

Abstract
This study delineates specific functions of Gαi2 and Gαi3 in T cell mobilization during the development of graft-versus-host disease (GVHD) and reveals reciprocal effects of these two G proteins on the onset and morbidity of the disease. A deletion of Gαi2 hampered trafficking of pathogenic T cells from secondary lymphoid tissues to inflammatory sites and sufficiently prevented GVHD. In contrast, a severer disease was induced in mice adoptively transferred with Gαi3-deficient T cells than those mice transferred with wild-type T cells. In agreement with this, pathogenic Gαi2−/− T cells displayed a defect in response to CXCL10, CXCL11, and CCL5, whereas lack of Gαi3 augmented T effector cell chemotaxis induced by CXCL10 and CXCL11 and resulted in their preference of homing to the liver and colon. Absence of either Gαi also abrogated sphingosince-1-phosphate (S1P)-mediated inhibition of T cell chemokinesis and facilitated T cell homing and expansion in the spleen and mesenteric lymph nodes at the early phase of GVHD development, which is another key determinant in the severity and early onset of the disease in the mice infused with Gαi3−/− T cells. These observations underscore interplay between Gαi2 and Gαi3 and potentially provide a novel strategy to prevent GVHD by blocking T cell homing at early stages and T effector cell trafficking at later time points.
Keywords: Gαi proteins, Graft-versus-host disease, T cell trafficking
Introduction
Graft-versus-host disease (GVHD) results when donor T cells attack immunocompromised hosts, which represents major morbidity of the clinical transplantation of allogeneic bone marrow or peripheral blood stem cells used to treat patients with various lymph-hemopoietic malignancies [1, 2]. The disease occurs in two phases following the transfer of donor lymphocytes [3, 4]. In the first phase, donor T cells enter peripheral lymphoid tissues including the spleen, lymph nodes, and Peyer's patch, a process that depends on homeostatic chemokines like CXCL12, CCL19 and CCL21 and adhesion molecules such as L-selectin and αlβ2 integrin [5, 6]. Upon arrival at the lymphoid tissues, donor Tcells recognize alloantigens expressed on host antigen-presenting cells (APC), become activated, and differentiate into T effector cells. These T effectors up-regulate tissue homing chemokine receptors CCR2, CCR5, and CXCR3 as well as produce inflammatory cytokines like IFN-γ and TNF-α. The inflammatory cytokines enter the circulation and induce the production of chemokines in target organs by host cells. Following this initial phase of chemokine production, a second wave of chemokine production is induced by recruited donor T cells in target organs. The recruited T cells produce the CCR5 ligands CCL3, CCL4, and CCL5 that sustain these cells in the target organs and ultimately cause damage to the infiltrated organs [7, 8].
In both these phases, chemokines act as one of the key players in the control of migration of donor T cells in and out of lymphoid tissues, and to inflammatory sites [7, 9]. These chemokine receptors activate heterotrimeric G proteins that are composed of α, β, and γ subunits. The GTP-binding proteins are classified into four major families, i.e. Gαi/o, Gαq, Ga12/13, and Gαs [10, 11]. In the Gαi/o family, some Gαi proteins can be irreversibly uncoupled from their receptors by pertussis toxin (PTX), an inhibitor that is widely used to link Gαi proteins to the signaling implicated in lymphocyte trafficking [12-15]. The primary PTX substrates in T lymphocytes are Gαi2 and Gαi3 as shown by in vitro PTX labeling and cDNA cloning [16, 17]. Our study has demonstrated that these two G proteins can be functionally additive, distinct, overlapping or antagonistic in chemotactic responses in vitro in a receptor-dependent fashion [18]. For instance, Gαi3 inhibited Gαi2 in CXCR3-mediated signaling by interacting with the receptor without activation and dissociation, with which it prevented Gαi2 interaction with the receptor [18]. Distinguished from the CXCR3 receptor, the CXCR4 receptor depended on both Gαi2 and Gαi3 for a full migratory response, while the CCR7 receptor required only one of the two Gαi proteins [18].
To extend our in vitro study to in vivo, we show here that GVHD is abolished in immunocompromised mice adoptively transferred with Gαi2−/− T cells. Impairment of T effector cell trafficking to susceptible organs is responsible, at least in part, for a lack of the disease in the mice. Conversely, the disease is significantly exacerbated in recipients of Gαi3-deficient T cells compared with those of WT T cells. Without Gαi3, pathogenic T cells traveled to target organs much more efficiently than without Gαi2, presumably in association with altered migratory responses to CXCL10, CXCL11 and CCL5 in the presence of Gαi2 or Gαi3 alone. In addition, absence of either Gαi protein increased T cell chemokinesis mediated by sphingosince-1-phosphate (S1P), but was without effects on its chemotactic responses. The increased chemokinesis facilitated accumulation and expansion of donor Gαi3−/− T cells in the secondary lymphoid tissues, critically contributing to the severity and early death of the recipient. The opposite effects of Gαi2 and Gαi3 on the onset of GVHD argue strongly coordinated regulation of chemokine receptors' function by Gαi2 and Gαi3 to ensure migration of a T cell at a right time and in a right place in vivo.
Results
Reciprocal effects of Gαi2 and Gαi3 on acute GVHD
To assess potential differences of Gαi2- and Gαi3-deficient Tcells in their ability to induce acute GVHD, T cells isolated from full-MHC-mismatched allogeneic (H-2b) WT, Gαi2-KO and Gαi3-KO mice were intraperitoneally administrated into severe combined immunodeficient (SCID) BALB/c mice (H-2d). As can be seen in Fig. 1A, all recipients gained weight in the first week post transfusion, which continued to be the case for another week in the mice receiving either WT or Gαi2-KO T cells, with the latter gaining more than the former. WT T cell-infused mice began developing signs of GVHD at week 3, including hunch, sluggishness, ruffled fur, and a continuous decrease in body weight over time (Fig. 1A). Some mice started dying at week 5, and about 40% of mice died in 8 weeks or reached a terminally ill stage (body weight loss >15%) and had to be sacrificed in accordance with institutional guidelines (Fig. 1B). Strikingly, a body weight loss was not observed in recipients of Gαi2-KO T cells. Instead, these animals gained some weight over the course of the 8-week observation and did not develop any sign of the disease up to 8 months when the mice were sacrificed (Fig. 1A and B). Conversely, mice receiving Gαi3-KO T cells developed GVHD in 2 weeks after transfusion. The disease was progressing much more rapidly than the mice receiving WT Tcells, forcing 85% the mice to be sacrificed in 7 weeks. The death rate was two or eight times higher than that of the mice receiving WT Tcells or Gαi2−/− Tcells, respectively (Fig. 1A and B).
Figure 1.
