

Abstract
The adenosine class of G protein-coupled receptors mediates the important role of extracellular adenosine in many physiological processes and is antagonized by caffeine. We have determined the crystal structure of the human A2A adenosine receptor in complex with a high affinity subtype-selective antagonist, ZM241385, to 2.6 Å resolution. Four disulfide bridges in the extracellular domain combined with a subtle repacking of the transmembrane helices relative to the adrenergic and rhodopsin receptor structures defines a pocket distinct from that of other structurally determined GPCRs. The arrangement allows for the binding of the antagonist in an extended conformation perpendicular to the membrane plane. The binding site highlights an integral role for the extracellular loops, together with the helical core in ligand recognition by this class of GPCRs, and suggests a role for ZM241385 in restricting the movement of a tryptophan residue important in the activation mechanism of the class A receptors.
Introduction
Extracellular adenosine plays an important role in physiology and initiates most of its effects through the activation of four guanine nucleotide binding protein (G protein) coupled receptor (GPCR) subtypes, A1, A2A, A2B and A3 (1, 2). Each of these four receptors plays an essential role in responding to adenosine in the central nervous system (3, 4) regulating pain (5), cerebral blood flow (6), basal ganglia functions (7), respiration (8) and sleep (9). These receptor subtypes are primarily coupled to the cAMP second messenger system and each has its own unique pharmacological profile. The A2A adenosine subtype is linked to Gs and Golf proteins and upon receptor activation, the intracellular levels of cAMP increase. At least three of the four adenosine receptor subtypes (A1, A2A and A2B) are blocked by naturally occurring methylxanthines, such as caffeine, with modest affinity. Interestingly, strong epidemiological evidence suggests that coffee drinkers have a lower risk of Parkinson’s disease (10). This effect has been linked to caffeine’s interaction with the A2A adenosine receptor, which controls locomotor behavior in basal ganglia together with dopamine D2 and metabotropic glutamate mGluR receptors (11, 12). Development of more selective compounds for adenosine receptor subtypes could provide a class of therapeutics for treating numerous human maladies, such as pain (5), Parkinson’s disease (7, 13), Huntington disease (14), asthma (15), seizures (16) and many other neurological disorders (14, 17).
We have determined the crystal structure of the human A2A adenosine receptor in complex with the subtype selective high affinity antagonist (4-(2-[7-amino-2-(2-furyl)-[1,2,4]triazolo-[2,3-a][1,3,5]triazin-5-ylamino] ethyl)-phenol (ZM241385) (18, 19). The basis for this compound’s selectivity over the adenosine A1 and A3 receptors can now be analyzed in the context of its molecular interactions with the A2A receptor along with previously reported mutagenesis data. With an additional human GPCR structure, it is now possible to extend the analysis of structural differences as they pertain to receptor pharmacology, ligand recognition and receptor activation across the members of the class A receptor family.
Structure determination
GPCRs possess numerous thermodynamic conformations (20, 21), implying an inherent structural flexibility (22-24). This flexibility manifests itself as thermal instability upon detergent extraction from lipid membranes and is one of the primary challenges in generating crystals of GPCRs (25, 26). In order to overcome this obstacle with the human A2A adenosine receptor, we applied the T4L fusion strategy (24, 27, 28), where most of the third cytoplasmic loop (Leu2095.70 - Ala2216.23) was replaced with lysozyme from T4 bacteriophage and the carboxyl-terminal tail (Ala317 - Ser412) was deleted to improve the likelihood of crystallization. The resulting recombinant construct (A2A-T4L-ΔC) was further stabilized during purification with (i) sodium chloride, which has a beneficial effect on adenosine receptor stability, (ii) a saturating concentration of the nonspecific adenosine receptor antagonist theophylline (ZM241385 was exchanged from theophylline in the last purification step) and (iii) including cholesteryl hemisuccinate throughout the purification. Purified A2A-T4L-ΔC bound to ZM241385 was crystallized using the in meso crystallization methodology where the lipid phase consisted of a mixture of monoolein and cholesterol (29).
Diffraction data from thirteen of the best crystals were combined to yield a 2.6 Å dataset (Table 1). Phases were obtained by molecular replacement using the coordinates of the β2-adrenergic receptor (β2AR) fused to T4-lysozyme (PDB accession number, 2RH1). The final refined model includes residues Ile3 to Gln310 of the human A2A adenosine receptor, residues 2 to 161 of T4-lysozyme, five lipid hydrocarbon chains modeled as stearic acid, eight sulfate ions and the antagonist ZM241385 bound in the ligand binding cavity (Figure 1). The experimental electron density for the amino (Met1 - Pro2) and carboxyl (Glu311 - Ala316) termini did not support modeling of these regions. In addition, the tip of second extracellular loop (Gln148 - Ser156) was not modeled due to weak experimental electron density. Although cholesterol does have a significant stabilizing effect on the A2A adenosine receptor and was included in the crystallization trials, in contrast to the β2-adrenergic receptor structure which has cholesterol bound in a pocket referred to as the cholesterol consensus motif (30), the A2A adenosine receptor structure has phospholipid bound in the same area.
Table 1.
