

Abstract
[3H]-Cimetidine (3HCIM) specifically binds to an unidentified site in the rat brain. Because recently-described ligands for this site have pharmacological activity, 3HCIM binding was presently characterized. 3HCIM binding was saturable, heat-labile, and distinct from the histamine H2 receptor. To test the hypothesis that 3HCIM binds to a cytochrome P450 (CYP), the effects of non-selective and isoform-selective CYP inhibitors were studied. The heme inhibitor KCN and the non-selective CYP inhibitor metyrapone both produced complete, concentration-dependent inhibition of 3HCIM binding (Ki = 1.3 mM and 11.9 μM, respectively). Binding was largely unaffected by inhibitors of CYPs 1A2, 2B6, 2C8, 2C9, 2D6, 2E1, and 19A1, but was eliminated by inhibitors of CYP2C19 (tranylcypromine) and CYP3A4 (ketoconazole). Synthesis and testing of CC11 (4(5)-(benzylthiomethyl)-1H-imidazole) and CC12 (4(5)-((4-iodobenzyl)-thiomethyl)-1H-imidazole) confirmed both drugs to be high affinity inhibitors of 3HCIM binding. On recombinant human CYPs, CC12 was a potent inhibitor of CYP2B6 (IC50 = 11.7 nM), CYP2C19 (51.4 nM), and CYP19A1 (140.7 nM), and had a range of activities (100 – 494 nM) on nine other isoforms. Although the 3HCIM binding site pharmacologically resembles some CYPs, eight recombinant human CYPs and three recombinant rat CYPs did not exhibit 3HCIM binding. Inhibition by KCN and metyrapone suggests that 3HCIM binds to a heme-containing brain protein (possibly a CYP). However, results with selective CYP inhibitors, recombinant CYPs, and a CYP antibody did not identify a 3HCIM-binding CYP isoform. Finally, CC12 is a new, potent inhibitor of CYP2B6 and CYP2C19 which may be a valuable tool for CYP research.
Introduction
Cimetidine (Fig. 1), a well known H2 receptor antagonist, is used to reduce gastric acid secretion in the treatment of various gastrointestinal disorders. In general, H2 antagonists are considered to be relatively safe drugs, with less than 3% of the population reporting side effects. Most of these are minor (Goodman, et al., 2001). In laboratory animals and/or man, however, cimetidine can produce significant additional effects including mental confusion, seizures, antinociception, and adverse drug interactions (Cannon, et al., 2004, Goodman, et al., 2001). The antinociceptive and seizure-producing effects of cimetidine are actions on the brain which are not mediated by the H2-receptor (Cannon, et al., 2004). Adverse drug interactions with cimetidine have been attributed to its well-known ability to act as a low-potency inhibitor of the CYP superfamily (see Sorkin and Darvey, 1983).
Figure 1.
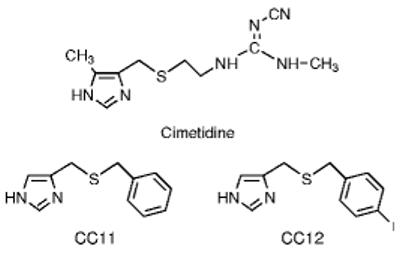
Chemical structures of cimetidine, CC11, and CC12.
Brain membranes were reported to exhibit high affinity binding of 3[H]-cimetidine (3HCIM) nearly 30 years ago (Burkard, 1978). Although early studies concluded that this binding represented brain H2 receptors, subsequent work showed this not to be true. For example, imidazole-containing H2 ligands have high affinity for the brain 3HCIM-binding site, but non-imidazole H2 ligands lack such activity (Warrander, et al., 1983;Rising, et al., 1980; Smith, et al., 1980). Furthermore, Warrander et al. (1983) reported the existence of imidazole-containing compounds with high affinity for the 3HCIM binding site, but lacking affinity for the H2 receptor. Thus, cimetidine has a high affinity, biological target in the brain which is distinct from the H2 receptor and remains to be identified. Liver, kidney and gastric mucosa have also been reported to contain a 3HCIM binding site (Reilly, et al., 1983;Warrander, et al., 1983;Chansel, et al., 1982;Rising, et al., 1980).
Since several of cimetidine’s side effects are not mediated by H2 receptors, the identity of the 3HCIM binding site is of biological interest. For example, we recently suggested that the 3HCIM binding site might represent an antinociceptive target. In support of this idea, CC12 (Fig. 1), a new derivative of cimetidine, showed high affinity for 3HCIM binding sites and behaved in vivo as a competitive antagonist of several antinociceptive agents (Hough, et al., 2007). Presently, we have characterized the pharmacological nature of 3HCIM binding in brain homogenates and tested the hypothesis that this binding site is a member of the CYP superfamily.
Methods
Materials
Thio-TEPA was purchased from Fisher Scientific (Atlanta, GA). Quercetin, DL-aminoglutethimide, ticlopidine, and diethyldithiocarbamate were purchased from Krackeler Scientific Inc. (Albany, NY). Cimetidine and fluvoxamine were purchased from Tocris Bioscience (Ellisville, MO). We thank Prof. R. Leurs (Leiden Amsterdam Center for Drug Design, Amsterdam) and Prof. M. Wentland (Rennselaer Polytechnic Institute, Troy, NY) for kindly providing samples of burimamide, and zolantidine, respectively. All other compounds were purchased from Sigma-Aldrich (St. Louis, MO).
Synthesis of CC11 and CC12
For synthesis of CC11, (1H-imidazol-4-yl)-methanol (3.44 grams, 0.035 moles) and benzyl mercaptan (1.25 grams, 0.010 moles) were dissolved in glacial acetic acid (12 ml) and heated at reflux under N2 for 4 days. The reaction was cooled to room temperature and acetic acid evaporated in vacuo. The residue was dissolved in dichloromethane and the pH adjusted to 9-10 with aqueous 10% sodium hydroxide solution. The dichloromethane layer was separated, the aqueous layer extracted twice with dichloromethane, the dichloromethane extracts combined, dried over MgSO4, filtered, and evaporated to give 2.24 grams of crude product, a brown viscous oil. Purification by flash silica chromatography using CH2Cl2/ Methanol (95:5) gave 1.5 grams of a colorless viscous oil which became a waxy solid upon standing. The free base (100 mg) was dissolved in 4 ml of 4N HCl in dioxane and stirred overnight at room temperature. Organic volatiles were evaporated in vacuo and dried on high vacuum for 1 hour at 50°C yielding CC11 (4(5)-(benzylthiomethyl)-1H-imidazole hydrochloride, Fig. 1). NMR (300 MHz, DMSO-d6) δ (9.05, s, 1H), (7.55,s, 1H), (7.37-7.20, m, 5H), (3.78, s, 4H). Synthesis of CC12 (4(5)-((4-iodobenzyl)thiomethyl)-1H-imidazole, Fig. 1) was performed as recently described (Hough, et al., 2007).
Tissue preparation
Sprague Dawley rats were euthanized with an overdose of pentobarbital or CO2, and brains were rapidly removed. In some cases frozen brains were purchased (Taconic Farms, Germantown, NY). Crude whole brain fractions were prepared by homogenizing (polytron) in 10 volumes of homogenate buffer (100 mM Tris-HCl, 0.5 mM EDTA, pH 7.4) and centrifugation (26,000 x g for 15 min). Supernatant fractions were removed and the pellets were resuspended in the same volume of buffer with a glass-teflon homogenizer. Resuspensions were recentrifuged, supernatants discarded, and pellets stored at -80°C. On the day of assay, pellets were resuspended in assay buffer (100 mM Tris-HCl, pH 7.4), centrifuged at 26,000 x g for 10 min, supernatants were discarded, pellets again resuspended in assay buffer and analyzed for 3HCIM-binding activity. For some experiments, whole brains were homogenized in sucrose buffer (250 mM sucrose, 50 mM Tris-HCl, 4 mM EDTA, pH 7.4) with a glass-teflon homogenizer and spun at 100,000 x g for 1 hr. Supernatants were discarded and pellets stored at -80°C. On the day of assay, pellets were re suspended in assay buffer and assayed for 3HCIM-binding activity.