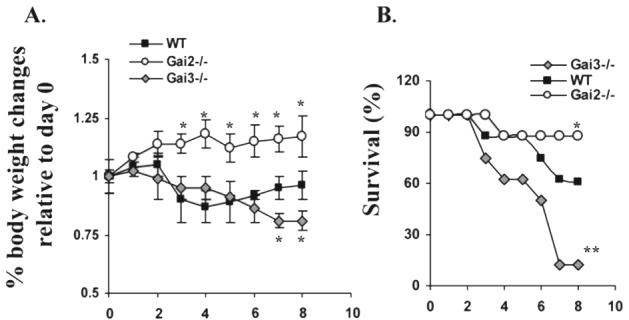
Reciprocal effects of Gαi2 and Gαi3 on GVHD development. SCID BALB/c mice were each administrated intraperitoneally with 107 T cells isolated from WT, Gαi2−/−, and Gαi3−/− mice on 129Sv/C57BL/6 background. Animals were monitored for weight (A) and survival (B) weekly. Note: significant acceleration of GVHD-associated morbidity in recipient of Gαi3−/− T cells (**p<0.01) and reduction of the disease in recipient of Gαi2−/− T cells (*p<0.05) as compared to mice receiving WT T cells. Results are from three (Gαi3−/− T cells) or four (Gαi2−/− T cells) independent experiments with 7∼12 mice per group in each experiment. The total number of mice was 31, 28 and 30 receiving WT, Gαi2−/− and Gαi3−/− T cells, respectively. Mean ± SD of weight changes relative to day 0 is shown in (A) (*p<0.05).
Histological examination revealed that a large number of T cells accumulated in the spleen and lymph nodes in the SCID mice transferred with either Gαi2−/− or Gαi3−/− T cells, concomitant with 1∼2 times enlargement of the spleen and a 2∼6-fold increase in the size of the lymph nodes, when compared with those mice receiving WT T cells (data not shown). Yet, little inflammation and lymphocyte infiltration were observed in the intestine, colon, lung, and liver in recipients of Gαi2 KO T cells (Fig. 2A and B; and data not shown), consistent with no GVHD development in the mice. In contrast, severe inflammation and lymphocyte infiltration were evident in the liver and colon in the mice infused with Gαi3-deficient T cells (Fig. 2A and B). As can be seen in Fig. 2A (v and vi), almost all portal areas were expanded with extensive mononuclear chronic inflammatory cell infiltrates in the liver. On higher resolution, some of the bile ducts were surrounded by infiltrated lymphocytes, causing bile duct damage and endothelitis (Fig. 2A, vii). By comparison, the inflammation was much less in the recipients of WT T cells, involving only some of the portal areas with focal duct damage (Fig. 2A, i and ii). The liver of mice receiving Gαi2-deficient T cells showed minimal chronic inflammation in some of the portal areas with no significant bile duct damage (Fig. 2A, iii and iv). In the colon, focal crypt epithelial cell degeneration (apoptosis) and crypt abscesses, typical pathological changes for acute GVHD, could be easily identified in the recipients of Gαi3−/−T mice and to a far lesser degree in the mice adoptively transferred with WT T cells, but was hardly seen in the mice receiving Gαi2−/− T cells (Fig. 2B). The pathological examination confirmed that the liver and colon lesions caused the early death of the animals reconstituted with Gαi3−/− T cells.
Figure 2.
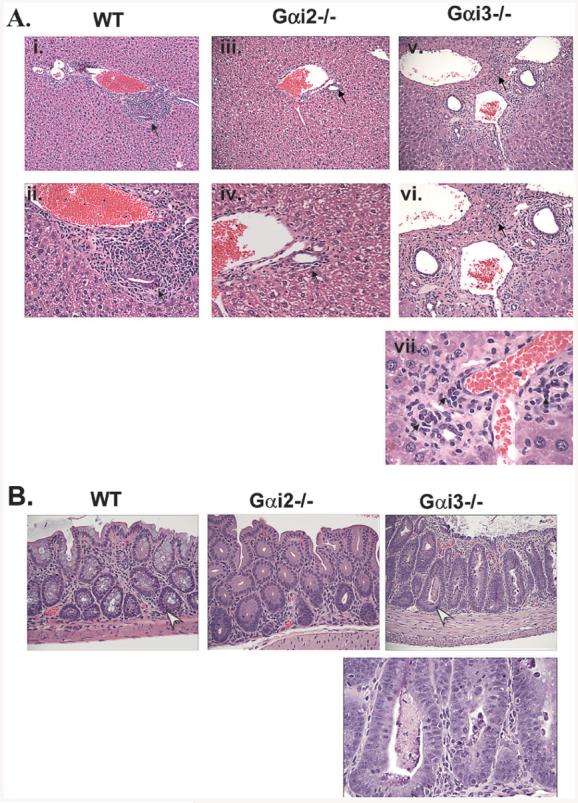
Histological examination of diseased liver and colons. (A) Shown are representative H & E-stained sections of the liver from mice reconstituted with WT T cells (i and ii) for 35 days, Gαi2−/− T cells (iii and iv) for 35 days or Gαi3−/− T cells (v, vi, and vii) for 21 days described in Fig. 1. Arrows indicate bile duct damage. The panels ii, iv, and vi (200X) and vii (600X) are the enlargement of i, iii, or v (100X), respectively. (B) Representative H&E-staining of colonic mucosa of the mice described in (A). Arrows indicate crypt abscesses and apoptosis in Gαi3−/− T cell-infused mice or apoptosis in mice adoptively transferred with WT T cells. The magnifications are 200X for the top three sections and 600X for the enlarged section of the colon from Gαi3−/− T cell-infused mice in the lower panel.
Altered donor T cell distribution
We next addressed whether the striking difference in the severity and onset of GVHD in the absence of Gαi2 or Gαi3 was attributed to altered T cell migration. The amount of donor T cells including CD4+ and CD8+ cells in various tissues was estimated in the recipients at varying times after cell transfer, as this model involves both CD4+ and CD8+ T cell immune responses [7]. One day after the transfer, donor Gαi3−/− T cells were most recovered from mesenteric lymph nodes, the spleen and blood among the three groups of mice (Fig. 3, the top left panel). Gαi2−/− cells were found at significantly greater quantities than WT T cells in mesenteric lymph nodes on day 1 as well (Fig. 3). The relatively high levels of Gαi3−/− Tcells and to a much lesser degree, of Gαi2−/− Tcells in the secondary lymphoid tissues were attributed to increased homing and/or sequestration of T cells in these tissues, because no cell division occurred yet at this time point. Moreover, more WT T cells than either Gαi2−/− or Gαi3−/− donor T cells were recovered from the peritoneal lavage in 6 and 12 h after cell transfer, which were inversely correlated to the number of corresponding T cells accumulated in the second lymphoid tissues (Fig. 3, the top right panel). Donor T cells in other tissues including colon, intestine, liver, and lung were hardly detected at this time point.
Figure 3.