Data collection and refinement statistics
| A2A-T4L-ΔC | |
|---|---|
| Data collection (APS GM/CA CAT ID-B, 10 μm beam) | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 47.7, 76.9, 86.6 |
| (°) | 101.3 |
| No. of reflections processed | 64,526 (8165) |
| No. unique reflections | 18,465 (356) |
| Resolution (Å) | 20.0 - 2.6 (2.8 - 2.6) |
| Rsym | 9.8 (38.9) |
| Mean I/σ(I) | 7.0 (2.3) |
| Completeness (%) | 96.8 (93.9) |
| Multiplicity | 3.5 (2.3) |
| Refinement | |
| Resolution (Å) | 20.0 - 2.6 |
| No. reflections (reference set) | 18,461 (937) |
| Rcrys / Rfree | 19.6 / 23.1 |
| No. atoms | 3769 |
| Protein | 3521 |
| Ions, lipids, ligand and other | 165 |
| Water oxygen | 83 |
| B-values (Å2) | |
| All atoms | 70.6 |
| Protein | 69.4 |
| Ligand | 66.7 |
| Lipid | 94.4 |
| R.m.s deviations from ideality | |
| Bond lengths (Å) | 0.002 |
| Bond angles (°) | 0.78 |
| Ramachandran plot statistics (%) (excl. Gly, Pro): | |
| Most favored regions | 92.8 |
| Additionally allowed regions | 7.2 |
| Generously allowed regions | 0.0 |
| Disallowed regions | 0.0 |
Highest resolution shell is shown in parenthesis.
Rsym = 100 Σn (ΣI |Ii - Î|)/Σn (Σ Ii)
Rcrys = 100 Σhkl |Fobs - Fcalc| Σhkl Fobs.
Rfree = test set 5%.
Figure 1.

Crystal structure of A2A-T4L-ΔC. A. Overall topology of A2A-T4L-ΔC. The transmembrane part of A2A-ΔC structure is colored brown (helices I - VIII) and the T4L is in cyan. The structure is viewed perpendicular to the plasma membrane. ZM241385 is colored light blue and the four lipid molecules bound to the receptor are colored as red. The four disulfide bonds are yellow. The sulfate ions are omitted. The extracellular loops (ECL1-3) are colored as green and the intracellular loops are colored as blue. The membrane boundaries are adapted from the OPM database (http://opm.phar.umich.edu/) using β2AR-T4L (2RH1) as a model. B. Rotated 180° around the x-axis. The images were created with PyMOL.
Biochemical characterization of A2A-T4L-ΔC
We have verified the functionality of A2A-T4L-ΔC by comparing its binding properties to A2A-T4 and A2A-WT (31). The A2A-T4L-ΔC, A2A-T4 and A2A-WT constructs expressed in Sf9 cells bind [3H]ZM241385 with similar affinity as the same constructs transiently expressed in HEK293 as judged by radioligand saturation experiments. This finding was corroborated in competition binding assays, as the two A2A-T4L constructs had IC50 values similar to A2A-WT for ZM241385 (Figure 2; Figure 2S and Table 2S in SOM). However, A2A-T4L and A2A-T4L-ΔC displayed significantly higher affinity for the subtype-selective agonist CGS21680 as compared to the A2A-WT construct, possibly indicating a shift toward the activated state induced by the incorporation of the T4L moiety. A comparable construct of the β2AR behaved in a similar fashion (24); however, unlike β2AR, the A2A-WT has no associated basal activity (G protein signalling in the absence of agonist). The inclusion of a high concentration of sodium chloride in the assay medium induced a substantial decrease in the agonist affinity for all of the tested constructs (Figure 2b) but did not appreciably affect antagonist affinity. Interestingly, pKi values for the agonist in the presence of sodium chloride were virtually identical for all constructs tested (Table 2S in SOM) suggesting that sodium chloride induced a shift in receptor equilibrium to an inactive state (32). In addition, sodium chloride induced a 10 °C increase in thermal stability for A2A-T4L-ΔC solubilized in n-Dodecyl-β-D-maltoside (Figure 3S in SOM). Thus, radioligand binding experiments support the conclusion that the construct used for crystallization is a functional receptor with an increased affinity for agonist, and wild-type affinity for antagonist.
Figure 2.

Ligand binding characteristics of A2A-WT, A2A-T4L and A2A-T4L-ΔC. A. Saturation binding isotherm for the binding of [3H]ZM241385 to different A2A-WT, A2A-T4L or A2A-T4L-ΔC receptors confined in membranes of Sf9 cells. The indicated preparations of A2A receptors were incubated with different concentrations of [3H]ZM241385 in the absence (filled shapes and solid lines) and presence (open shapes and dashed lines) of 1 M NaCl as described in SOM. The figure shown represents data combined from two separate experiments performed in triplicate. The equilibrium constant (Kd) values of [3H]ZM241385 in the absence and the presence of 1 M NaCl were 2.1 ± 0.7 nM, 1.3 ± 0.2 nM for A2A-WT; 2.0 ± 0.3 nM, 0.9 ± 0.1 nM for A2A-T4L and 1.8 ± 0.2 nM, 1.0 ± 0.1 nM for A2A-T4L-DC, respectively. B. One point binding assay demonstrating the binding of [3H]ZM241385 to membranes (5 μg / assay point) of HEK 293T cells transfected with A2A-WT, A2A-T4L or A2A-T4L-ΔC. [3H]ZM241385 was used at a concentration equivalent to the previously observed equilibrium constant (Kd). Lower panels - the ability of increasing concentrations of C. the agonist CGS21680 or D. the antagonist ZM241385 to compete with [3H]ZM241385 binding at A2A-WT (circles), A2A-T4L (triangles), A2A-T4L-ΔC (squares) constructs in HEK293T cells was tested in the absence (filled shapes and solid lines) or presence (open shapes and dashed lines) of 1 M NaCl. The figure shown represents data combined from three separate experiments performed in duplicate.