3HCIM radioligand binding
3HCIM binding experiments were performed following Smith et al. (1980). Resuspended crude membrane pellets (360-470 μg of rat brain protein) or 2 pmol of recombinant CYPs (rCYPs, 5.4-14.3 μg of protein, Supersomes™, BD Biosciences, Woburn, MA) were incubated in a total volume of 0.1 ml containing 50 nM 3HCIM (22-25 Ci/mmol, Amersham Biosciences Corp., Piscataway, NJ), appropriate concentrations of competing ligand, and assay buffer for 60 min on ice, in polystyrene test tubes. Burimamide (30 μM) or cimetidine (10 μM) was used to evaluate non-specific binding. Following incubation, samples were rapidly filtered through GF/B filters and rinsed three times with 1.5 ml of ice-cold assay buffer. Filters were placed in 5 ml of Ecoscint scintillation fluid and counted in a scintillation counter. Protein content was determined using the bicinchoninic acid method (Pierce Chemical, Rockford, IL). For competition studies, percent specific binding was calculated using the following formula: (drug – non-specific)/(total – non-specific) x 100, where drug indicates the amount of binding in the presence of competing ligand. For antibody studies, anti-CYP2C8/9/18/19 antibody (0.1-0.5 μg of protein, clone 1-68-11, Calbiochem, La Jolla, CA) and rat brain homogenate (resuspended 100,000 x g pellet, 308 μg) were preincubated for 20 min at 37°C in a volume of 60 μl, chilled (5 min) and assayed as described for 3HCIM binding in a final volume of 0.1 ml.
Quantification of recombinant CYP activities
Activities of CYP2B6, CYP2C19, and CYP2C11 were quantified by measurement of 9-anthraldehyde oxidation, similar to the methods of Marini, et. al. (2003). Briefly, recombinant CYP2B6 (rCYP2B6, 2 pmol, 26 μg protein, BD Biosciences) was preincubated for 15 min at 37°C with 9-anthraldehyde (5 μM final) and inhibitors in 100 mM potassium phosphate buffer, pH 7.4 at a volume of 766 μl. The reaction was initiated by the addition of an NADPH-regenerating system (NADPH-RS, 0.4 mM NADP+, 0.85 mM glucose-6-phosphate, 0.1 U/ml glucose-6-phosphate dehydrogenase, 0.85 mM MgCl2, BD Biosciences) and incubated for 60 min at 37°C in a final volume of 1 ml. The reaction was stopped by the addition of NaOH (1 ml, 0.5 N). Ethyl acetate (4 ml) was then added, the mixture was shaken for 5 min, and centrifuged at 1,300 x g for 5 min. HCl (1 ml, 0.5 N) was added to an aliquot of the resulting aqueous phase (1.5 ml) and extracted with ethyl acetate as before. Fluorescence of this final organic phase was measured with a Perkin-Elmer spectrofluorimeter at 255 nm (excitation) and 458 nm (emission) wavelengths and 9-anthracene carboxylic acid levels calibrated from a standard curve. Measurement of rCYP2C19 (2 pmol, 9.4 μg of protein) activity was performed identically to that of rCYP2B6 except that the preincubation was at room temperature, 9-anthraldehyde concentration was 25 μM, NADPH-RS was approximately threefold higher in concentration, inhibitors were added at the start of the reaction, and incubation time was 45 min. Measurement of rCYP2C11 (0.5-2 pmol, 1.15-4.6 μg of protein) activity was performed identically to that of rCYP2B6, except that higher concentrations of substrate (9-anthraldehyde, 100 μM) and NADPH-RS (2-fold) were employed, no preincubation was performed, and incubation time was 45 min (see Figs. 7 and 8). Recombinant CYP19A1 activity was quantified by a high throughput screening method using dibenzylfluorescein (0.2 μM) as substrate, exactly as described by Stresser et al. (2000). Pilot CYP inhibitor studies (Table 2, not in brackets) were performed by commercial assays (MDS Pharma Services, Taipei, Taiwan, see www.mdsps.com).
Figure 7.

Lack of 3HCIM specific binding to rCYP2C11. Left: Sf9 microsomes expressing rCYP2C11 (0.5 or 2.0 pmol enzyme [1.35 or 5.4 μg protein, respectively] per tube, or nontransfected microsomes (control, 10 μg protein) were incubated with 3HCIM (50 nM) and filtered as in Figure 3. 3HCIM binding (left ordinate, mean ± SEM from triplicate determinations) is shown from a single experiment. Right: To ensure that CYP2C11 was properly filtered by the binding assay, CYP2C11 (50 pmol enzyme, 135 μg protein) or liver membranes (100,000 x g pellet, 387 μg) were incubated as in the left panel but in the absence of 3HCIM. Samples were filtered with either filter paper present or absent, followed by washing as described, or not filtered at all (100% controls). Filtrates or control samples (860 μl) were analyzed for CYP2C11 activity as described. Control enzyme activities of the unflitered CYP2C11 sample (13.5 μg protein) and the unfiltered liver microsome sample (38.7 μg protein) were 5.7 nmol/min-mg and 1.3 nmol/min-mg, respectively. Right ordinate shows percent of control enzyme activity detected (mean ± SEM) of 2-3 determinations from one experiment. These experiments show that rCYP2C11 is viable (right), is filtered in the radioligand-binding assay (right), but does not specifically bind 3HCIM (left).
Figure 8.

Effects of a CYP2C8/9/18/19 antibody on brain 3HCIM binding and recombinant CYP2C11 enzymatic activity. Open bar (3HCIM binding): Resuspended 100,000 x g pellets (308 μg protein) from rat brain were preincubated with the antibody ( μg IgM, ordinate) in 0.1M Tris-HCl, pH 7.4 for 20 min at 37°C in a volume of 60 μl. Following preincubation, 3HCIM, unlabelled cimetidine (to evaluate nonspecific binding) and buffer were added to a final volume of 100 μl and specific binding was measured as in Figure 3. Control 3HCIM binding activity (0 μg IgM) was 0.34 pmol/mg. Solid bars (2C11 activity): Recombinant CYP2C11-containing sf9 microsomes (2 pmol, 4.6 μg protein of CYP) in 0.1 M potassium phosphate buffer, pH 7.4 were preincubated for a total of 20 min in a final volume of 60 μl. Following preincubation, an NADPH-RS and buffer were added to a final volume of 1ml and the 9AA oxidation assay commenced as described. Control (0 μg IgM) CYP2C11 activity was 4.34 pmol / (min x mg protein). For both data sets, data points represent the mean fractional inhibition of activity ± SEM of triplicate determinations.
Table 2.