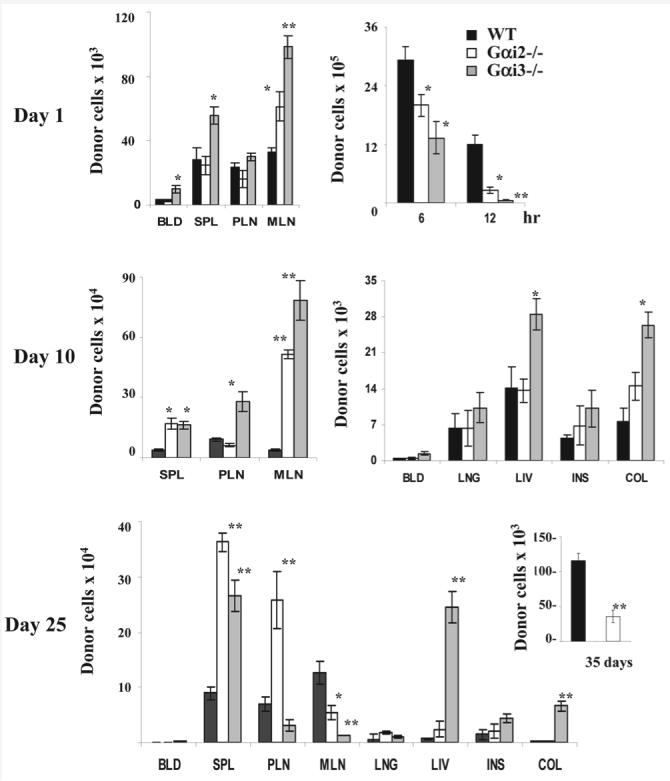
Effects of Gαi2 and Gαi3 on donor T cell tissue distribution at indicated times after adoptive transfer. T cells isolated from WT, Gαi2−/− or Gαi3−/− mice were labeled with CFSE and injected into SCID BALB/c mice at a dose of 107 T cells/mouse. The total numbers of donor T cells in different tissues were obtained on the basis of percentages of CD3+ cells at days 1 (top), 10 (middle), 25 (low), and 35 (inset in the lower panel). Donor T cells in the cavity lavage were also shown after 6 and 12 h injection (top right panel). BLD, blood; SPL, spleen; PLN, peripheral lymph nodes; MLN, mesenteric lymph nodes; LNG, lung; LIV, liver; INS, intestine; and COL, colon. Results are the mean ± SD of three independent experiments each with three mice per time point (n=9 at each time point). *p<0.05 or **p<0.01 as analyzed by a Wilcoxon signed rank test.
Gαi3−/− T cells continuously accumulated at the highest levels at day 10 among all three groups of mice in the liver and colon, and in particular, in peripheral and mesenteric lymph nodes where a 3- or 19-fold increase was seen, respectively, as compared to WT T cells (Fig. 3, middle). This drastic increase in the number of Gαi3−/− T cells may reflect both increased T cell homing and more robust proliferation of Gαi3−/− T cells. About 90% Gαi3-deficient T cells in the spleen, the largest T cell pool, had undergone at least six cycles of cell division, indicative of a more than sixfold decrease in CFSE fluorescent intensity (Fig. 4A). However, only 70% of WT T cells had done so (Fig. 3, top panel). This difference was not due to altered potential of T cell proliferation in absence of Gαi3, because when Gαi3−/− and WT T cells were stimulated with irradiated splenocytes prepared from CB-17 SCID mice, no difference or a slight decrease in T cell proliferation was observed in the absence compared to presence of Gαi3 (data not shown), similar to early investigation [19, 20]. Presumably, increased homing of Gαi3−/− T cells to lymphoid tissues may accelerate their proliferation as a consequence of a relatively high ratio of T cells to APC in lymphoid tissues. As with Gαi2−/− T cells, we did observe a slight increase in their proliferation relatively to WT T cells when the cells were stimulated with irradiated splenocytes prepared from CB-17 SCID mice (data not shown), consistent also with earlier investigation [20, 21]. Hence, the slight increase (∼10%) in proliferation, in line with enhanced lymphoid homing, may additively contribute to the increased number of Gαi2−/− T cells in the secondary lymphoid tissues (Fig. 3 and 4A). Notably, majority of T cells presented in non-lymphoid tissues such as the liver, lung, colon, and intestine were CFSE-dim at day 10 (Fig. 4A). This was also the case at day 25 (data not shown), confirming that T cells trafficked to their destination only after fully activated [3, 7, 22].
Figure 4.

Differential proliferation of donor T cells in the presence or absence of a Gαi protein. Mice reconstituted with CFSE-labeled T cells prepared from indicated mice as described in Fig. 3 were sacrificed at days 10 (A) or 25 (B). CFSE fluorescence intensity was analyzed by flow cytometry on gated CD3+ T cells in different tissues. The numbers within each histogram indicate percentages of CFSE-bright and CFSE-dim cell populations, respectively. One representative result of nine mice with similar results is shown for each tissue. SPL, spleen; PLN, peripheral lymph nodes; MLN, mesenteric lymph nodes; LNG, lung; LIV, liver; INS, intestine; and COL, colon.
As activated T cells were continuously traveling from the lymphoid tissues to target organs over time, the amount of T cells at day 25 declined dramatically in lymphoid tissues from day 10, concurrent with elevated levels of these cells in target organs, especially in the liver and colon (Fig. 3, the lower panel). Gαi3−/− T cells recovered in the liver and colon were 32 or 36 times that of WT T cells, respectively, which is likely to account for the early mortality of the mice. Remarkably, in spite of the relatively high number and full activation, Gαi2−/− T cells in the spleen and peripheral lymph nodes, but not those in the mesenteric lymph nodes were refractory to leave for the target organs, resulting in their massive accumulation in lymphoid tissues, and reduced presence in the target organs, in agreement with lack of GVHD development in the mice. Unlike Gαi-deficient T cells, WT T cells rose slowly but steadily in number at day 25 from those attained at day 10 in the lymphoid tissues. Significant proportions (∼26%) of WT T cells in these mice appeared still proliferating in the peripheral lymph nodes (Fig. 4B). The delayed proliferation and a relatively slow increase in the number of T cells in the secondary lymphoid tissues were consistent with the late onset of GVHD in the mice compared to those mice harboring Gαi3−/− T cells (Fig. 1). Accumulation of activated or fully divided T cells to the liver was not apparent until day 35 in the recipients of WT T cells, and ultimately exceeded that of Gαi2−/− T cells (Fig. 3, inset in the low panel). The results suggest that trafficking of pathogenic Tcells out of the lymphoid tissues to the target organs is the key to the disease development. However, the levels of initial T cell homing to the secondary lymphoid tissues determined the severity and onset time of the disease.
To directly compare migratory capacity of T effector cells towards susceptible organs like the liver and colon in the presence or absence of Gαi2 or Gαi3, competitive homing assays were carried out. Briefly, pathogenic Gαi2- and Gαi3-deficient T cells and WT control T cells were isolated from SCID mice that had received these cells 3 weeks ago. The isolated Gαi2- or Gαi3-deficent T cells were labeled with a red fluorescent dye TRITC, mixed with an equal number of CFSE-labeled WT T cells, or vice versa, and i.v. administrated into early GVHD-diseased hosts induced by WT T cells. After 1 or 18 h, trafficking of Gαi2- or Gαi3-deficient T cells to inflammatory organs relative to WT T cells was compared in the same animal by homing index. As shown in Fig. 5A, the homing index of Gαi2-deficient T cells was significantly greater in circulation than that of WT or Gαi3-deficient T cells one hour after T cell administration. This, in keeping with reduced homing indices of Gαi2-deficient T cells in bone marrow and liver, suggests that lack of Gαi2 hampered T cell homing to these tissues. We were unable to detect fluorescently labeled T cells in the colon samples at this time point. In contrast to Gαi2−/− T cells, homing index of circulating Gαi3−/− T cells was smaller than that of WT T cells, concomitant with increased distribution of Gαi3-deficient T cells in the spleen and liver, implicating reciprocal roles of these two G proteins in the control of T effector cell trafficking in the tissues. While adoptively transferred T effector cells were continuously migrating to inflammatory tissues, the number of Gαi3−/− T cells in the liver and colon accumulated to a significantly higher level than that of WT or Gαi2−/− T cells at 18 h after T cell transfer (Fig. 5B). The result clearly suggests preference of Gαi3-KO T effectors homing to the liver and colon over WT or Gαi2-KO counterparts.
