

Abstract
Fibroblasts play a major role in processes such as wound repair, scarring, and fibrosis. Differentiation into myofibroblasts, characterized by upregulation of smooth muscle α-actin (smα) in response to profibrotic agents such as TGFβ is believed to be an important step in fibrosis. Therefore, elucidating mechanisms of myofibroblast differentiation might reveal novel targets in treating diseases such as idiopathic pulmonary fibrosis (IPF). MK2 is a kinase substrate of p38 MAP kinase that mediates some effects of p38 activation on the actin cytoskeleton. Using mouse embryonic fibroblasts (MEF) from MK2 knockout (MK2−/−) mice, we demonstrate that disrupting expression of MK2 expression reduces filamentous actin and stress fibers. It also causes MK2−/− MEF to express less smα than their corresponding wild-type (WT) MEF at baseline and in response to TGFβ. Furthermore, TGFβ causes downregulation of smα in MK2−/− MEF, instead of upregulation observed in WT MEF. Expression of other fibroblast markers, such as collagen, is not altered in MK2−/− MEF. Our results further suggest that downregulation of smα in MK2−/− MEF is not due to lack of activation of serum responsive promoter elements, but probably due to reduced smα message stability in these cells. These results indicate that MK2 plays a key role in regulation of smα expression, and that targeting MK2 might present a therapeutic approach in managing conditions such as pulmonary fibrosis.
Keywords: fibrosis, smooth muscle actin, cytoskeleton, MAPKAPK2, fibroblast, stress fibers
Differentiation of fibroblasts into myofibroblasts is an important event in many conditions such as wound repair and fibrosis. For example, in pulmonary fibrosis (PF) myofibroblasts occur in areas of active fibrosis and are responsible for production and deposition of extracellular matrix proteins [Vyalov et al., 1993]. Myofibroblasts derive from fibroblasts through the action of growth factors, such as, TGFβ [Desmouliere et al., 1993; Ronnov-Jessen and Petersen, 1993; Yokozeki et al., 1997; Roy et al., 2001]. Several signaling pathways have been proposed to mediate the actions of TGFβ on fibroblasts including the MAP kinase p38. Furthermore inhibition of p38 reduced pulmonary [Underwood et al., 2000; Matsuoka et al., 2002] and renal [Stambe et al., 2004] fibrosis in animal models. Recently the role of differentiation of fibroblasts into myofibroblasts in the pathogenesis of pulmonary hypertension has also been highlighted [Stenmark et al., 2002; Short et al., 2004]. We launched this project to investigate the role of MAP kinase activated protein kinase 2 (MAPKAPK2 or MK2), which is a downstream effector of p38, in fibrosis, and the mechanism by which its activation by TGFβ leads to differentiation of myofibroblasts.
PF can arise in response to drugs and asbestos but the interstitial idiopathic pulmonary fibrosis (IPF) variant of the disease arises by unknown mechanisms. It has long been postulated that an inflammatory reaction is an important component or initiator of fibrosis. Several cytokines have been shown to be altered in fibrosis in animal models as well as human patients. For example, TGFβ [Khalil et al., 1991], IL-1β and TNFα [Piguet et al., 1993; Pan et al., 1996] have been reported to be elevated in the lungs of IPF patients. The range of cytokines altered during fibrosis led researchers to propose that fibrosis is the result of an imbalance between pro-inflammatory and profibrotic cytokines, and hence a dysregulated rather than a simple inflammatory reaction [Wallace et al., 1995; Ando et al., 1999; Ziesche et al., 1999]. Nevertheless, the role of inflammation in fibrosis continues to be questioned based on the fact that fibrosis can arise without clear evidence of inflammation, and that the levels of altered cytokines cannot be correlated with severity of fibrosis. While the range of cytokines and factors that become altered in the development of fibrosis continues to be investigated, it is clear that some of these factors, such as TGFβ initiate responses in model systems that mimic the human disease. Indeed, the key to understanding the etiology of PF might evolve from a better understanding of signaling events in fibroblasts in response to factors such as TGFβ leading to myofibroblast differentiation.