Architecture of the human A2A adenosine receptor
The residues constituting the transmembrane α-helices are: Gly51.31-Trp321.58 (helix I); Thr412.39-Ser672.65 (helix II); His753.23-Arg1073.55 (helix III); Thr1194.40-Leu1404.61 (helix IV); Asn1755.36-Ala2045.65 (helix V); Arg2226.24-Phe2586.60 (helix VI); Leu2697.34-Arg2917.56 (helix VII) (33). A small non-transmembrane helix is located at the membrane-cytoplasm interface and comprises Arg2968.47-Leu3088.59 (helix VIII). The A2A adenosine receptor does not contain the canonical palmitoylation site(s) found in the majority of GPCRs; instead, helix VIII is stabilized by interactions with helix I (34). The residues defining intracellular and extracellular loops (ICLs and ECLs) are: Leu331.59-Val402.38 (ICL1); Ile1083.56-Gly1184.39 (ICL2); Leu2085.69-Ala2216.23 (ICL3); Thr682.66-Cys743.22 (ECL1); Leu1414.62-Met1745.35 (ECL2); Cys2596.61-Trp2687.33 (ECL3). In our structure ICL3 has been replaced by 160 residues from T4L lysozyme (see Supplementary Figure S1). Additionally, The N-linked glycan associated with Asn1544.75 has been removed enzymatically to improve crystallization.
Our crystallographic model of A2A-T4L-ΔC bound to ZM241385 reveals three features distinct from the previously reported GPCR structures. First, the organization of the extracellular loops is markedly different from β1AR, β2AR and bovine/squid rhodopsins (23, 24, 30, 35-37). Secondly, ZM241385 binds in an extended conformation perpendicular to the plane of the membrane and co-linear with transmembrane helix VII interacting with both ECL2 and ECL3. This is somewhat incongruous with earlier molecular modeling studies based on β2AR and rhodopsin homology models where ZM241385 and other antagonists were docked into a binding site emulating that of β2AR and rhodopsin (for examples see (38, 39), and references therein). Finally, a subtle divergence in the helical positions and orientations relative to rhodopsin and β2AR redefines the antagonist binding cavity so that it is located closer to helices VI and VII and allowing only limited interactions with helices III and V.
Helical position and binding pocket diversity
Among the class A GPCRs, the sequence identity is highest within the α-helical transmembrane regions and ranges from 20-50% (40, 41). Not surprisingly, the helical arrangement is similar among the human β2AR, turkey β1AR and squid/bovine rhodopsins structures determined to date. However, shifts in the relative positions of the various helices results in a root mean square deviation (RMSDs) between 2.0 to 2.5 Å (depending on how the alignment is carried out and which structures are being compared) that has structural, and biochemical implications. Most of the structural divergence arises in the extracellular portions of helices I, II, III and V, where the variation in the positions of helices II, III and V appears to redefine the location of the ligand binding pocket (42). However, comparisons between ground-state rhodopsin bound to retinal and β2AR bound to carazolol show minimal differences as the relative helical shifts are smaller (Figure 3a and 3b) (27). The position of the retinal and carazolol binding pocket is very similar and making most contact with helices III, V, VI (Figure 3a and Figure 4). The binding pocket of the A2A adenosine receptors is shifted closer to helices VI and VII which contribute the majority of the binding interactions associated with helical regions, as judged by occluded surface area calculations (43, 44) (Figure 3b and Figure 4). A concomitant shift of helices II and V (7 Å and 3 Å, respectively) toward the binding pocket, and a lateral shift of helix III toward helix V by 3 Å, compensates for the absence of ligand interactions in this region by increasing protein packing interactions (Figure 3a and 3b).
Figure 3.

Slight changes in helical positions alter the orientation of the ligand binding pocket. A. A surface rendering of the binding pocket for ZM241385 in the A2A adenosine receptor. Helical positions for A2A adenosine (tan), β2AR (pdbid: 2RH1) (blue) and rhodopsin (pdbid: 1U19) (green) are shown after alignment with the FatCat server (38). Ligands for each receptor are shown to illustrate the differences in binding orientation and the differences in the adenosine A2A binding pocket. B. A top view of the helical bundle illustrating the maximal helical positional shifts of A2A relative to β2AR.
Figure 4.