Figure 5.

Enhanced trafficking of Gαi3-deficient T effectors to inflammatory sites in early GVHD hosts. Gαi2- or Gαi3-KO T cells and WT T cells activated by MHC-mismatched hosts were isolated, stained with a fluorescent dye TRITC or CFSE, respectively, mixed at a 1:1 ratio, and injected into SCID mice that had been primed by WT T cells for three weeks as detailed in the Materials and methods. Distribution of Gαi2- or Gαi3-KO T cells relative to WT T cells in different tissues was analyzed at 1 or 18 h after T cell transfer. Homing efficiency in the presence or absence of a specific Gαi protein is expressed as homing indices where homing index for WT T cells is normalized to 1 (dash line). Data shown are the means ± SD for seven mice at each time point in two separate experiments. BM, bone marrow; PLN, peripheral lymph nodes; MLN, mesenteric lymph nodes; *p<0.05, **p<0.01 compared to WT control T cells.
Abrogation of S1P-mediated inhibition of T cell homing to lymphoid tissues in absence of Gαi
Differential distribution of naive donor T cells in lymphoid tissues at day 1 after transfer in the absence of either Gαi suggests indispensable roles for these two G proteins in spatial and temporal regulation of homing and/or sequestration of naive T cells to lymphoid tissues, consistent with well documented roles of homeostatic chemoattractants CCL21, CCL19, and CXCL12 (SDF-1) in lymphoid tissue homing in a PTX-sensitive manner [23]. Although our previous investigation demonstrated comparable chemotaxis towards an increasing concentration gradient of CCL21 or CC19 in the presence or absence of Gαi2 or Gαi3 (Fig. 6C), we found that a deletion of Gαi2 or Gαi3 significantly impaired chemotactic responses to SDF-1 (Fig. 6B) [18]. The impaired migration was however contradicting increased numbers of donor Gαi2- and Gαi3-deficient T cells in lymphoid tissues on day 1 post T cell transfer (Fig. 3).
Figure 6.

Chemotactic responses of non-stimulated T cells in the presence or absence of a Gαi protein. (A) T cells freshly isolated from WT, Gαi2−/− and Gαi3−/− mice were added to the upper chamber and S1P at the indicated concentrations was added to the lower chamber. After 4-h incubation, the migrated cells in the lower chamber were counted and expressed as mean chemotactic indexes ± SD of cell migration toward S1P relative to control medium as defined in the Materials and methods. (B and C) T cells isolated from indicated mice, along with 500 nM S1P, were added to the upper chamber and varying concentrations of CXCL12 (B) and CCL21 (C) were added to the lower chamber. Migration was assayed as in (A) and data are presented as mean chemotactic indexes ± SD of cell migration toward CXCL12 or CCL21 relative to control medium. Cumulative data from at least six (A) or three (B and C) experiments with each in triplicate are shown. Statistic significance (*p<0.05 or **p<0.01) in the presence vs. absence of a specific Gαi protein.
T cell egress from the thymus and secondary lymphoid tissues is primarily regulated by the S1P1 receptor that is coupled exclusively to PTX-sensitive proteins [24, 25]. Our data showed that absence of either Gαi2 or Gαi3 weakened the chemotactic response over the S1P concentrations tested, but the effect was lacking statistic significance when compared to that of WT T cells (Fig. 6A), implicating that lack of either Gαi2 or Gαi3 is unlikely to increase their sequestration in the secondary lymphoid tissues. The nearly normal migratory response of Gαi2-KO and Gαi3-KO T cells explains no gross defect in thymocyte and T cell egress in these two KO mouse strains [19, 26].
Early studies have suggested that high plasma concentrations of S1P (0.3∼3 μM) may play a role in limiting T cell homing evoked by homeostatic chemokines SDF-1, CCL21 and CCL19 [27]. Presumably, disruption of this limitation could facilitate T cell migration toward the secondary lymphoid tissues. To test this possibility, a modified migration assay was employed in which a high concentration of S1P (500 nM) was added to the upper chamber, along with WT, Gαi2−/−, or Gαi3−/− T cells, and varying concentrations of CXCL12, CCL19 and CCL21 were added to the lower chamber. As shown in Fig. 6B, addition of S1P to the upper chamber suppressed chemokinesis (random walk) significantly in WT T cells, resulting in a decrease in the number of the cells recovered in the lower chamber compared to medium controls, as shown by a negative chemotactic index. Yet, deletion of either Gαi2 or Gαi3 rendered T cells refractory to S1P-induced inhibition of chemokinesis. Moreover, S1P failed to inhibit SDF-1-induced chemotactic responses in the absence of either Gαi2 or Gαi3, while it significantly suppressed WT T cell migration stimulated under similar conditions. Beyond that, SDF-1-induced chemotaxis was increased significantly in the presence relative to absence of S1P in Gαi3-deficient T cells (Fig. 6B). S1P was also ineffective in suppression of chemotaxis in these cells when stimulated with CCL21 (Fig. 6C) or CCL19 (data not shown). Taken together, abrogation of S1P-mediated inhibition in T cell homing to lymphoid tissues may contribute to the increased numbers of Gαi2-KO and Gαi3-KO T cells in these tissues (Fig. 3).
Distinct functions of Gαi2 and Gαi3 in regulation of T effector cell migration
Despite significant increases in the number of Gαi2-KO or Gαi3-KO Tcells in the lymphoid tissues relative to that of WT Tcells, a severe GVHD was seen only in the recipients of Gαi3-KO T cells, not in those of Gαi2-KO T cells. This, in line with preferred travel of Gαi3-KO T effector cells to the liver and colon over Gαi2-KO and WT T effectors, argues strongly that sufficient migration of activated T cells to inflammatory sites is the key to an onset of the disease. It is thus imperative to us to look into effects of Gαi2 and Gαi3 on chemotactic responses of activated T cells toward several chemokines that are well documented in the onset of GVHD. Our previous studies demonstrated that a lack of Gαi2 abolished CXCR3-mediated migration of ConA-activated T cells, whereas deletion of Gαi3 enhanced the migration [18]. This also held the truth for alloreactive T cells. Lack of Gαi3 significantly augmented T cell migration induced by CXCL10 or CXCL11, whereas absence of Gαi2 completely abolished T cell migration towards these two chemokines (Fig. 7A and B). Thus, Gαi3 antagonizing Gαi2 function in CXCR3-mediated signaling would account, at least in part, for the reciprocal effects of Gαi2 and Gαi3 on GVHD development, as the CXCR3 receptor is one of the key chemokine receptors in directing migration of activated T cells to inflammatory cites [28, 29].