Inhibition of human CYP isoforms by CC12 and cimetidine.
| CYP Isoform |
Species | % Inh. at 200 nM a or CC12 IC50 (μM) b |
Cimetidine Ki (μM)c |
Cimetidine Reference c |
Tested for 3HCIM Binding?f |
|---|---|---|---|---|---|
| 2B6 | Human | 100%a [0.0117]d | --- | --- | Yes |
| 2C19 | Human | 0.051b [0.0514]d | 14 | (Cohen, et al., 2003) | Yes |
| 19A1 | Human | 88%a [0.1407]d | --- | --- | Yes |
| 3A5 | Human | 64%a | --- | --- | Yes |
| 2A6 | Human | 62%a | --- | --- | Yes |
| 1A2 | Human | 0.120b | 86 | (Martinez, et al., 1999) | No |
| 2C9 | Human | 0.128b | 140 | (Miners, et al., 1988) | Yes |
| 3A7 | Human | 57%a | --- | --- | No |
| 3A4 | Human | 0.217b | 82 | (Kerlan, et al., 1992) | No |
| 2E1 | Human | 41%a | ---e | --- | No |
| 2C8 | Human | 33%a | --- | --- | Yes |
| 2D6 | Human | 0.494b | 38 | (Madeira, et al., 2004) | No |
| 2C18 | Human | N.T. | --- | --- | Yes |
| 2C11 | Rat | N.T. | --- | --- | Yes |
| 2C6 | Rat | N.T. | --- | --- | Yes |
| 2B1 | Rat | N.T. | --- | --- | Yes |
Percent inhibition of enzyme activity in the presence of 200 nM CC12 in duplicate.
IC50 values were estimated by non-linear regression from pilot studies with three concentrations of CC12 in duplicate.
Ki values for cimetidine taken from the literature cited.
IC50 values in brackets are from Fig. 6.
CYP2E1 has also been reported to be inhibited by cimetidine, however a Ki value has not been reported (Rendic, 2002).
All enzymes tested lacked specific 3HCIM-binding activity. N.T., not tested.
Data analysis
Data analysis was performed with GraphPad Prism Software (San Diego, CA). Data from saturation curves were fit to a one-site rectangular hyperbola to estimate KD and Bmax. Inhibitors of 3HCIM binding were evaluated by fitting to sigmoidal dose-response curves with variable slopes to estimate IC50 values. The effects of CC12 on CYP activities were evaluated by fits to one-site competition curves. Ki values were calculated by use of the Cheng-Prusoff equation.
Results
Biochemical characterization of the 3HCIM-binding site
Saturation experiments with increasing concentrations of 3HCIM (1 to 600 nM) resulted in a concentration-dependent increase in specific binding (Fig. 2). Non-linear regression of the saturation curve yielded a Bmax of 0.941 ± 0.027 pmol/mg of protein and a KD of 66.7 ± 5.2 nM (Fig. 2). At 50 nM 3HCIM, non-specific binding accounted for 22.5 ± 0.7% of the total binding. Additional experiments confirmed that specific binding was linear with protein content, that incubation time allowed for equilibrium binding, and that boiling of the homogenate eliminated specific binding (data not shown). Similar to previously published reports, the H2 receptor antagonists ranitidine (Smith, et al., 1980) and zolantidine, did not inhibit 3HCIM binding at H2-receptor relevant concentrations (IC50s > 30 μM, also not shown).
Figure 2.

Saturation of the 3HCIM-binding site in the rat brain. Whole brain crude membrane homogenates (390 μg) were incubated in triplicate with varying concentrations of 3HCIM (abscissa) for 60 min, and then filtered as described. Non-specific binding was evaluated with 10 μM cimetidine. KD and Bmax values were estimated by non-linear regression. Inset: the same data are shown in Scatchard format. Examples of the mean total and non-specific binding, at 50 nM 3HCIM, were 5,633 cpms (0.24 pmol) and 1,588 cpms (0.07 pmol), respectively. Ordinate represents the mean ± SEM of triplicate determinations from a single experiment. Similar results were obtained from two other experiments.
Pharmacological characterization of the 3HCIM-binding site
Cimetidine is a well-documented low-potency CYP inhibitor (Sorkin and Darvey, 1983). To investigate whether the 3HCIM-binding site resembles a CYP-like protein, the effects of the non-selective CYP inhibitors metyrapone and cyanide were determined on 3HCIM binding (Fig. 3). Both drugs produced concentration-dependent inhibition (pIC50 values of -4.68 ± 0.04 [IC50 = 20.8 μM] and -2.65 ± 0.05 [IC50 = 2.2 mM], for metyrapone and cyanide, respectively).
Figure 3.

Inhibition of 3HCIM binding by metyrapone and KCN. Whole brain crude membrane homogenates (360-470 μg protein) were incubated in triplicate with 50 nM 3HCIM as in Figure 2 with increasing concentrations of metyrapone and KCN (abscissa). Non-specific binding was evaluated with 30 μM burimamide or 10 μM cimetidine, both giving the same results. Each sample was measured in triplicate, from two individual experiments (total of 6 replicates), with the ordinate representing the mean ± SEM. IC50 values were estimated by non-linear regression (variable slope) and are provided.
A series of selective inhibitors of various CYP isoforms were chosen to further characterize 3HCIM binding (see Table 1 for inhibitors, CYP targets and Ki values). Figure 4 shows the effects of these compounds on the 3HCIM-binding site. Fluvoxamine (an inhibitor of CYP1A2), thio-TEPA (CYP2B6), quercetin (CYP2C8) sulfaphenazole (CYP2C9), quinidine (CYP2D6), diethyldithiocarbamate (CYP2E1), and DL-aminoglutethimide (CYP19A1) all produced less than 50% inhibition of 3HCIM binding at 100 μM (Fig. 4), a concentration well above all of the reported CYP-inhibitory potencies (Table 1). In contrast, ketoconazole (CYP3A4) and tranylcypromine (CYP2C19) both completely displaced 3HCIM binding with pIC50s of 4.96 ± 0.15 (IC50 = 11.1 μM) and 4.72 ± 0.06 (IC50 = 18.9 μM), and Hill coefficients of -0.8 ± 0.2 and -0.8 ± 0.1, respectively (Fig. 4). The potency of ketoconazole as an inhibitor of 3HCIM binding (calculated Ki = 6.4 μM, Table 1) is greater than 300-fold lower than this drug’s published affinity as a CYP3A4 inhibitor (Ki = 0.02 μM, Table 1). Tranylcypromine, however, competed with 3HCIM binding at a concentration comparable to its potency as an inhibitor of CYP2C19 activity (Ki = 10.8 vs. 8 μM, respectively, Table 1). However, ticlopidine (another inhibitor of CYP2C19) had no effect on 3HCIM binding (Fig. 4).
Table 1.
Effects of selective CYP inhibitors on 3HCIM binding in brain homogenates. Compounds thought to be isoform-selective CYP inhibitors were screened for their activities on 3HCIM. For each inhibitor, the CYP targeted and the literature Ki value is given.
| CYP Inhibitor | CYP Isoform Inhibited |
Reported Ki (μM) |
Reference | Ki on 3HCIM Binding (μM) a |
|---|---|---|---|---|
| Thio-TEPA | 2B6 | 2.8 | (Turpeinen, et al., 2004) | >100 |
| Tranylcypromine | 2C19 | 8 | (Inaba, et al., 1985) | 10.8 |
| Ticlopidine | 2C19 | 0.18 | (Ko, et al., 2000) | >100 |
| DL-Aminogluthimide | 19A1 | 0.6 | (Foster, et al., 1985) | >100 |
| Fluvoxamine | 1A2 | 0.18 | (Brosen, et al., 1993) | >100 |
| Sulfaphenazole | 2C9 | 0.12 | (Miners, et al., 1988) | >100 |
| Ketoconazole | 3A4 | 0.02 | (Back and Tjia, 1991) | 6.4 |
| Diethyldithiocarbamate | 2E1 | 7 | (Baranova, et al., 2005) | >100 |
| Quercetin | 2C8 | ∼1-3 | (Dierks, et al., 2001) | >100 |
Ki values calculated from IC50 values in Figure 4.
Figure 4.

Inhibition of 3HCIM binding in rat brain homogenates by selective CYP inhibitors. Binding was performed and analyzed as in Figure 3. Each data point represents the mean ± SEM of a total of 5-6 determinations from two individual experiments. Estimated IC50 values are given for ketoconazole (Keto) and tranylcypromine (Tran).