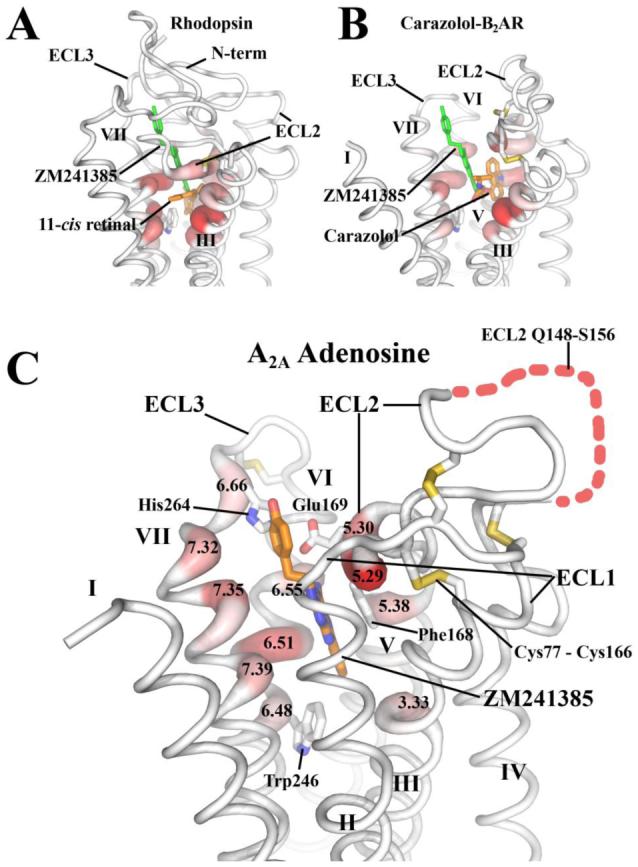
Normalized occluded surface (NOS) area changes due to ligand binding. Increases in occluded surface area are represented as thickened red areas of the protein backbone chain (38). A. Rhodopsin (pdbid: 1U19) with retinal (orange) is shown along with the position of ZM241385 (green) for comparison. Retinal makes extensive contact with helices III, V, VI and VII deep in the binding pocket. B. β2AR bound to carazolol (orange) (pdbid: 2RH1) is shown along with the position of ZM241385 (green) for comparison. Carazolol also makes extensive contacts with helices III, V, VI and VII deep in the binding pocket but is responsible for minimal changes in NOS of Trp 2866.48 the canonical “toggle switch”. C. A2A adenosine receptor bound to ZM241385 (orange carbon) has a very different binding orientation relative to rhodopsin and β2AR having minimal interaction with helices III and V, but extensive interactions with helices VI and VII as well as residues in a ECL2 and ECL3. ZM241385 also forms significant contacts with Trp2466.48. All interacting positions on the receptor are displayed as thick red areas and labeled by their corresponding Ballesteros-Weinstein designation (33).
Conformational equilibrium and receptor activation
A common feature of the class A GPCRs is the presence of a tryptophan residue (at position 6.48) on helix VI whose rotameric position is thought to control the equilibrium between the active and inactive states of each receptor (45). Based on the position of retinal in the rhodopsin structure it was proposed that ligand interactions with this key residue could modulate receptor equilibrium (46). Interestingly, the contact area between ligand and the “toggle switch” tryptophan residue at position 6.48 varies considerably among the solved receptor structures. For instance, rhodopsin and β2AR have a similar binding mode as noted; however, retinal in rhodopsin has a contact area of 36 Å2, whereas carazolol bound to β2AR lacks any direct contact with Trp2866.48 (27). Ground-state rhodopsin has virtually no basal activity, whereas β2AR has a relatively high basal activity which is suppressed somewhat by carazolol as an inverse agonist (23, 47). The observed increase in contact area may have direct implications for inverse agonist efficacy or suppressed basal activity by limiting the range of motion of the “toggle switch” tryptophan. The competitive antagonist ZM241385 has a 14 Å2 contact area with Trp2466.48 despite an altered binding mode relative to rhodopsin (Figure 4c). Although it remains to be established biochemically, this finding is suggestive of the ability of this ligand to stabilize the A2A adenosine receptor in an inactive state.
Interactions between the cytoplasmic end of helix III (conserved D/ERY (Asp3.49 Arg3.50 Tyr3.51 sequence motif) and helix VI (Glu6.30) have been proposed to constitute an “ionic-lock” that may play a role in restraining the fully inactive conformation of rhodopsin and other class A receptors (36, 48, 49). Of particular note is that with the exception of the rhodopsins, none of the GPCR structures solved to date have the ionic lock interaction, including the A2A adenosine receptor. Instead, as in β1AR and β2AR, the D/ERY motif in the A2A adenosine receptor participates in interactions that restrain the conformation of ICL2. In the A2A adenosine receptor, Asp1013.49 forms a hydrogen bond with Tyr1123.60 in ICL2 and Thr412.39 at the base of helix II (Figure 5a). Similar hydrogen bonding interactions were reported in the turkey β1AR structure (37), but not in any of the β2AR structures where Asp1303.49 forms a hydrogen bond with Ser1433.62 although there is a tyrosine at the 3.60 position (Figure 5b and 5c) (23, 24, 30). This discrepancy is caused by a short helical section in the ICL2 loop of both β1AR and the A2A adenosine receptor that is not present in any of the β2AR structures (Figure 5). It has been proposed that ICL2 serves as a control switch facilitating G protein activation through a select set of interactions (50). Interestingly the basal activity profile among the β1AR, β2AR and the A2A adenosine receptors correlates with the presence of this short helix in ICL2 and the presence of hydrogen bonding interactions between tyrosine at position 3.60 in ICL2 and Asp at position 3.49. In β1AR and A2A adenosine receptor, both of which have low basal activity, this interaction is present (51, 52). In contrast, β2AR exhibits high basal activity and lacks helical structure within its ICL2 resulting in altered interactions with the DRY motif (51). Instead of participating in an “ionic lock’ as in rhodopsin, the arginine residue in the D/ERY motif may play a role in stabilizing the deprotonated state of the adjacent aspartate or glutamate residue, which would strengthen the polar interactions between the D/ERY motif and both ICL2, and helix II. This set of interactions may have direct implications in G protein activation (48).