Figure 7.
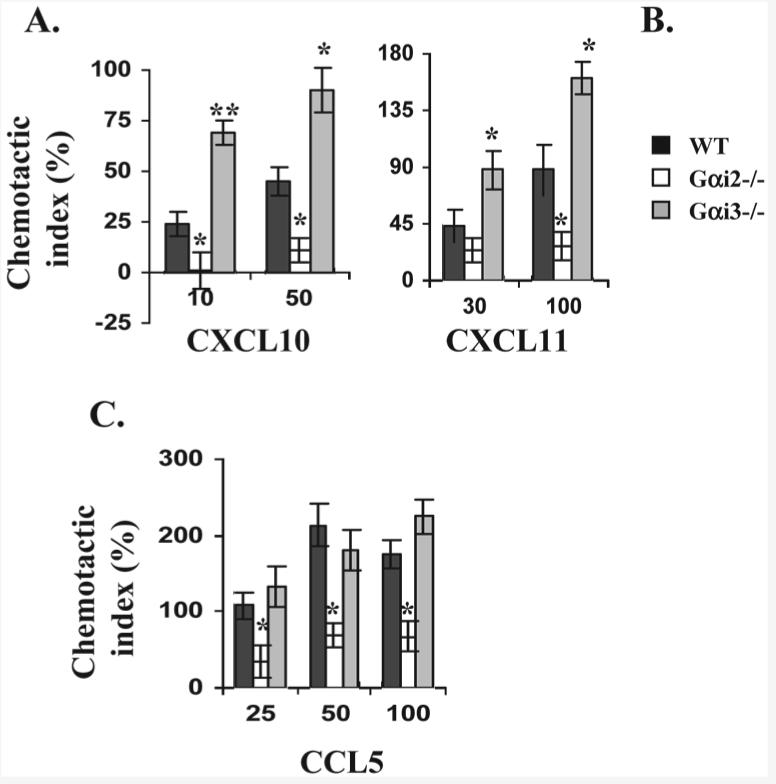
Chemotactic responses of activated T cells in the presence vs. absence of Gαi2 or Gαi3. T cells isolated from the indicated mice were either stimulated for 6 days with irradiated splenocytes isolated from SCID BALB/c mice (A and B) or stimulated with ConA for 4 days (C). Chemotaxis of the resultant cells was assayed as in Fig. 6A. Data are presented as mean chemotactic indexes ± SD of cell migration toward CXCL10 (A), CXCL11 (B), and CCL5 (C) relative to control medium. Cumulative data from at least six (A) or three (B and C) experiments with each in triplicate are shown. Statistic significance (*p<0.05 or **p<0.01) in the presence vs. absence of a specific Gαi protein.
The chemotactic response to CXCL9 was too low to detect in these alloreactive T cells. In general, the effects of Gαi2 or Gαi3 on T cell migration stimulated by various chemokines are receptor-specific, irrespective of whether the cells were activated by ConA or by irradiated splenocytes prepared from CB-17 SCID mice, except that the overall responses were smaller in the latter than the former. Hence, chemotaxis in ConA-stimulated T cells was shown for chemokines inducing a relatively low response like CCL5 (Fig. 7C), although both ConA-stimulated and alloreactive T cells were used. Unlike CXCR3 agonists, CCL5, another important chemokine involved in T cell recruitment to the liver [30], induced comparable chemotaxis in both Gαi3−/− and WT T cells at all concentrations tested (Fig. 7C). However, absence of Gαi2 impaired CCL5-stimulated chemotaxis significantly, indicating that CCL5 depends on Gαi2 for conveying a full migratory signal (Fig. 7C). This, in line with the defect in CXCR3-mediated signaling, may be the reason behind insufficient migration of Gαi2−/− T cells to the liver (Fig. 3).
The varying chemotactic responses observed in the presence vs. absence of a Gαi protein were not ascribed to expression levels of the specific receptors (data not shown). The CXCR3 receptor was expressed similarly on alloreactive T cells in the presence or absence of Gαi2 or Gαi3, resembling what was described in ConA-stimulated T cells [18]. The CCR5 receptor was also indistinguishable in these three groups of T cells. Our flow cytometric analysis data also revealed unaltered levels of the CCR7 receptor, but decreased expression of the CXCR4 receptor on T cells in the absence compared to presence of Gαi2 or Gαi3 [18]. Moreover, these cells expressed Gαi2 or Gαi3 at levels similar to those WT Tcells as previously described (data not shown), ruling out that the differential chemotactic responses are attributed to altered expression of Gαi2, Gαi3 or a specific receptor.
Discussion
Our previous studies have demonstrated that Gαi3 antagonizes Gαi2 in CXCR3-mediated signaling by interacting with the receptor without activation and dissociation [18]. This translated into accelerated migration of T cells stimulated with all three CXCR3 agonists in the absence of Gαi3. In contrast, deletion of Gαi2 ablated T cell migration towards these agonists [18]. A severer GVHD in immunocompromised mice adoptively transferred with Gαi3−/− T cells than those mice transferred with WT T cells, but lacking such a disease in mice receiving Gαi2−/− T cells, argues strongly that Gαi3 inhibits Gαi2-mediated signaling in vivo and is critical in directing activated Tcells from lymphoid tissues to target organs. Our data showed that activated Gαi3−/− T cells were capable of migrating to target organs more efficiently than Gαi2−/− Tcells, which contributes, at least in part, to earlier mortality of the recipients, whereas activated Gαi2−/− T cells were inadequately trapped in the lymphoid tissues, unable to elicit GVHD. Apart from reciprocal regulation of pathogenic T cell trafficking, lack of either Gαi3 or Gαi2 resulted in more efficient T cell homing to lymphoid tissues than their presence, suggesting similarity of Gαi2 and Gαi3 in directing trafficking of naive T cells. These observations provide compelling evidence that T cell ingress and egress of various tissues at different activation stages are coordinately controlled by a specific array of chemokines and receptors as well as different heterotrimeric G proteins at multiple levels. Gαi2- and Gαi3-KO mice provide a unique system to address the effects of multiple chemokine receptors on spatial and temporal regulation of T cell trafficking in vivo during the disease development.
A body of evidence has shown increased production of chemokines such as CXCL9, CXCL10, CXCL11, CCL3, CCL4, CCL5, MIP-1α and β, MCP-1 and MCP-2, and so on, at varying stages during GVHD development [7, 22]. This study details the effects of Gαi2 and Gαi3 on the activities of the corresponding receptors and addresses how they are involved in eliciting GVHD at varying stages. Among them, the chemokine receptor CXCR3 is a key receptor in guiding donor T cells to inflammatory areas and T cells lacking CXCR3 fail to elicit GVHD or reject allograft [28, 29]. Our current study extends these observations by showing that more efficient migration of activated T cells exacerbates GVHD in the absence of Gαi3, concomitant with enhanced activity of the CXCR3 receptor. On the other hand, abrogation of CXCR3 signaling by Gαi2 deletion hampers T effector cell migration, preventing the disease. The lack of GVHD in mice receiving Gαi2−/− T cells may be also ascribed partially to aberrant chemotaxis of these T cells to CCL5, an inflammatory chemokine being essential in attracting and sustaining T effector cells to the liver [31]. We detected little migratory responses over background even in ConA-activated T cells when stimulated with chemokines MIP-1β (CCL4), MIP-1α (CCL3), MCP-1 and MCP-2 at concentrations ranging from 10 to 200 ng/mL (data not shown) [7]. The role of these chemokines in the reciprocal effects of Gαi2- or Gαi3-deficency on the onset of GVHD is not known at present. In addition, although lack of Gαi2 or Gαi3 has little impact on differentiation of T regulatory cells and their function (our unpublished data) [26], its influence over infiltration of T regulatory cells versus effector cells may differ, which can also contribute to the described disparity of the disease induced by Gαi2- and Gαi3-KO T cells, a possibility that requires further investigation.