High affinity ligands for 3HCIM-binding proteins
The search for high affinity ligands led to the study of the cimetidine analogs CC11 (Warrander, et al., 1983) and CC12 (Hough, et al., 2007, Fig. 1). In agreement with earlier results (Warrander, et al., 1983), CC11 produced complete, concentration-dependent inhibition of 3HCIM binding, yielding a pIC50 of 8.05 ± 0.03 (IC50 = 9.0 nM) and a Hill coefficient of -1.4 ± 0.1 (Fig. 5). CC12 was also shown to be a potent inhibitor of 3HCIM binding, producing a pIC50 of 7.78 ± 0.11 (IC50 = 16.5 nM) and a Hill coefficient of -1.1 ± 0.2 (Fig. 5).
Figure 5.

Inhibition of 3HCIM binding by CC11 and CC12. Experiments were performed and analyzed as in Figure 3. Each data point represents the mean ± SEM of a total of 8-9 (CC11) or 10-12 (CC12) determinations from 3-4 experiments. IC50 values for CC11 and CC12 are provided. CC12 data in this figure are re-drawn from Hough, et al (2007) to permit comparison with the CC11 results.
Because CC12 is a potent inhibitor of 3HCIM binding, the effects of CC12 on several human CYP isoforms were measured in pilot studies. Five CYP isoforms previously reported to be inhibited by cimetidine (Table 2) were evaluated. Preliminary results indicated that CC12 produced concentration-dependent inhibition of all five enzymes, with estimated IC50 values between 51 and 494 nM (Table 2).
Because of the unexpectedly wide range of activities of CC12, and increased availability of commercial CYP testing, CC12 was subjected to preliminary screening on seven additional CYP human isoforms (Table 2). A single concentration of CC12 (200 nM) inhibited the activities of these enzymes as well (33 to 100%). Thus, CC12 exhibited some affinity for all twelve CYP isoforms.
Due to the apparently high affinity of CC12 for CYP2B6, CYP2C19, and CYP19A1 in these preliminary studies, additional detailed experiments were performed to validate these findings. CC12 produced concentration-dependent inhibition of CYP2B6, 2C19, and 19A1 activities, yielding pIC50 values of 7.93 ± 0.08 (IC50 = 11.7 nM), 7.29 ± 0.09 (IC50 = 51.4 nM), and 6.85 ± 0.06 (IC 50 = 140.7 nM), respectively (Fig. 6).
Figure 6.

CC12 inhibition of CYP2B6, 2C19, and 19A1 activity. Sf9 microsomes expressing rCYP2B6 (2 pmol), 2C19 (2 pmol), or 19A1 (0.4 pmol) were assayed in the presence of varying concentrations of CC12 (abscissa) as described. Enzymatic activity (ordinate) is reported as the mean (± SEM) activity from three separate experiments, each determined in triplicate (or in one case duplicate). Basal activities (mean ± SEM, in the absence of competing ligand) were 1.362 ± 0.050, 0.296 ± 0.021, and 0.016 ± 0.001 nmol/(min x mg of protein) for CYP2B6, 2C19, and 19A1, respectively. Estimated IC50 values for CC12 on each enzyme are given.
3HCIM binding potential of recombinant candidate CYPs
Although CC12 binds to the 3HCIM-binding site and also inhibits the activities of CYPs 2B6, 2C19, and 19A1, and at low concentrations, only the selective CYP2C19 inhibitor, tranylcypromine, competed with 3HCIM-binding at relevant concentrations. Even though results with ticlopidine (another 2C19 inhibitor, Table 1) failed to substantiate the relevance of CYP2C19, rat homologs of the human CYP2C19 were sought. An NCBI BLAST search of the human 2C19 amino acid sequence was performed against the rat genome. This BLAST identified two rat proteins (CYP2C11 and CYP2C6) as homologues of human CYP2C19. Thus, experiments were performed to directly test the hypothesis that CYP2C11 or CYP2C6 accounts for the specific binding of 3HCIM in the rat brain.
When 3HCIM was incubated with sf9 microsomes containing rCYP2C11 (Fig 7, left), no specific binding was measured. Companion studies were performed to ensure that the recombinant enzyme was viable and was properly filtered in the radioligand binding assays (Fig. 7, right). To ensure that optimal binding conditions for the recombinant enzymes were met, variations in assay conditions, including adding higher enzyme amounts (4 pmol) or coincubating the enzyme (2-6 pmol/tube) with brain homogenate during radioligand binding assays, were employed, and had no effect on binding activity. Furthermore, preincubation of the enzyme with 3HCIM and an NADPH-RS failed to produce a reliable specific radioligand binding signal. Additional studies similar to those in Fig. 7 found that eight human CYPs (2C19, 2A6, 2B6, 2C8, 2C9, 2C18, 3A5, and 19A1) and two other rat CYPs (2C6 and 2B1, rat homologues of CYP2C19 and CYP2B6, respectively) did not exhibit specific binding of 3HCIM (data not shown, see Table 2).
A CYP2C8/9/18/19 antibody was also employed to further evaluate the similarities between 3HCIM binding in the rat brain and CYP2C19. As shown (Fig. 8), antibody treatment caused concentration-dependent inhibition of rCYP2C11 activity, but had no effect on rat brain 3HCIM binding.
Discussion
Presently we have confirmed that 3HCIM specifically binds to rat brain homogenates and have found that this binding is heat-labile, suggesting it is proteinaceous in nature. The dissociation constant of the binding (KD = 67 nM, Fig. 2) is similar to several previously reported values (42 nM, Burkard, 1978, 52.2 nM, Rising, et al., 1980, and 40 nM, Devoto, et al., 1980). Additional KD values from 15.7 nM to 400 nM (as reviewed in Warrander, et al., 1983) have been reported. As discussed by Warrander et al. (1983), variations in assay conditions (including incubation temperature and time, buffering conditions, concentrations of radioligand studied, type of drug added to evaluate non-specific binding, and species) may contribute to this variability. Such variations in conditions may also affect the measurement of receptor density. The Bmax value currently reported (0.94 pmol/mg of protein, Fig. 2) is consistent with several other laboratory results (0.68, Rising, et al., 1980 and 1.3 pmol/mg, calculated from Burkard, 1978). However, other reported values vary from 0.23 to 3.9 pmol/mg (see Warrander et al., 1983). As compared with many receptors for biogenic amines in the brain, this density is high, yet higher levels have been found for the cannabinoid CB1 receptor in the brain (1.85 pmol/mg, Devane, et al., 1988). The high density of 3HCIM-binding sites (much greater than H2 receptor expression levels, Gajtkowski, et al., 1983), reinforces the conclusion that these sites are not the H2 receptor. The present findings, showing that pharmacologically active concentrations of ranitidine and zolantidine (both non-imidazole-containing H2 receptor antagonists) fail to inhibit 3HCIM binding, are similar to earlier results (Warrander, et al., 1983;Gajtkowski, et al., 1983;Chansel, et al., 1982;Smith, et al., 1980), further supporting the conclusion that the binding is not to the histamine H2 receptor. Furthermore, other histamine receptors or histamine binding sites seem to be excluded by the finding that very high concentrations of histamine are required to inhibit 3HCIM binding (IC50 values were 0.8 mM in unpublished data from this lab, and >1 mM from Smith et al., 1980).
Cimetidine is a well-known inhibitor of several human CYP isoforms, although affinities are low (Ki values = 14-140 μM, Table 1). These Ki values are much higher than cimetidine’s dissociation constant for the 3HCIM-binding site in the rat brain (KD = 67 nM, Fig. 2). Despite this discrepancy, it seems possible that 3HCIM could bind to a CYP at concentrations lower than those required to inhibit enzyme function. The Ki values for cimetidine on human CYPs are also much higher than therapeutic plasma levels of cimetidine, even though these levels can modify drug metabolism (Madeira, et al., 2004). This suggests the possibility that cimetidine may bind to additional CYP isoforms.