Figure 5.

A Comparison of interactions between helix III (E/DRY motif) and ICL2 for human A2A-T4L-ΔC, human β2AR-T4L (pdbid: 2RH1) and turkey β1AR (pdbid 2VT4). A. A2A-T4L-ΔC interactions. The DRY motif does not participate in any stabilizing ionic interactions similar to β2AR and β1AR. Instead Arg1023.50 may play a role in shifting the pKa of the adjacent Asp1013.49 allowing this residue to make stronger hydrogen bonding interactions with helix II and ICL2. B. Turkey β1AR participates in similar interactions as A2A-T4L-ΔC without the hydrogen bond to helix II. C. β2AR does not contain a helical segment in ICL2 and has a modified set of interactions. D. The canonical “ionic lock” in rhodopsin.
Extracellular loops: mediation of ligand entry and binding
The extracellular surface properties of the A2A adenosine receptor is largely dictated by its second extracellular loop (ECL2), which is considerably different from that of β1AR, β2AR and rhodopsin (Figure 1 and Figure 4). The ECL2 of the A2A adenosine receptor lacks the prominent secondary structural elements, such as β-sheet and α-helix, which were observed in the rhodopsin and β-adrenergic receptors, respectively. Instead, the ECL2 of the A2A adenosine receptor is mainly a spatially constrained random coil possessing three disulfide linkages with ECL1 (Figure 4C). Two of the three disulfide bonds (Cys712.69 - Cys1595.20 and Cys743.22 - Cys1464.67) are unique to the A2A adenosine receptor while the third (Cys773.25 - Cys1665.27) is conserved among many class A GPCRs. In addition, a fourth intraloop disulfide bond is formed in ECL3 between Cys2596.61 and Cys2626.64 with the sequence CPDC which creates a kink in the loop that constrains the position of ECL3 and orients His2646.66 at the top of the ligand binding site. Importantly, the extensive disulfide bond network forms a rigid, open structure exposing the ligand binding cavity to solvent, possibly allowing free access for small molecule ligands. In addition, the family conserved disulfide bridge (Cys773.25 - Cys1665.27) is adjacent to a short helical segment that presents two crucial residues for ligand binding interactions (Phe1685.29 and Glu1695.30). The missing tip of the loop (Gln148 - Ser156) is spatially distinct from the ligand binding site, and probably does not directly interact with the binding cavity. Mutation of Cys2626.64 to Gly did not affect binding to radioligand agonist or antagonist indicating that the kink in ECL3 may not be a prerequisite for receptor function or that the other disulfide bonds are sufficient to constrain extracellular loop architecture (53). Mutational studies on the A1 adenosine receptor indicate that these cysteine residues (Cys803.25 - Cys1695.27 in the A1 receptor) (see Figure 1S in SOM) are critical for expression due to a complete loss of radiolabeled antagonist binding in the absence of this disulfide bond.
Analysis of the ligand binding cavity
To date, structural and biophysical data on the class A GPCRs with diffusible ligands has been dominated by the biogenic amine receptors, such as the adrenergic, dopamine, and serotonin families. These amine ligands are all positively charged at physiologic pH and are known to interact with a key negatively charged aspartate residue (Asp3.32) on helix III. Indeed, in all three of the available β-adrenergic structures, each co-crystallized ligand interacts with this residue and binds in a pocket quite similar to that of retinal in rhodopsin. Analysis of the binding sites of the three available GPCR structures indicates two possibilities for the other members of the class A family: i) other ligands will bind in a spatially similar binding site, with the ligand specificity dictated by sequence differences within the binding pocket, or ii) ligands for other receptors may bind in a completely different fashion interacting with other positions on the receptor. In contrast to the β-adrenergic ligands and retinal, ZM241385 occupies a significantly different position in the transmembrane network (Figure 4) where its orientation is almost perpendicular to the membrane plane (Figures 4c and 6). The bicyclic triazolotriazine core of ZM241385 is anchored by an aromatic stacking interaction with Phe1685.29, an aliphatic hydrophobic interaction with Ile2747.39 and a hydrogen bonding interaction with Asn2536.55 (Figure 6). Adjacent to Phe1685.29 a polar residue (Glu1695.30) interacts with the exocyclic amino group (N15 atom) linked to the bicyclic core of ZM241385 (Figure 6b). Mutation of Glu1695.30 to alanine reduces the affinity for both antagonists and agonists and causes a 1000-fold reduction in agonist efficacy (54). However, mutating this position to glutamine did not have a substantial impact on antagonist binding affinity, suggesting hydrogen bonding as the predominant means of interacting with N15 of ZM241385 as opposed to Coulombic interactions (Figure 6b). Early studies indicate that mutation of Asn2536.55 to alanine, which would disrupt an important polar contact with the exocyclic N15 atom of ZM241385, results in a complete loss of both agonist and antagonist binding (55). The structure also shows that Ile2747.39 forms a hydrophobic contact with the C12 atom of ZM241385; accordingly mutation of Ile2747.39 to alanine results in negligible antagonist binding and a 30-fold reduction in agonist potency (55). No mutagenesis data is available for Phe1685.29 or Leu249 both of which anchor the bicyclic ring of ZM241385 through π stacking and hydrophobic interactions respectively although their involvement in ligand binding has been proposed (56). The phenolic hydroxyl group extending from the ethylamine