Our study stresses that migration of not only Tcell effectors but also naive T cells is critically involved in the severity and mortality of GVHD. Increasing numbers of donor Gαi3-deficient T cells and to a lesser extent, Gαi2-deficient T cells were observed in the secondary lymphoid tissues at one day after transfusion, in spite of impeded CXCR4-mediated signaling in these cells. Increased T cell homing to lymphoid tissues probably results from a loss of S1P-mediated inhibition in T cell chemokinesis that facilitates T cell homing elicited by the CCR7 and CXCR4 receptors, with preference of Gαi3−/− T cells to Gαi2−/− T cells (Fig. 5). S1P is present at 0.1∼1 μM in blood and body fluids and at a decreased level (10∼100 nM) in the lymphoid tissues. This elevating S1P concentration gradient directs T cell egress from the lymphoid tissues to blood. Distinguished from egress, the major effect of plasma S1P concentrations on Tcells is to retain them in circulation by dampening their chemotactic responses to various chemokines [27]. Reduced chemokinesis may be the underlying mechanism whereby a high concentration of S1P dampens chemotaxis induced by CXCR4 and CCR7 ligands. This possibility is supported by the ability of high levels of S1P to inhibit chemotaxis induced by a broad range of chemokines [27]. Moreover, deletion of either Gαi2 or Gαi3 abolished S1P-mediated suppression in T cell chemokinesis, which was accompanied by increasing Tcell homing mediated by CCR7- and CXCR4 ligands. On the contrary, increased circulating T cells owing to accelerated egress and diminished homing was observed in S1P1 receptor-transgenic mice that over-express the S1P1 receptor in T cells [32]. These mice are relatively insensitive to T cell-mediated delayed-type hypersensitivity (DTH) responses as compared to that in WT mice.
Despite importance of Gαi2 and Gαi3 in directing of lymphocyte trafficking, gene target deletion of either Gαi2 or Gαi3 gives rise to no gross defect in T cell homing or egress [19, 26]. Gαi2-deficient mice exhibit impairment in lymph node development in several anatomic locations and in Peyer's patch formation [19, 33]. The mice also develop inflammatory bowel disease, due at least in part to a lack of Tcell response toTGF-β that results in a Th1-skewed hyperimmune response in the colon [19, 21]. One unanswered question was that inflammation was not observed in all tissues in the mice, with the notable exception of the colon, although T cells in Gαi2−/− mice were unresponsive to TGF-β, apparently inconsistent with the systemic inflammation developed in the mice lacking TGF-β or a TGF-β receptor [34, 35]. Our current study suggests that failure of T cells to reach target organs can make the impaired TGF-β response futile. Indeed, trafficking of pathogenic T cells to the colon seems far less reliant on Gαi2, because of comparable homing of Gαi2-deficeint and WT T cells to the colon.
Ample studies addressing the role of chemokines in GVHD using mice with gene-targeted deletion of individual chemokine receptors or ligands suggest significant redundancy in the chemokine system. In this regard, multiple chemokines can bind to a particular receptor or conversely, multiple receptors can interact with a particular chemokine. The interplay between Gαi2 and Gαi3 proteins introduces another layer of complexity in the control of cell migration velocity, in addition to the directionality that chemokines and their receptors provide [9]. Gαi2- and Gαi3-KO mouse strains allow us insight into the distinct, overlapping, antagonistic, and additive effects of Gαi2 and Gαi3 on the function of multiple chemokine receptors in vivo, providing more comprehensive description of cell mobilization during inflammation and immune responses as compared to mice lacking individual chemokines or their receptors.
Materials and methods
Animals
Female CB-17 SCID mice (H-2d) were purchased from Taconic Farms (Germantown, NJ) and used as recipients at the age of 5∼6 weeks. These SCID mice do not have Tand B lymphocytes and are commonly used for inducing acute GVHD. Gαi2-KO, Gαi3-KO, and WT control mice on the mixed 129Sv/C57BL/6 background were generated by gene targeting and backcrossed with C57BL/6 (B6) mice for at least five generations as described [19, 36]. Both healthy female and male donor mice were used at 4–6 weeks of age unless otherwise indicated. The mice were housed in conventional cages at the animal facilities of the Massachusetts General Hospital in accordance with institutional guidelines.
Reagents and antibodies
A green fluorochrome CFSE was purchased from Molecular Probes (Eugene, OR) and BioMag goat anti-rat IgG magnetic beads used to purify T cells were from Polysciences (Warrington, PA). The mAb used in flow cytometric analysis were of rat origin and directed at mouse CD3 (PE), CXCR4 (PE), and CXCR3 (Alexa Fluor 488) all from PharMingen (San Jose, CA). PE-conjugated rat anti-mouse CCR7 (PE), CCR5 (PE) or CCR9 (PE) Abs were obtained from BioLegend (San Diego, CA) or R&D systems (Minneapolis, MN), respectively.
Induction of GVHD
To prepare donor T cells, single-cell suspensions prepared from lymph nodes (LN) and spleens of WT, Gαi2-KO and Gαi3-KO mice were treated with a cocktail of rat anti-mouse mAb against CD19, CD32, and CD16 followed by three consecutive depletions of Ab-bound cells with BioMag goat anti-rat IgG per the manufacturer's instruction. The resulting T cells were intraperitoneally administrated into CB-17 SCID mice at a dose of 1 × 107 T cells per mouse. The recipient mice were weighed and observed twice a week for signs of acute GVHD such as hunching, reduced mobility, mottled fur texture, and/or a loss of skin integrity.
Migration assays
Freshly isolated T cells were directly assayed for migratory responses towards increasing concentration gradients of indicated chemokines. For the study of migratory responses in activated T cells, T cells were activated either with 2.0 μg/mL Con A for 3∼6 days as described [18] or by coculturing with irradiated splenocytes prepared from CB-17 SCID mice for 5∼7 days in a complete RPMI medium (10% FBS, 2 mM L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, and 50 μM β-mercaptoethanol). The cells to be tested were added to the upper chamber and chemokines at indicated concentrations were added in triplicate to the lower chamber in Aim-V medium (Gibco) in a 48-well micro-chemotaxis chamber (Neuroprobe, Bethesda, MD). After 4∼5 h migration at 37°C with 5% CO2, the cells in the lower chambers were harvested and counted in a Coulter Z1 cell counter (Beckman Coulter, CA) and compared with or without chemokine additions using a chemotactic index (CI). CI was calculated by the formula: CI = (number of cells migrating to a chemokine) – (number of cells migrating to the medium control)/number of cells migrating to a medium × 100%.