The existence of several cimetidine-CYP interactions seems to support the hypothesis that the brain 3HCIM-binding site is a CYP, even though this has never been tested. Inhibition of the binding by cyanide and metyrapone (Fig. 3, discussed further below), is also consistent with (but not proof of) a CYP-binding mechanism. Because of the lack of selectivity of these inhibitors, and the multiplicity of CYP isoforms (currently 88 CYP isoforms are known in the rat, Nelson, 2007), two additional approaches were taken to identify candidate 3HCIM-binding CYP isoforms. The first approach was to study the effects of selective CYP inhibitors on 3HCIM binding (Fig. 4, Table 1). By initially focusing on CYP isoforms known to be inhibited by cimetidine (Table 1), it was predicted that if the 3HCIM-binding site resembles any of these CYP isoforms, then selective inhibitors of one or more of these enzymes should compete with 3HCIM binding at concentrations equivalent to (or less than) those reported to inhibit CYP activities. The findings that pharmacologically active concentrations of fluvoxamine, sulconazole, quinidine, and diethyldithiocarbamate all failed to inhibit 3HCIM binding (Fig. 4, Table 1) suggest that the 3HCIM-binding site does not resemble the human CYPs 1A2, 2C9, 2D6, or 2E1, respectively. Moreover, the binding site does not resemble CYP3A4, as ketoconazole’s 3HCIM-binding constant (Ki = 6.4 μM, Fig. 4, Table 1) is much larger than the comparable value on CYP3A4 enzyme activity (Ki = 0.02 μM). In contrast, tranylcypromine’s affinity for the binding site (Ki = 10.8 μM) is virtually identical with its activity at CYP2C19 (Ki = 8 μM, Fig. 4, Table 1), suggesting that the 3HCIM-binding site may resemble a CYP2C19-like protein. This similarity was not sustained by results with other CYP2C19 inhibitors, since ticlopidine was inactive on the binding site (Fig. 4, Table 1). Because ticlopidine has been suggested to be a mechanism-based suicide inhibitor of CYP2C19 (Ha-Duong et al., 2001), additional experiments (i.e. pre-incubation of brain membranes with ticlopidine at 37°C with NADPH-RS) ensured that this drug did not require metabolic activation in order to inhibit 3HCIM binding (data not shown).
Since pharmacological screening failed to clearly implicate a 3HCIM-binding candidate CYP, other targets for tranylcypromine and ketoconazole were considered. Although these drugs are widely used as isoform-selective inhibitors (e.g. Erickson, et al., 1999;Chauret, et al., 1997), both have documented actions at additional CYP isoforms. In addition to its activity on CYP2C19, tranylcypromine has been reported to inhibit CYPs 2B6 and 19A1 (Stresser, 2004), but failure of thio-TEPA (an inhibitor of 2B6) and DL-aminoglutethimide (an inhibitor of 19A1) to displace 3HCIM binding (Fig. 4, Table 1) indicates that the binding site does not resemble these enzymes. Tranylcypromine is also a potent inhibitor of CYP2A6 (IC50 = 0.05 μM, Draper, et al., 1997), but the binding site seems to not resemble this isoform because tranylcypromine’s affinity for 3HCIM binding is much lower (IC50 = 10.8 μM, Table 1). Ketoconazole is a well-known inhibitor of CYP3A4, but this drug has also been reported to inhibit CYP2C8 at concentrations (IC50 = 4.4 μM, (Stresser, 2004)) similar to its observed affinity for 3HCIM binding (IC50 = 11.1 μM, Fig. 4, Table 1), suggesting the possible relevance of this isoform. However, the inability of quercetin (another 2C8 inhibitor) to influence 3HCIM binding (Fig. 4, Table 1) seems to exclude a role for CYP2C8-like activity. Thus, despite these documented additional actions of tranylcypromine and ketoconazole, no clear 3HCIM-binding CYP candidate emerged from the pharmacological screening.
High affinity ligands were synthesized and tested on 3HCIM binding and CYP activities as a second approach towards identifying 3HCIM-binding proteins. Warrander, et al. (1983) showed that the 3HCIM-binding site was not the H2 receptor by demonstrating a series of cimetidine analogs which competed with 3HCIM binding, but which lacked H2-receptor activity. One compound (herein named CC11) from that study had high affinity for the binding site (Ki = 5 nM, Warrander, et al., 1983), but the synthesis, rationale, and possible relevance to CYPs were not given. CC11 was synthesized and tested presently to confirm this drug’s high affinity for 3HCIM-binding (Ki = 5.2 nM, Fig. 5). Although not mentioned by Warrander et al. (1983), earlier literature reported CC11 to be a weak inhibitor of aldrin epoxidation (a CYP-mediated reaction) in rat liver microsomes (Wilkinson, et al., 1972).
CC12 (the para-iodo derivative of CC11, Fig. 1) was described for the first time in 2007 (Hough, et al., 2007), and the activity of this drug on CYP enzymes has not been previously studied. Originally synthesized to investigate the biological relevance of 3HCIM binding, the compound is a potent ligand for the binding site (Ki = 9.5 nM, as in Fig. 5) and also behaves in vivo as an antagonist of several analgesic drugs including morphine and the cimetidine derivative improgan (Hough et al., 2007). The mechanisms accounting for this latter activity are not known (Hough, et al., 2007). CC12 was found to be a moderately potent H3 antagonist (Ki = 50 nM), but the drug lacks activity in vitro on a variety of other CNS receptors (Hough, et al., 2007). Chemically, CC12 belongs to the family of phenyl-imidazole CYP inhibitors which commonly interact with the heme moiety of CYP, yielding a “type II” difference spectrum (Correia and Ortiz de Montellano, 2005). It is remarkable that within this family, the nature of the aromatic heteroatom and the structure of the side chain still permit a wide variety of potencies and selectivities for various isoforms. Drugs from this class include metyrapone, ketoconazole and letrozole (Correia and Ortiz de Montellano, 2005). However, even when compared with many of these other compounds, CC12 is a very high potency CYP inhibitor (on CYP2B6, IC50 = 11.7 nM, equivalent to Ki = to 5.4 nM, see Stresser et al., 2000 for Km value). Other compounds reported to act on CYP2B isoforms include miconazole (Ki = 50 nM, Zhang, et al., 2002), sulconazole (Ki = 40 nM, Zhang, et al., 2002), and 4-(4-chlorophenyl)imidazole (IC50 = 70 nM, Spatzenegger, et al., 2001). Additional studies are needed to assess the potency, selectivity and utility of CC12 as a tool for CYP research.
Because CC12 has high affinity for the 3HCIM-binding site (Ki = 9.5 nM, Fig. 5), this drug should be a potent inhibitor of any CYP isoform capable of 3HCIM-binding. Although CC12 showed inhibitory activity on a total of twelve CYPs (estimated IC50 values = 11.7-494 nM, Table 2, Fig. 6), this drug showed high affinity for only three isoforms: CYP2B6, 2C19, and 19A1 (IC50 values = 11.7, 51.4, and 140.7 nM, respectively, Fig. 6). The lack of 3HCIM-binding affinity by inhibitors of CYP2B6 and 19A1 (Fig. 4, Table 1) suggests little relevance of these isoforms to the binding site. However, two inhibitors of CYP2C19 (CC12 and tranylcypromine) also inhibited 3HCIM binding, whereas another inhibitor (ticlopidine) did not, suggesting the possible significance of CYP2C19-like proteins.