chain of ZM241385 forms a hydrogen bond with an ordered water molecule. The phenyl ring forms hydrophobic interactions with Leu2677.32 and Met2707.35 that would suggest hydrophobicity rather than aromaticity as means of interaction with the phenolic substituent. Indeed, a ZM241385 derivative, with a cycloalkyl substituent (LUF5477) instead of phenylmethylene, also has high affinity for the A2A adenosine receptor. In a recent study on new antagonists for the A2A adenosine receptor it was demonstrated that tremendous substituent flexibility exists in this area of the pharmacophore (57). This observation correlates well with the directionality of the phenylethylamine substituent in ZM241385 as it is directed towards the more solvent exposed extracellular region (ECL2 and ECL3) rather than towards the transmembrane domain of the receptor as was previously proposed (38, 39). The other substituent in ZM241385 is the furan ring, a feature that occurs in many A2A adenosine receptor antagonists. This moiety is located deep in the ligand binding cavity and directed towards helices V and VII, where it hydrogen bonds to Asn2536.55 and forms a water-mediated interaction with His2506.52 (Figure 6A). Hydrophobic interactions of the furan ring system include His2506.52 with C23 and Leu2496.51 with the C22 and C21 atoms of ZM241385. Mutation of His2506.52 to alanine completely abolishes ligand binding, whereas mutation to phenylalanine or tyrosine residues modestly affects agonist binding but not antagonist binding (55, 58); replacement with an asparagine slightly increases ligand affinity (58). The furan ring is approximately 3 Å away from the highly conserved Trp2466.48 an important residue in receptor activation as discussed above (59). We speculate that the hydrophobic interactions between ZM241385’s furan ring and this residue will hinder the structural rearrangements necessary for activation, constraining the receptor in an inactive state.
Figure 6.

Ligand binding cavity of A2A-T4L-ΔC with ZM241385 bound. A. Residues within 5 Å of the ZM241385 are shown in stick representation. Nitrogen atoms are colored blue, oxygen atoms are colored red, and sulfur atoms are colored yellow. Only the interacting helices, ECL3 and the interacting part of ECL2 are shown. The two disulfide bridges in close proximity to the binding cavity are shown as orange sticks. ZM241385 is positioned co-linear with respect to the transmembrane helices V, VI and VII, and the binding cavity is elongated to the ECL3 and helical ends of TM VI and VII. For comparison to retinal chromophore or beta-blockers binding site, see figure 3 for details. The Phe1685.29 from ECL2 forms various aromatic stacking interactions with the bicyclic core of ZM241385. Trp2466.48 associated with stabilizing the antagonist structure is at 3 Å distance from the furan ring of ZM241385. The binding cavity includes four ordered water molecules shown as light blue dots. B. Schematic representation of the interactions between A2A-T4L-ΔC and ZM241385 at the ligand binding cavity combined with mutation analysis for adenosine agonist/antagonists interactions. Mutations that are reported to disrupt antagonist and/or agonist binding are within blue squares: Glu1695.30, His2506.25, Asn2536.55 and Ile2747.39.
The A2A adenosine ligand bound structure suggests that there is no general, family conserved receptor binding pocket in which selectivity is achieved through different amino acid side chains. Rather, the pocket itself can vary in position and orientation, yielding more opportunity for receptor diversity and ligand selectivity. Perhaps as a result of this shift the empirically determined positions of the substituents (phenyl/furanyl) extending from the aromatic core of ZM241385 deviate significantly from what was suggested in molecular modeling studies in which the ligand binding site of retinal and/or beta-blockers was used as the starting point (38, 39). The position of these substituents can be seen as the rationale for A2A receptor selectivity and this may help in the design of new chemical entities with increased selectivity for this important drug target.
Supplementary Material
Acknowledgments
This work was supported in part by the NIH Roadmap Initiative grant P50 GM073197 for technology development, Protein Structure Initiative grant U54 GM074961 for production, and Pfizer. A.P.IJ. and R.J.L. thank the Dutch Top Institute Pharma for financial support through the GPCR forum program (D1-105). The authors thank Jeffrey Velasquez for help on molecular biology, Tam Trinh and Kirk Allin for help on baculoviral expression, Angela Walker for assistance with manuscript preparation and Raymond Benoit for laboratory management. The authors acknowledge Yuan Zheng, The Ohio State University, and Martin Caffrey, University of Limerick, for the generous loan of the in meso robot (built with support from the National Institutes of Health [GM075915], the National Science Foundation [IIS0308078], and Science Foundation Ireland [02-IN1-B266]); and Janet Smith, Robert Fischetti and Nukri Sanishvili at the GM/CA-CAT beamline at the Advanced Photon Source, for assistance in development and use of the minibeam and beamtime. The GM/CA-CAT beamline (23-ID) is supported by the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104). Atomic coordinates and structure factors have been deposited in the Protein Data Bank with identification code XXXX.
Footnotes
One sentence summary: The structure of a human adenosine receptor adds to the repertoire of GPCR structural information and shows that the ligand binding pocket can vary position and orientation among receptors.
