation and non-st-elevation myocardial infarction) Circulation. 2006;113(5):732–761. doi: 10.1161/CIRCULATIONAHA.106.172860. [DOI] [PubMed] [Google Scholar]
- 2.Spertus JA, Eagle KA, Krumholz HM, Mitchell KR, Normand SL. American College of Cardiology and American Heart Association methodology for the selection and creation of performance measures for quantifying the quality of cardiovascular care. Circulation. 2005;111:1703–1712. doi: 10.1161/01.CIR.0000157096.95223.D7. [DOI] [PubMed] [Google Scholar]
- 3.Cannon CP, Battler A, Brindis RG, et al. American College of Cardiology key data elements and definitions for measuring the clinical management and outcomes of patients with acute coronary syndromes. A report of the American College of Cardiology Task Force on Clinical Data Standards (Acute Coronary Syndromes Writing Committee) J. Am. Coll. Cardiol. 2001;38:2114–2130. doi: 10.1016/s0735-1097(01)01702-8. [DOI] [PubMed] [Google Scholar]
- 4.Ellison KE, Gandhi G. Optimising the use of β-adrenoceptor antagonists in coronary artery disease. Drugs. 2005;65:787–797. doi: 10.2165/00003495-200565060-00006. [DOI] [PubMed] [Google Scholar]
- 5.Thattassery E, Gheorghiade M. β-blocker therapy after acute myocardial infarction in patients with heart failure and systolic dysfunction. Heart Fail. Rev. 2004;9:107–113. doi: 10.1023/B:HREV.0000046365.53467.f4. [DOI] [PubMed] [Google Scholar]
- 6.Chen ZM, Pan HC, Chen YP, et al. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. 2005;366:1622–1632. doi: 10.1016/S0140-6736(05)67661-1. [DOI] [PubMed] [Google Scholar]
- 7.McDonald CG, Majumdar SR, Mahon JL, Johnson JA. The effectiveness of β-blockers after myocardial infarction in patients with Type 2 diabetes. Diabetes Care. 2005;28:2113–2117. doi: 10.2337/diacare.28.9.2113. [DOI] [PubMed] [Google Scholar]
- 8.Lanfear DE, Jones PG, Marsh S, Cresci S, McLeod HL, Spertus JA. β2-adrenergic receptor genotype and survival among patients receiving β-blocker therapy after an acute coronary syndrome. JAMA. 2005;294:1526–1533. doi: 10.1001/jama.294.12.1526. [DOI] [PubMed] [Google Scholar]
- 9.Lemberger T, Saladin R, Vazquez M, et al. Expression of the peroxisome proliferator-activated receptor α gene is stimulated by stress and follows a diurnal rhythm. J. Biol. Chem. 1996;271:1764–1769. doi: 10.1074/jbc.271.3.1764. [DOI] [PubMed] [Google Scholar]
- 10.Finck BN, Han X, Courtois M, et al. A critical role for PPARα-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc. Natl Acad. Sci. USA. 2003;100:1226–1231. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panagia M, Gibbons GF, Radda GK, Clarke K. PPAR-α activation required for decreased glucose uptake and increased susceptibility to injury during ischemia. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H2677–H2683. doi: 10.1152/ajpheart.00200.2004. [DOI] [PubMed] [Google Scholar]
- 12.Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J. Clin. Invest. 2005;115:547–555. doi: 10.1172/JCI200524405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huss JM, Levy FH, Kelly DP. Hypoxia inhibits the peroxisome proliferator-activated receptor α/retinoid X receptor gene regulatory pathway in cardiac myocytes: a mechanism for O2-dependent modulation of mitochondrial fatty acid oxidation. J. Biol. Chem. 2001;276:27605–27612. doi: 10.1074/jbc.M100277200. [DOI] [PubMed] [Google Scholar]
- 14.Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor α (PPARα) in the cellular fasting response: the PPARα-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA. 1999;96:7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dewald O, Sharma S, Adrogue J, et al. Downregulation of peroxisome proliferator-activated receptor-α gene expression in a mouse model of ischemic cardiomyopathy is dependent on reactive oxygen species and prevents lipotoxicity. Circulation. 2005;112:407–415. doi: 10.1161/CIRCULATIONAHA.105.536318. [DOI] [PubMed] [Google Scholar]
- 16.Wallhaus TR, Taylor M, DeGrado TR, et al. Myocardial free fatty acid and glucose use after carvedilol treatment in patients with congestive heart failure. Circulation. 2001;103:2441–2446. doi: 10.1161/01.cir.103.20.2441. [DOI] [PubMed] [Google Scholar]
- 17.Panchal AR, Stanley WC, Kerner J, Sabbah HN. β-receptor blockade decreases carnitine palmitoyl transferase I activity in dogs with heart failure. J. Card. Fail. 1998;4:121–126. doi: 10.1016/s1071-9164(98)90252-4. [DOI] [PubMed] [Google Scholar]