The lack of 3HCIM-binding activity in rCYP2C11 or rCYP2C6 indicates that these proteins (which are highly homologous to human CYP2C19) do not account for 3HCIM-binding in the rat brain. Furthermore, the findings that rCYP2A6, 2B6, 2C8, 2C9, 2C18, 2C19, 3A5, and19A1, as well as the 2B6 rat homologue, rCYP2B1, all failed to bind 3HCIM (Table 2) suggest these enzymes do not resemble 3HCIM-binding proteins.
The conclusion that the rat brain 3HCIM-binding site does not resemble any of the CYPs presently tested assumes that these methods would have detected 3HCIM binding to the rCYPs. Calculations show that the amount of enzymes added to the binding assay (2 pmol) should have yielded a specific binding signal of 0.85 pmol (43% of the CYPs should have bound according to a KD of 67 nM, Fig. 2). Such a result would have been easily detectable, since it corresponds to a signal approximately twice that of the Bmax value for rat brain homogenate (Fig. 2). It was also necessary to show that the CYP proteins, supplied in insect microsomal fractions, could be filtered in the radioligand binding assay. This was confirmed by the lack of enzyme activity in the filtrate of the binding assay (Fig. 7, right).
The conclusion that the brain binding site does not resemble any of the currently-tested CYPs could also depend on specific differences in rat brain and insect cell preparations. For example, the recombinant enzyme systems currently studied are coexpressed with high levels of cytochrome P450 reductase and cytochrome b5. If these proteins prevented 3HCIM binding, then coincubation of the insect microsomes with brain homogenates should have decreased 3HCIM binding in the homogenates. Alternatively, if the recombinant enzymes (in the presence of an NADPH-RS) did not contain the necessary components and/or cofactors to bind 3HCIM (as might be present in brain homogenates), then coincubation of the homogenate with the recombinant enzymes should have increased binding activity. Since neither of these results was obtained, it is concluded that differences in the brain and insect cell preparations are not relevant.
Other potential differences between the rat brain homogenate and the rCYPs that might affect 3HCIM binding include post-translational modifications and potential alleleic variations of CYPs. Although the potential significance of these differences is difficult to assess, the present use of a CYP2C8/9/18/19 antibody known to inhibit CYP2C11 activity (Park, et al., 1989) and that is immunoreactive with 2C6 (Waxman, et al., 1987) provided another approach. The finding that this antibody (at concentrations which inhibited rCYP2C11 activity, Fig. 8) did not alter 3HCIM binding activity in rat brain fractions reinforces the conclusion that the 3HCIM-binding site is not CYP2C6 or 2C11.
Cimetidine’s potency as a CYP inhibitor has been reported to be increased following the drug’s preincubation with rat liver microsomes and an NADPH-RS (Levine, et al., 1998). Under these conditions, it has been suggested that cimetidine’s higher potency can be explained by the formation of a metabolite-intermediate complex with CYP isoforms (Levine, et al., 1998). If this hypothesis is correct, then one reason why 3HCIM binding to rCYPs was not detected could be that conditions for forming this complex were present in rat brain homogenates, but not in insect cell microsomes. Accordingly, mimicking these preincubation conditions should have resulted in an increase in 3HCIM binding of rat homogenates, thereby permitting the detection of specific 3HCIM binding to rCYPs. The finding that the NADPH-RS preincubation had no effect on 3HCIM binding suggests that metabolic activation is not required for 3HCIM binding in either the brain or in the recombinant systems.
The present results show that the 3HCIM binding site in rat brain is not CYP2C11, 2C6 or 2B1, but other CYP isoforms, and even non-CYP proteins remain to be tested. The present search for 3HCIM-binding CYPs in the rat has depended heavily on testing the commercially-available human isoforms. Rat CYPs with high (>70%) homology to CYP2C19 (CYP2C6 and 2C11) lacked 3HCIM-binding activity. However, three additional rat CYPs (2C7, 2C24, and 2C79) also have high homologies to CYP2C19 (Nelson, 2007) and remain viable candidates for 3HCIM-binding activity. None of these are commercially available, and one of these (2C79) has only been described in silico. It is not known if the CYP2C8/9/18/19 antibody used presently (Fig. 8) cross-reacts with these additional 2C isoforms. These isoforms remain to be cloned and tested for 3HCIM binding activity.
Another limitation of the present study rests on the assumption that rat CYPs with high homologies to a human CYP will share ligand binding profiles. However, CYPs are considered to be rather promiscuous, in that multiple enzymes from different families and subfamilies, can have overlapping substrate/inhibitor profiles (Rendic, 2002). For example, mutations of only three amino acid residues can alter CYP2C19 substrate selectivity to more closely resemble that of CYP2C9 (Jung, et al., 1998). Thus, it is possible that the only homologies relevant to 3HCIM and CC12 binding are those in the substrate and inhibitor recognition sites. Subtle rat-human differences might also explain why the rat binding protein shows 2C19-like activity with some, but not all 2C19 inhibitors. Because crystal structures and homology models of several CYPs with inhibitors have been described (Otyepka, et al., 2007), it may be possible to employ this approach in future searches for 3HCIM-binding CYPs.
The classic demonstration of the existence of a CYP in tissues or of a drug-CYP interaction is the carbon monoxide (CO)-difference spectrum, in which dithionite-reduced microsomes combine with CO to yield the characteristic 450 nm spectrum (Guengerich, 1991). Thus, the ideal experiment to demonstrate high affinity binding of cimetidine to brain CYPs would employ cimetidine-induced alterations in the CO-difference spectra from brain. Unfortunately, brain CYPs are not in high enough abundance to yield a reproducible CO-difference spectrum (X. Ding, personal communication).
If the 3HCIM-binding protein in brain is not a CYP, then inhibition of this binding by cyanide (Fig. 3) suggests the importance of other heme-containing proteins. Cyanide is a well-known inhibitor of CYPs with affinities similar to values found presently (IC50 values = 1-2 mM, Kitada, et al., 1977). However, this reagent also inactivates other iron-containing (especially heme-containing) non-CYP proteins (Dixon and Webb, 1964). A query of the rat proteome (UniProt Knowledgebase) using the Integr8 Web Portal (http://www.ebi.ac.uk/integr8/EBI-Integr8-HomePage.do) with the gene ontology parameter “heme-binding” (GO:0020037) returns 115 unique heme-binding proteins. Approximately one-half of these are CYPs. Notable among the remaining half are several electron transport proteins (e.g. cytochrome-related), oxygen transporters (hemoglobin, myoglobin, cytoglobin, neuroglobin), and other oxidases and peroxidases. The list also includes specialized enyzmes such as soluble guanylate cyclase, and three forms of nitric oxide synthase (NOS). Some of these hemoproteins can be excluded as potential 3HCIM binding targets because they are soluble, and would not have been present in the membrane fractions used presently. Two other metalloenzymes (e-NOS and n-NOS) are unlikely to bind 3HCIM because CC12 lacked activity on these enzymes (Hough et al. 2007). In contrast to the case with cyanide, a literature review of metyrapone shows that its experimental use has been limited to CYP inhibition (e.g. Masubuchi, et al., 1998;Ignarro, et al., 1985). Thus, inhibition of 3HCIM binding by metyrapone suggests that the binding site may be a rat CYP isoform which was not presently tested. Recently we used the high affinity of CO for heme in unpublished pilot studies and found that 3HCIM binding in brain homogenates was reduced by approximately 30% following CO treatment under reducing (dithionite) conditions. These findings, along with cyanide results, suggest the relevance of heme-containing proteins (CYP or non-CYP).