References and Notes
- 1.Fredholm BB, Chen JF, Masino SA, Vaugeois JM. Annu Rev Pharmacol Toxicol. 2005;45:385. doi: 10.1146/annurev.pharmtox.45.120403.095731. [DOI] [PubMed] [Google Scholar]
- 2.Fredholm BB, Jzerman API, Jacobson KA, Klotz KN, Linden J. Pharmacol Rev. 2001;53:527. [PMC free article] [PubMed] [Google Scholar]
- 3.Dunwiddie TV, Masino SA. Annu Rev Neurosci. 2001;24:31. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- 4.Jacobson KA, Gao ZG. Nat Rev Drug Discov. 2006;5:247. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sawynok J, Liu XJ. Prog Neurobiol. 2003;69:313. doi: 10.1016/s0301-0082(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 6.Shi Y, et al. J Cereb Blood Flow Metab. 2008;28:111. doi: 10.1038/sj.jcbfm.9600511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwarzschild MA, Agnati L, Fuxe K, Chen JF, Morelli M. Trends Neurosci. 2006;29:647. doi: 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Lahiri S, Mitchell CH, Reigada D, Roy A, Cherniack NS. Respir Physiol Neurobiol. 2007;157:123. doi: 10.1016/j.resp.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basheer R, Strecker RE, Thakkar MM, McCarley RW. Prog Neurobiol. 2004;73:379. doi: 10.1016/j.pneurobio.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 10.Hernan MA, Takkouche B, Caamano-Isorna F, Gestal-Otero JJ. Ann Neurol. 2002;52:276. doi: 10.1002/ana.10277. [DOI] [PubMed] [Google Scholar]
- 11.Ferre S. J Neurochem. 2008;105:1067. doi: 10.1111/j.1471-4159.2007.05196.x. [DOI] [PubMed] [Google Scholar]
- 12.Ferre S, et al. Front Biosci. 2008;13:2391. doi: 10.2741/2852. [DOI] [PubMed] [Google Scholar]
- 13.Schapira AH, et al. Nat Rev Drug Discov. 2006;5:845. doi: 10.1038/nrd2087. [DOI] [PubMed] [Google Scholar]
- 14.Blum D, Hourez R, Galas MC, Popoli P, Schiffmann SN. Lancet Neurol. 2003;2:366. doi: 10.1016/s1474-4422(03)00411-3. [DOI] [PubMed] [Google Scholar]
- 15.Brown RA, Spina D, Page CP. Br J Pharmacol. 2008;153(Suppl 1):S446. doi: 10.1038/bjp.2008.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.During MJ, Spencer DD. Ann Neurol. 1992;32:618. doi: 10.1002/ana.410320504. [DOI] [PubMed] [Google Scholar]
- 17.Benarroch EE. Neurology. 2008;70:231. doi: 10.1212/01.wnl.0000297939.18236.ec. [DOI] [PubMed] [Google Scholar]
- 18.Ongini E, Dionisotti S, Gessi S, Irenius E, Fredholm BB. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:7. doi: 10.1007/pl00005326. [DOI] [PubMed] [Google Scholar]
- 19.Poucher SM, et al. Br J Pharmacol. 1995;115:1096. doi: 10.1111/j.1476-5381.1995.tb15923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cohen BE, et al. Proc Natl Acad Sci U S A. 2005;102:965. doi: 10.1073/pnas.0409469102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobilka BK, Deupi X. Trends Pharmacol Sci. 2007;28:397. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Jaakola VP, Prilusky J, Sussman JL, Goldman A. Protein Eng Des Sel. 2005;18:103. doi: 10.1093/protein/gzi004. [DOI] [PubMed] [Google Scholar]
- 23.Rasmussen SG, et al. Nature. 2007;450:383. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 24.Rosenbaum DM, et al. Science. 2007;318:1266. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 25.Magnani F, Shibata Y, Serrano-Vega MJ, Tate CG. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0804396105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. Proc Natl Acad Sci U S A. 2008;105:877. doi: 10.1073/pnas.0711253105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cherezov V, et al. Science. 2007;318:1258. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engel CK, Chen L, Prive GG. Biochim Biophys Acta. 2002;1564:38. doi: 10.1016/s0005-2736(02)00398-x. [DOI] [PubMed] [Google Scholar]
- 29.See Supporting Material at Science online (SOM).
- 30.Hanson MA, et al. Structure. 2008;16:897. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.A2A-T4 refers to a construct in which the third cytoplasmic loop (residues Leu2085.69 to Ala2216.23) was replaced with the lysozyme from T4 bacteriophage and the full carboxy terminus intact (Ala317 to Ser412). A2A-WT refers to the wild-type construct without the T4 lysozyme. All constructs have a FLAG purification tag in the amino terminus and ten histidine residues in the carboxy terminus.