Analysis of donor T cells in the tissues of recipient mice
Purified donor T cells were labeled with CFSE at a final concentration of 10 μM in RPMI supplemented with 10% FBS (complete medium) per the manufacturer's instructions. CB-17 SCID recipient mice were intraperitoneally administrated each with 1 × 107 T cells and the date was recorded as day 0. Recipient mice were sacrificed on days 1, 10, 25, and 35 post donor cell transfer. Blood, liver, lung, intestine, colon, spleen, mesenteric lymph nodes (MLN) and peripheral lymph nodes (PLN, including inguinal and axillary lymph nodes) were procured from the recipient mice. Single-cell suspensions were prepared by mincing the tissues against a 40-μm cell strainer (BD Bioscience, Bedford, MA). The resulting cells were collected, counted and washed twice. Aliquots of 106 cells were stained with PE-conjugated anti-CD3 Ab and analyzed by a FACSCalibur (BD Bioscience) cytometer equipped with Cellquest software (San Jose, CA). CFSE fluorescence intensity was analyzed on gated donor T (CD3+) cells.
Competitive homing experiments
To compare homing efficiency of pathogenic T cells in the absence of either Gαi2 or Gαi3, T cells prepared from Gαi2-KO, Gαi3-KO and age-matched WT mice were injected into CB-17 SCID mice as above. Donor Gαi-deficient T cells and WT control T cells were isolated from corresponding recipient mice three weeks later. Then, Gαi2- or Gαi3-deficient T cells were labeled with 10 μM red fluorescent dye tetramethylrhodamine isothiocyanate (TRITC) for 10 min in complete medium at 37°C with 5% CO2, and WT control T cells were labeled at room temperature with 10 μM green dye CFSE as above or vice versa [37]. Equal amounts of different dye-labeled cells were mixed and intravenously administrated, at a dose of 1 × 107 cells per mouse, into CB-17-SCID mice that had received 1 × 107 WT T cells 3 weeks ago to induce inflammation in the target tissues. The mice were sacrificed 1 or 18 h after T cell transfer and single-cell suspensions of recipient tissues were analyzed by flow cytometry. Homing efficiency of Gαi2- and Gαi3-KO T cells relative to WT T cells was calculated as homing index (HI) using a formula: HI = %TRITC+organ/%CFSE+organ: TRITCinput/CFSEinput as described [37].
Histological examination
Tissues including liver, colon, intestines, lung, skin, and spleen were removed within 24 h of any overt sign of terminal illness the mice showed, such as weight loss in excess of 15%. These tissues were fixed with 10% formalin, embedded in paraffin, and sectioned at 4 μm. Sections were stained with hematoxylin (H) and eosin (E) by standard methods and subjected to histological examination.
Statistical analysis
The Student's two-tailed t-test was used to analyze the significance of experimental group and relevant controls unless otherwise indicated.
Acknowledgements
We thank members in Dr. Wu's group for stimulating discussion. This work is supported by the National Institutes of Health grants AI050822 and AI070785, Research Scholar Grant RSG-01–178–01-MGO from the American Cancer Society, and Senior Research Award (1657) from the Crohn's & Colitis Foundation of America (to M.X.W.) and by the Intramural Research Program of the NIH, NIEHS (to L.B). B.T. is supported in part by a NIH T32 training grant (AR07098–31) from the Department of Dermatology, Harvard Medical School.
Abbreviations
- PLN
peripheral lymph node
- PTX
pertussis toxin
- S1P
sphingosince-1-phosphate
- TRITC
tetramethylrhodamine isothiocyanate
Footnotes
Conflict of interest: The authors declared no financial or commercial conflict of interest.
References
- 1.Appelbaum FR. Haematopoietic cell transplantation as immunotherapy. Nature. 2001;411:385–389. doi: 10.1038/35077251. [DOI] [PubMed] [Google Scholar]
- 2.Ho VT, Soiffer RJ. The history and future of T-cell depletion as graft-versus-host disease prophylaxis for allogeneic hematopoietic stem cell transplantation. Blood. 2001;98:3192–3204. doi: 10.1182/blood.v98.12.3192. [DOI] [PubMed] [Google Scholar]
- 3.Wysocki CA, Panoskaltsis-Mortari A, Blazar BR, Serody JS. Leukocyte migration and graft-versus-host disease. Blood. 2005;105:4191–4199. doi: 10.1182/blood-2004-12-4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Panoskaltsis-Mortari A, Price A, Hermanson JR, Taras E, Lees C, Serody JS, Blazar BR. In vivo imaging of graft-versus-host-disease in mice. Blood. 2004;103:3590–3598. doi: 10.1182/blood-2003-08-2827. [DOI] [PubMed] [Google Scholar]
- 5.Pribila JT, Quale AC, Mueller KL, Shimizu Y. Integrins and T cell-mediated immunity. Annu. Rev. Immunol. 2004;22:157–180. doi: 10.1146/annurev.immunol.22.012703.104649. [DOI] [PubMed] [Google Scholar]
- 6.Li B, New JY, Yap EH, Lu J, Chan SH, Hu H. Blocking L-selectin and alpha4-integrin changes donor cell homing pattern and ameliorates murine acute graft versus host disease. Eur. J. Immunol. 2001;31:617–624. [PubMed] [Google Scholar]
- 7.New JY, Li B, Koh WP, Ng HK, Tan SY, Yap EH, Chan SH, Hu HZ. T cell infiltration and chemokine expression: relevance to the disease localization in murine graft-versus-host disease. Bone Marrow Transplant. 2002;29:979–986. doi: 10.1038/sj.bmt.1703563. [DOI] [PubMed] [Google Scholar]
- 8.Wysocki CA, Burkett SB, Panoskaltsis-Mortari A, Kirby SL, Luster AD, McKinnon K, Blazar BR, Serody JS. Differential roles for CCR5 expression on donor T cells during graft-versus-host disease based on pretransplant conditioning. J .Immunol. 2004;173:845–854. doi: 10.4049/jimmunol.173.2.845. [DOI] [PubMed] [Google Scholar]
- 9.Simon SI, Green CE. Molecular mechanics and dynamics of leukocyte recruitment during inflammation. Annu. Rev. Biomed. Eng. 2005;7:151–185. doi: 10.1146/annurev.bioeng.7.060804.100423. [DOI] [PubMed] [Google Scholar]
- 10.Wilkie TM, Gilbert DJ, Olsen AS, Chen XN, Amatruda TT, Korenberg JR, Trask BJ, et al. Evolution of the mammalian G protein alpha subunit multigene family. Nat. Genet. 1992;1:85–91. doi: 10.1038/ng0592-85. [DOI] [PubMed] [Google Scholar]