The present studies suggest that the 3HCIM binding site in brain is a heme-containing protein, but its identity as a CYP could not be confirmed. This protein needs to be identified for several reasons, including the biological activity of inhibitors of this binding (e.g. CC12), the pharmacological activities of cimetidine which are not mediated by the H2 receptor, and the well-known (but poorly understood) nature of cimetidine-CYP drug interactions. Furthermore, cimetidine’s affinity for this binding site is more than 10-fold higher than the affinity for any other known protein (including the H2 receptor, see Gajtkowski, et al., 1983), arguing for its potential importance. Several approaches may be needed to identify this site, including cloning and testing of additional rat CYP isoforms and non-CYP hemoproteins, modeling ligand-CYP active sites, or protein purification.
Acknowledgements
We thank Amanda B. Carpenter and Konstantina Svokos for excellent technical assistance. We also thank Dr. Melissa VanAlstine for discussions and pilot results. Dr. Mark Fleck and Dr. Xinxin Ding provided helpful comments on the manuscript and the project.
Supported by grants (DA-03816, DA-07307) from the National Institute on Drug Abuse.
List of non-standard abbreviations
- CYP
cytochrome P450
- rCYP
recombinant cytochrome P450
- CC11
4(5)-(benzylthiomethyl)-1H-imidazole hydrochloride
- CC12
4(5)-((4-iodobenzyl)thiomethyl)-1H-imidazole
- 3HCIM
[3H]-cimetidine
- NADPH-RS
NADPH regenerating system
References
- Back DJ, Tjia JF. Comparative effects of the antimycotic drugs ketoconazole, fluconazole, itraconazole and terbinafine on the metabolism of cyclosporin by human liver microsomes. Br J Clin Pharmacol. 1991;32:624–626. doi: 10.1111/j.1365-2125.1991.tb03963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranova J, Anzenbacherova E, Anzenbacher P, Soucek P. Minipig cytochrome P450 2E1: comparison with human enzyme. Drug Metab Dispos. 2005;33:862–865. doi: 10.1124/dmd.104.003392. [DOI] [PubMed] [Google Scholar]
- Brosen K, Skjelbo E, Rasmussen BB, Poulsen HE, Loft S. Fluvoxamine is a potent inhibitor of cytochrome P4501A2. Biochem Pharmacol. 1993;45:1211–1214. doi: 10.1016/0006-2952(93)90272-x. [DOI] [PubMed] [Google Scholar]
- Burkard WP. Histamine H2-receptor binding with 3H-cimetidine in brain. Eur J Pharmacol. 1978;50:449–450. doi: 10.1016/0014-2999(78)90153-x. [DOI] [PubMed] [Google Scholar]
- Cannon KE, Fleck MW, Hough LB. Effects of cimetidine-like drugs on recombinant GABAA receptors. Life Sci. 2004;75:2551–2558. doi: 10.1016/j.lfs.2004.05.020. [DOI] [PubMed] [Google Scholar]
- Chansel D, Oudinet JP, Nivez MP, Ardaillou R. Histamine H2 receptors in rat renal glomeruli. Biochem Pharmacol. 1982;31:367–375. doi: 10.1016/0006-2952(82)90184-8. [DOI] [PubMed] [Google Scholar]
- Chauret N, Gauthier A, Martin J, Nicoll-Griffith DA. In vitro comparison of cytochrome P450-mediated metabolic activities in human, dog, cat, and horse. Drug Metab Dispos. 1997;25:1130–1136. [PubMed] [Google Scholar]
- Cohen LH, Remley MJ, Raunig D, Vaz AD. In vitro drug interactions of cytochrome P450: an evaluation of fluorogenic to conventional substrates. Drug Metab Dispos. 2003;31:1005–1015. doi: 10.1124/dmd.31.8.1005. [DOI] [PubMed] [Google Scholar]
- Correia MA, de Montellano Ortiz PR. Inhibition of Cytochrome P450 Enzymes. In: de Montellano Ortiz PR., editor. Cytochrome P450 Structure, Mechanism, and Biochemistry. Kluwer Academic / Plenum Publishers; New York: 2005. pp. 247–322. [Google Scholar]
- Devane WA, Dysarz FA, III, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- Devoto P, Marchisio AM, Carboni E, Spano PF. Detection of 3H-cimetidine specific binding in rat anterior pituitary. Eur J Pharmacol. 1980;63:91–93. doi: 10.1016/0014-2999(80)90121-1. [DOI] [PubMed] [Google Scholar]
- Dierks EA, Stams KR, Lim HK, Cornelius G, Zhang H, Ball SE. A method for the simultaneous evaluation of the activities of seven major human drug-metabolizing cytochrome P450s using an in vitro cocktail of probe substrates and fast gradient liquid chromatography tandem mass spectrometry. Drug Metab Dispos. 2001;29:23–29. [PubMed] [Google Scholar]
- Dixon M, Webb EC. Enzymes. Academic Press Inc.; New York: 1964. Enzyme inhibitors; pp. 337–341. [Google Scholar]
- Draper AJ, Madan A, Parkinson A. Inhibition of coumarin 7-hydroxylase activity in. 1997. [DOI] [PubMed]
- Erickson DA, Mather G, Trager WF, Levy RH, Keirns JJ. Characterization of the in vitro biotransformation of the HIV-1 reverse transcriptase inhibitor nevirapine by human hepatic cytochromes P-450. Drug Metab Dispos. 1999;27:1488–1495. [PubMed] [Google Scholar]
- Foster AB, Jarman M, Leung CS, Rowlands MG, Taylor GN, Plevey RG, Sampson P. Analogues of aminoglutethimide: selective inhibition of aromatase. J Med Chem. 1985;28:200–204. doi: 10.1021/jm00380a009. [DOI] [PubMed] [Google Scholar]
- Gajtkowski GA, Norris DB, Rising TJ, Wood TP. Specific binding of 3H-tiotidine to histamine H2 receptors in guinea pig cerebral cortex. Nature. 1983;304:65–67. doi: 10.1038/304065a0. [DOI] [PubMed] [Google Scholar]
- Goodman LS, Gilman AG, Hardman JG, Limbird LE. Agents used for control of gastric acidity and treatment of peptic ulcers. In: Hardman JG, Limbird LE, Gilman AG, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. McGraw-Hill; New York: 2001. pp. 1005–1020. [Google Scholar]
- Guengerich FP. Reactions and significance of cytochrome P-450 enzymes. JBC. 1991;266:10019–10022. [PubMed] [Google Scholar]
- Ha-Duong NT, Dijols S, Macherey A-C, Goldstein JA, Dansette PM, Mansuy D. Ticlopidine as a selective mechanism-based inhibitor of human cytochrome P450 2C19. Biochemistry. 2001;40:12112–12122. doi: 10.1021/bi010254c. [DOI] [PubMed] [Google Scholar]
- Hough LB, Nalwalk JW, Phillips JG, Kern B, Shan Z, Wentland MP, de E,I, Janssen E, Barr T, Stadel R. CC12, a high-affinity ligand for [3H]cimetidine binding, is an improgan antagonist. Neuropharmacology. 2007;52:1244–1255. doi: 10.1016/j.neuropharm.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro LJ, Harbison RG, Wood KS, Wolin MS, McNamara DB, Hyman AL, Kadowitz PJ. Differences in responsiveness of intrapulmonary artery and vein to arachidonic acid: mechanism of arterial relaxation involves cyclic guanosine 3′:5′-monophosphate and cyclic adenosine 3′:5′-monophosphate. J Pharmacol Exp Ther. 1985;233:560–569. [PubMed] [Google Scholar]