- 32.Gao ZG, Ijzerman AP. Biochem Pharmacol. 2000;60:669. doi: 10.1016/s0006-2952(00)00360-9. [DOI] [PubMed] [Google Scholar]
- 33.Amino-acid numbering based on human Adenosine A2a primary sequence (accession number P29274).; NOTE: In addition to numbering residue positions in the primary amino acid sequence, the residues have numbers in superscripts (X.YY) that indicate their position in each transmembrane helix (X, helix number, from 1 to 8)), relative to the most conserved reference residue in that helix (YY). This residue is arbitrarily assigned the number 50, numbers decreasing towards N-terminus and increasing towards C-terminus However, the numbering is not used in loop regions beyond residues X.20 and/or X.80 or T4L
- 34.In this crystal form, the crystallographic contacts are mostly driven by the T4L protein where receptor-to-lysozyme and lysozyme-to-lysozyme mainly form the lattice contacts. A relatively large receptor-to-receptor crystallographic interface (∼520 Å2) forms anti-parallel receptor dimers (supplementary figure). The total surface interface between receptor and T4L moieties is ∼1300 Å2, whereas lysozyme-to-lysozyme is ∼200 Å2. The largest contact interface(∼500 Å2) between receptor and T4L is non-crystallographic, and is located in the cytoplasmic site, where receptor is fused to the T4L,. The other receptor-to-lysozyme surface interfaces are crystallographic (260 Å2). In comparison to the previously solved β2AR-T4L fusion proteins, the T4L domain is significantly tilted from the membrane plane, and creates more surface interactions than seen in human β2AR-T4L contructs that were solved in different space groups.
- 35.Murakami M, Kouyama T. Nature. 2008;453:363. doi: 10.1038/nature06925. [DOI] [PubMed] [Google Scholar]
- 36.Palczewski K, et al. Science. 2000;289:739. [Google Scholar]
- 37.Warne T, et al. Nature. 2008;454:486. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinelli A, Tuccinardi T. Med Res Rev. 2008;28:247. doi: 10.1002/med.20106. [DOI] [PubMed] [Google Scholar]
- 39.Yuzlenko O, Kiec-Kononowicz K. J Comput Chem. 2008 doi: 10.1002/jcc.21001. [DOI] [PubMed] [Google Scholar]
- 40.Joost P, Methner A. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-11-research0063. RESEARCH0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vassilatis DK, et al. Proc Natl Acad Sci U S A. 2003;100:4903. doi: 10.1073/pnas.0230374100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The FatCat server ( http://fatcat.burnham.org/) was used for structural alignment of the TMs with the rhodopsin structure 1U19 as a reference taken directly from that server: “It simultaneously addresses the two major goals of flexible structure alignment; optimizing the alignment and minimizing the number of rigid-body movements (twists) around pivot points (hinges) introduced in the reference structure.”
- 43.Ratnaparkhi GS, Varadarajan R. Biochemistry. 2000;39:12365. doi: 10.1021/bi000775k. [DOI] [PubMed] [Google Scholar]
- 44.We used the program occluded surface (OS) that calculate the occluded surface and atomic packing of protein model structures: http://www.csb.yale.edu/userguides/datamanip/os/
- 45.It has been speculated that the general activation mechanism include following changes 6.47 (gauche + conformers) / 6.48 (trans - conformers) / 6.52 (trans - conformers) represent the active state (R*) and 6.47 (trans - conformers) / 6.48 (gauche + conformers) / 6.52 (gauche + conformers) represent inactive state (R).
- 46.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Science. 1996;274:768. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 47.Basal or constitutive activity is the spontaneous production of cellular response in the absence of a ligand. Inverse agonist shifts the equilibrium towards inactive state. Agonist shifts the conformation towards the active state. Neutral antagonist binds to receptors and block the active site but not shift the equilibrium. A typical GPCRs can “dial” almost any conformational equilibrium between fully inactive and fully active therefore agonist/inverse agonist are classified as weak/partial/full. Depending on a receptor and cellular environment, nature of an invert agonism and truly neutral antagonism is difficult to detect.
- 48.Vogel R, et al. J Mol Biol. 2008;380:648. doi: 10.1016/j.jmb.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 49.Okada T, et al. J Mol Biol. 2004;342:571. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 50.Burstein ES, Spalding TA, Brann MR. J Biol Chem. 1998;273:24322. doi: 10.1074/jbc.273.38.24322. [DOI] [PubMed] [Google Scholar]
- 51.Birnbaumer L, Levy FO, Zhu X, Kaumann AJ. Texas Heart Inst J. 1994;21:16. [PMC free article] [PubMed] [Google Scholar]
- 52.Zezula J, Freissmuth M. Br J Pharmacol. 2008;153(Suppl 1):S184. doi: 10.1038/sj.bjp.0707674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scholl DJ, Wells JN. Biochem Pharmacol. 2000;60:1647. doi: 10.1016/s0006-2952(00)00474-3. [DOI] [PubMed] [Google Scholar]
- 54.Kim J, et al. Mol Pharmacol. 1996;49:683. [PMC free article] [PubMed] [Google Scholar]
- 55.Kim J, Wess J, van Rhee AM, Schoneberg T, Jacobson KA. J Biol Chem. 1995;270:13987. doi: 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moro S, et al. Chem Commun (Camb) 2003:2949. doi: 10.1039/b303439a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mantri M, et al. J Med Chem. 2008;51:4449. doi: 10.1021/jm701594y. [DOI] [PubMed] [Google Scholar]
- 58.Jiang Q, Lee BX, Glashofer M, van Rhee AM, Jacobson KA. J Med Chem. 1997;40:2588. doi: 10.1021/jm970084v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Audet M, Bouvier M. Nat Chem Biol. 2008;4:397. doi: 10.1038/nchembio.97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.