- 18.Flavell DM, Jamshidi Y, Hawe E, et al. Peroxisome proliferator-activated receptor α gene variants influence progression of coronary atherosclerosis and risk of coronary artery disease. Circulation. 2002;105:1440–1445. doi: 10.1161/01.cir.0000012145.80593.25. [DOI] [PubMed] [Google Scholar]
- 19.Jamshidi Y, Montgomery HE, Hense HW, et al. Peroxisome proliferator — activated receptor α gene regulates left ventricular growth in response to exercise and hypertension. Circulation. 2002;105:950–955. doi: 10.1161/hc0802.104535. [DOI] [PubMed] [Google Scholar]
- 20.Zak I, Balcerzyk A, Sarecka B, Niemiec P, Ciemniewski Z, Dylag S. Contemporaneous carrier-state of two or three ‘proatherosclerotic’ variants of APOE, ICAM1, PPARA and PAI-1 genes differentiate CAD patients from healthy individuals. Clin. Chim. Acta. 2005;362:110–118. doi: 10.1016/j.cccn.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 21.Cresci S. The PPAR genes, cardiovascular disease and the emergence of PPAR pharmacogenetics. Expert Opin. Pharmacother. 2005;6:2577–2591. doi: 10.1517/14656566.6.15.2577. [DOI] [PubMed] [Google Scholar]
- 22.Flavell DM, Ireland H, Stephens JW, et al. Peroxisome proliferator-activated receptor α gene variation influences age of onset and progression of Type 2 diabetes. Diabetes. 2005;54:582–586. doi: 10.2337/diabetes.54.2.582. [DOI] [PubMed] [Google Scholar]
- 23.Spertus J, Safley D, Garg M, Jones P, Peterson ED. The influence of race on health status outcomes one year after an acute coronary syndrome. J. Am. Coll. Cardiol. 2005;46:1838–1844. doi: 10.1016/j.jacc.2005.05.092. [DOI] [PubMed] [Google Scholar]
- 24.Alpert JS, Thygesen K, Antman E, Bassand JP. Myocardial infarction redefined — a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J. Am. Coll. Cardiol. 2000;36:959–969. doi: 10.1016/s0735-1097(00)00804-4. [DOI] [PubMed] [Google Scholar]
- 25.Braunwald E. Unstable angina. A classification. Circulation. 1989;80:410–414. doi: 10.1161/01.cir.80.2.410. [DOI] [PubMed] [Google Scholar]
- 26.Antman EM, Cohen M, Bernink PJ, et al. The TIMI risk score for unstable angina/ non-ST elevation MI: A method for prognostication and therapeutic decision making. JAMA. 2000;284:835–842. doi: 10.1001/jama.284.7.835. [DOI] [PubMed] [Google Scholar]
- 27.Marsh S, King CR, Garsa AA, McLeod HL. Pyrosequencing of clinically relevant polymorphisms. Methods Mol. Biol. 2005;311:97–114. doi: 10.1385/1-59259-957-5:097. [DOI] [PubMed] [Google Scholar]
- 28.Finck BN, Lehman JJ, Leone TC, et al. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J. Clin. Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leone TC, Lehman JJ, Finck BN, et al. PGC-1α deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:E101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am. J. Hum. Genet. 2002;70:425–434. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrell FE., Jr . Regression Modeling Strategies. Springer; NY, USA: 2001. [Google Scholar]
- 32.R Development Core Team . A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2005. [Google Scholar]
- 33.Chen S, Tsybouleva N, Ballantyne CM, Gotto AM, Jr, Marian AJ. Effects of PPARα, γ and δ haplotypes on plasma levels of lipids, severity and progression of coronary atherosclerosis and response to statin therapy in the lipoprotein coronary atherosclerosis study. Pharmacogenetics. 2004;14:61–71. doi: 10.1097/00008571-200401000-00007. [DOI] [PubMed] [Google Scholar]
- 34.Flavell DM, Torra I Pineda, Jamshidi Y, et al. Variation in the PPARα gene is associated with altered function in vitro and plasma lipid concentrations in Type II diabetic subjects. Diabetologia. 2000;43:673–680. doi: 10.1007/s001250051357. [DOI] [PubMed] [Google Scholar]
- 35.Tai ES, Demissie S, Cupples LA, et al. Association between the PPARA L162V polymorphism and plasma lipid levels: the Framingham Offspring Study. Arterioscler. Thromb. Vasc. Biol. 2002;22:805–810. doi: 10.1161/01.atv.0000012302.11991.42. [DOI] [PubMed] [Google Scholar]
- 36.Gislason GH, Rasmussen JN, Abildstrom SZ, et al. Long-term compliance with β-blockers, angiotensinconverting enzyme inhibitors, and statins after acute myocardial infarction. Eur. Heart J. 2006;27:1153–1158. doi: 10.1093/eurheartj/ehi705. [DOI] [PubMed] [Google Scholar]
- 101.American Heart Association Heart Disease and Stroke Statistics — 2005 update. www.americanheart.org