- Inaba T, Jurima M, Mahon WA, Kalow W. In vitro inhibition studies of two isozymes of human liver cytochrome P-450. Mephenytoin p-hydroxylase and sparteine monooxygenase. Drug Metab Dispos. 1985;13:443–448. [PubMed] [Google Scholar]
- Jung F, Griffin KJ, Song W, Richardson TH, Yang M, Johnson EF. Identification of amino acid substitutions that confer a high affinity for sulfaphenazole binding and a high catalytic efficiency for warfarin metabolism to P450 2C19. Biochemistry. 1998;37:16270–16279. doi: 10.1021/bi981704c. [DOI] [PubMed] [Google Scholar]
- Kerlan V, Dreano Y, Bercovici JP, Beaune PH, Floch HH, Berthou F. Nature of cytochromes P450 involved in the 2-/4-hydroxylations of estradiol in human liver microsomes. Biochem Pharmacol. 1992;44:1745–1756. doi: 10.1016/0006-2952(92)90068-t. [DOI] [PubMed] [Google Scholar]
- Kitada M, Chiba K, Kamataki T, Kitagawa H. Inhibition by cyanide of drug oxidations in rat liver microsomes. Jpn J Pharmacol. 1977;27:601–608. doi: 10.1254/jjp.27.601. [DOI] [PubMed] [Google Scholar]
- Ko JW, Desta Z, Soukhova NV, Tracy T, Flockhart DA. In vitro inhibition of the cytochrome P450 (CYP450) system by the antiplatelet drug ticlopidine: potent effect on CYP2C19 and CYP2D6. Br J Clin Pharmacol. 2000;40:343–351. doi: 10.1046/j.1365-2125.2000.00175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Law EY, Bandiera SM, Chang TK, Bellward GD. In vivo cimetidine inhibits hepatic CYP2C6 and CYP2C11 but not CYP1A1 in adult male rats. J Pharmacol Exp Ther. 1998;284:493–499. [PubMed] [Google Scholar]
- Madeira M, Levine M, Chang TK, Mirfazaelian A, Bellward GD. The effect of cimetidine on dextromethorphan O-demethylase activity of human liver microsomes and recombinant CYP2D6. Drug Metab Dispos. 2004;32:460–467. doi: 10.1124/dmd.32.4.460. [DOI] [PubMed] [Google Scholar]
- Marini S, Grasso E, Longo V, Puccini P, Riccardi B, Gervasi PG. 4-Biphenylaldehyde and 9-anthraldehyde: two fluorescent substrates for determining P450 enzyme activities in rat and human. Xenobiotica. 2003;33:1–11. doi: 10.1080/0049825021000017894. [DOI] [PubMed] [Google Scholar]
- Martinez C, Albet C, Agundez JA, Herrero E, Carrillo JA, Marquez M, Benitez J, Ortiz JA. Comparative in vitro and in vivo inhibition of cytochrome P450 CYP1A2, CYP2D6, and CYP3A by H2-receptor antagonists. Clin Pharmacol Ther. 1999;65:369–376. doi: 10.1016/S0009-9236(99)70129-3. [DOI] [PubMed] [Google Scholar]
- Masubuchi Y, Saito H, Horie T. Structural requirements for the hepatotoxicity of nonsteroidal anti-inflammatory drugs in isolated rat hepatocytes. J Pharmacol Exp Ther. 1998;287:208–213. [PubMed] [Google Scholar]
- Miners JO, Smith KJ, Robson RA, McManus ME, Veronese ME, Birkett DJ. Tolbutamide hydroxylation by human liver microsomes. Kinetic characterisation and relationship to other cytochrome P-450 dependent xenobiotic oxidations. Biochem Pharmacol. 1988;37:1137–1144. doi: 10.1016/0006-2952(88)90522-9. [DOI] [PubMed] [Google Scholar]
- Nelson DR. The cytochrome P450 homepage. [4-27-2007]; doi: 10.1186/1479-7364-4-1-59. http://drnelson.utmem.edu/CytochromeP450.html [DOI] [PMC free article] [PubMed]
- Otyepka M, Skopalik J, Anzenbacherova E, Anzenacher P. What common structural features and variations of mammalian P450s are known to date? Biochim Biophys Acta. 2007;1770:376–389. doi: 10.1016/j.bbagen.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Park SS, Waxman DJ, Lapenson DP, Schenkman JB, Gelboin HV. Monoclonal antibodies to rat liver cytochrome P-450 2c/RLM5 that regiospecifically inhibit steroid metabolism. Biochem Pharmacol. 1989;38:3067–3074. doi: 10.1016/0006-2952(89)90017-8. [DOI] [PubMed] [Google Scholar]
- Reilly PE, Carrington LE, Winzor DJ. The interaction of cimetidine with rat liver microsomes. Biochem Pharmacol. 1983;32:831–835. doi: 10.1016/0006-2952(83)90584-1. [DOI] [PubMed] [Google Scholar]
- Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34:83–448. doi: 10.1081/dmr-120001392. [DOI] [PubMed] [Google Scholar]
- Rising TJ, Norris DB, Warrander SE, Wood TP. High affinity 3H-cimetidine binding in guinea-pig tissues. Life Sci. 1980;27:199–206. doi: 10.1016/0024-3205(80)90138-1. [DOI] [PubMed] [Google Scholar]
- Smith IR, Cleverley MT, Ganellin CR, Metters KM. Binding of [3H]cimetidine to rat brain tissue. Agents Actions. 1980;10:422–426. doi: 10.1007/BF01968040. [DOI] [PubMed] [Google Scholar]
- Sorkin EM, Darvey DL. Review of cimetidine drug interactions. Drug Intell Clin Pharm. 1983;17:110–120. doi: 10.1177/106002808301700205. [DOI] [PubMed] [Google Scholar]
- Spatzenegger M, Wang Q, He YQ, Wester MR, Johnson EF, Halpert JR. Amino acid residues critical for differential inhibition of CYP2B4, CYP2B5, and CYP2B1 by phenylimidazoles. Mol Pharmacol. 2001;59:475–484. doi: 10.1124/mol.59.3.475. [DOI] [PubMed] [Google Scholar]
- Stresser DM. High-throughput screening of human cytochrome P450 inhibitors using fluorometric substrates. In: Yan Z, Caldwell G, editors. Optimization in Drug Discovery. Humana Press Inc.; Totowa, NJ: 2004. pp. 215–229. [Google Scholar]
- Stresser DM, Turner SD, McNamara J, Stocker P, Miller VP, Crespi CL, Patten CJ. A high-throughput screen to identify inhibitors of aromatase (CYP19) Anal Biochem. 2000;284:427–430. doi: 10.1006/abio.2000.4729. [DOI] [PubMed] [Google Scholar]
- Turpeinen M, Nieminen R, Juntunen T, Taavitsainen P, Raunio H, Pelkonen O. Selective inhibition of CYP2B6-catalyzed bupropion hydroxylation in human liver microsomes in vitro. Drug Metab Dispos. 2004;32:626–631. doi: 10.1124/dmd.32.6.626. [DOI] [PubMed] [Google Scholar]
- Warrander SE, Norris DB, Rising RJ, Wood TP. 3H-Cimetidine and the H2-receptor. Life Sci. 1983;33:1119–1126. doi: 10.1016/0024-3205(83)90015-2. [DOI] [PubMed] [Google Scholar]
- Waxman DJ, Lapenson DP, Park SS, Attisano C, Gelboin HV. Monoclonal antibodies inhibitory to rat hepatic cytochromes P-450: P-450 form specificities and use as probes for cytochrome P-450-dependent steroid hydroxylations. Mol Pharmacol. 1987;32:615–624. [PubMed] [Google Scholar]
- Wilkinson CF, Hetnarski K, Yellin TO. Imidazole derivatives--a new class of microsomal enzyme inhibitors. Biochem Pharmacol. 1972;21:3187–3192. doi: 10.1016/0006-2952(72)90147-5. [DOI] [PubMed] [Google Scholar]
- Zhang W, Ramamoorthy Y, Kilicarslan T, Nolte H, Tyndale RF, Sellers EM. Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug Metab Dispos. 2002;30:314–318. doi: 10.1124/dmd.30.3.314. [DOI] [PubMed] [Google Scholar]