- 11.Fields TA, Casey PJ. Signalling functions and biochemical properties of pertussis toxin-resistant G-proteins. Biochem. J. 1997;321:561–571. doi: 10.1042/bj3210561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamm HE. How activated receptors couple to G proteins. Proc. Natl. Acad. Sci. USA. 2001;98:4819–4821. doi: 10.1073/pnas.011099798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamm HE, Gilchrist A. Heterotrimeric G proteins. Curr. Opin. Cell Biol. 1996;8:189–196. doi: 10.1016/s0955-0674(96)80065-2. [DOI] [PubMed] [Google Scholar]
- 14.Kaslow HR, Burns DL. Pertussis toxin and target eukaryotic cells: binding, entry, and activation. FASEB J. 1992;6:2684–2690. doi: 10.1096/fasebj.6.9.1612292. [DOI] [PubMed] [Google Scholar]
- 15.Kehrl JH. Heterotrimeric G protein signaling: roles in immune function and fine-tuning by RGS proteins. Immunity. 1998;8:1–10. doi: 10.1016/s1074-7613(00)80453-7. [DOI] [PubMed] [Google Scholar]
- 16.Beals CR, Wilson CB, Perlmutter RM. A small multigene family encodes Gi signal-transduction proteins. Proc. Natl. Acad. Sci. USA. 1987;84:7886–7890. doi: 10.1073/pnas.84.22.7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim SY, Ang SL, Bloch DB, Bloch KD, Kawahara Y, Tolman C, Lee R, et al. Identification of cDNA encoding an additional alpha subunit of a human GTP-binding protein: expression of three alpha i subtypes in human tissues and cell lines. Proc. Natl. Acad. Sci. USA. 1988;85:4153–4157. doi: 10.1073/pnas.85.12.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson BD, Jin Y, Wu KH, Colvin RA, Luster AD, Birnbaumer L, Wu MX. Inhibition of Gai2 activation by Gai3 in CXCR3-mediated signaling. J. Biol. Chem. 2007;282:9547–9555. doi: 10.1074/jbc.M610931200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rudolph U, Finegold MJ, Rich SS, Harriman GR, Srinivasan Y, Brabet P, Boulay G, et al. Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat. Genet. 1995;10:143–150. doi: 10.1038/ng0695-143. [DOI] [PubMed] [Google Scholar]
- 20.Huang TT, Zong Y, Dalwadi H, Chung C, Miceli MC, Spicher K, Birnbaumer L, et al. TCR-mediated hyper-responsiveness of autoimmune Galphai2(−/−) mice is an intrinsic naive CD4(+) T cell disorder selective for the Galphai2 subunit. Int. Immunol. 2003;15:1359–1367. doi: 10.1093/intimm/dxg135. [DOI] [PubMed] [Google Scholar]
- 21.Wu JY, Jin Y, Edwards RA, Zhang Y, Finegold MJ, Wu MX. Impaired TGF-{beta} responses in peripheral T cells of G{alpha}i2−/− mice. J. Immunol. 2005;174:6122–6128. doi: 10.4049/jimmunol.174.10.6122. [DOI] [PubMed] [Google Scholar]
- 22.Ichiba T, Teshima T, Kuick R, Misek DE, Liu C, Takada Y, Maeda Y, et al. Early changes in gene expression profiles of hepatic GVHD uncovered by oligonucleotide microarrays. Blood. 2003;102:763–771. doi: 10.1182/blood-2002-09-2748. [DOI] [PubMed] [Google Scholar]
- 23.Broxmeyer HE, Kohli L, Kim CH, Lee Y, Mantel C, Cooper S, Hangoc G, et al. Stromal cell-derived factor-1/CXCL12 directly enhances survival/antiapoptosis of myeloid progenitor cells through CXCR4 and G(alpha)i proteins and enhances engraftment of competitive, repopulating stem cells. J. Leukoc. Biol. 2003;73:630–638. doi: 10.1189/jlb.1002495. [DOI] [PubMed] [Google Scholar]
- 24.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 25.Windh RT, Lee MJ, Hla T, An S, Barr AJ, Manning DR. Differential coupling of the sphingosine 1-phosphate receptors Edg-1, Edg-3, and H218/Edg-5 to the G(i), G(q), and G(12) families of heterotrimeric G proteins. J. Biol. Chem. 1999;274:27351–27358. doi: 10.1074/jbc.274.39.27351. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Finegold MJ, Jin Y, Wu MX. Accelerated transition from the double-positive to single-positive thymocytes in G alpha i2-deficient mice. Int. Immunol. 2005;17:233–243. doi: 10.1093/intimm/dxh204. [DOI] [PubMed] [Google Scholar]
- 27.Graeler M, Shankar G, Goetzl EJ. Cutting edge: suppression of T cell chemotaxis by sphingosine 1-phosphate. J. Immunol. 2002;169:4084–4087. doi: 10.4049/jimmunol.169.8.4084. [DOI] [PubMed] [Google Scholar]
- 28.Duffner U, Lu B, Hildebrandt GC, Teshima T, Williams DL, Reddy P, Ordemann R, et al. Role of CXCR3-induced donor T-cell migration in acute GVHD. Exp. Hematol. 2003;31:897–902. doi: 10.1016/s0301-472x(03)00198-x. [DOI] [PubMed] [Google Scholar]
- 29.Hancock WW, Lu B, Gao W, Csizmadia V, Faia K, King JA, Smiley ST, et al. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J. Exp. Med. 2000;192:1515–1520. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welniak LA, Wang Z, Sun K, Kuziel W, Anver MR, Blazar BR, Murphy WJ. An absence of CCR5 on donor cells results in acceleration of acute graft-vs.-host disease. Exp. Hematol. 2004;32:318–324. doi: 10.1016/j.exphem.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 31.Murai M, Yoneyama H, Harada A, Yi Z, Vestergaard C, Guo B, Suzuki K, et al. Active participation of CCR5(+)CD8(+) T lymphocytes in the pathogenesis of liver injury in graft-versus-host disease. J. Clin. Invest. 1999;104:49–57. doi: 10.1172/JCI6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Graler MH, Huang MC, Watson S, Goetzl EJ. Immunological effects of transgenic constitutive expression of the type 1 sphingosine 1-phosphate receptor by mouse lymphocytes. J. Immunol. 2005;174:1997–2003. doi: 10.4049/jimmunol.174.4.1997. [DOI] [PubMed] [Google Scholar]
- 33.Han SB, Moratz C, Huang NN, Kelsall B, Cho H, Shi CS, Schwartz O, Kehrl JH. Rgs1 and Gnai2 regulate the entrance of B lymphocytes into lymph nodes and B cell motility within lymph node follicles. Immunity. 2005;22:343–354. doi: 10.1016/j.immuni.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 34.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- 36.Hornquist CE, Lu X, Rogers-Fani PM, Rudolph U, Shappell S, Birnbaumer L, Harriman GR. G(alpha)i2-deficient mice with colitis exhibit a local increase in memory CD4+ T cells and proinflammatory Th1-type cytokines. J. Immunol. 1997;158:1068–1077. [PubMed] [Google Scholar]
- 37.Weninger W, Crowley MA, Manjunath N, von Andrian UH. Migratory properties of naive, effector, and memory CD8(+) T cells. J. Exp. Med. 2001;194:953–966. doi: 10.1084/jem.194.7.953. [DOI] [PMC free article] [PubMed] [Google Scholar]