Myofibroblasts are identified by increased expression of smα, which is usually expressed in smooth muscle cells. Functionally, myofibroblasts are more contractile and structurally they are characterized by increased actin stress fibers both of which are believed to be due to the increased expression of smα (for review, see Hinz and Gabbiani [2003]). Thus the mechanism by which smα expression is regulated has received considerable attention. In this report we have focused on TGFβ, a classical profibrotic factor, which has been linked to remodeling. TGFβ belongs to large family of proteins that are important in a variety of diseases ranging from cancer to pulmonary hypertension. It has been shown to stimulate a pathway that proceeds from cell surface receptors to the nucleus through Smad proteins (for review, see Attisano and Wrana [2002]). Yet TGFβ can signal independently of Smad proteins by activating Ras and Erk [Hartsough and Mulder, 1995; Hartsough et al., 1996], Rho GTPase and JNK [Atfi et al., 1997], as well as p38 MAP kinase [Hanafusa et al., 1999]. The latter pathway has recently been shown to play an important role in mediating the effects of hypoxia on different cell types [Seko et al., 1997; Conrad et al., 1999, 2000; Kacimi et al., 2000; Carini et al., 2001; Das et al., 2001; Kayyali et al., 2001; Marais et al., 2001; Welsh et al., 2001]. We have previously described how p38 activation leads to actin stress fiber formation in hypoxic endothelial cells and demonstrated that it is mediated by MK2 and its substrate HSP27 [Kayyali et al., 2002]. p38 signaling leading to HSP27 phosphorylation has also been shown to mediate fibroblast-mediated wound contractions [Hirano et al., 2002].
In evaluating the role of the p38 pathway in mediating the effects of TGFβ on fibroblast differentiation we have focused on MK2, an immediate downstream effector of p38. This kinase has been shown to phosphorylate serum response factor (SRF) which activates smα expression [Heidenreich et al., 1999]. Using mouse embryonic fibroblasts (MEF) from wild-type (WT) mice and MK2−/− mice in which the expression of MK2 has been disrupted, we examined myofibroblast potential and responsiveness to TGFβ. Our results indicate that MK2 plays an important role in myofibroblast phenotypic expression. Fibroblasts that lack MK2 express very little smα at baseline or in response to TGFβ. Furthermore, disruption of MK2 expression reverses the responsiveness of fibroblasts to TGFβ, implicating its mediation of the effects of TGFβ under normal conditions and in disease processes such as fibrosis.
MATERIALS AND METHODS
Materials and Reagents
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin G potassium, streptomycin, fungizone, and glutamine were purchased from Invitrogen (Carlsbad, CA). All other reagents and drugs were obtained from Sigma (St. Louis, MO), with the exception of SB-203580-HCl (p38 MAPK inhibitor) and which was purchased from Calbiochem (La Jolla, CA). TGFβ was purchased from R&D Systems (Minneapolis, MN) and activated before use according to manufacturers instructions. On the day of the experiments, all drugs were diluted in serum-free medium.
Cell Culture and Transfection
Immortalized MEF from WT and MK2−/− knockout mice were prepared as described earlier [Kotlyarov et al., 1999]. In brief, primary embryonic fibroblasts from C57BL/6J WT and MK2−/− mice, were co-transfected with pSV40Tag encoding the SV40 large T antigen and the pREP8 plasmid, and then, histidinol-resistant colonies were selected and expanded. The cells were maintained in DMEM containing 10% FBS, penicillin, streptomycin, fungizone, and glutamine at 378C in humidified air containing 5% CO2. Fibroblasts were passaged in 0.25% trypsin-0.02% ethylenediaminetetraacetic acid (EDTA) solution, and 1 day prior to the experiments, cells were maintained in serum-free media.
For SRE promoter-reporter assays WT and MK2−/− MEF, were transfected with the SRE.L-luciferase vector which contains two copies of SRE.L, a derivative of c-fos serum response element (SRE) driving the expression of luciferase [Chihara et al., 1997]. The SRE.L vector was introduced alone in the case of WT MEF, and co-transfected with pMEpuro (a selection vector which confers resistance to puromycin to eukaryotic cells) in the case of MK2−/− MEF. The vectors were introduced (5 μg of SRE.L, and 0.5 μg pMEpuro) into MEF using lipofectamine. Stable transfected WT MEF cell lines were obtained by selection with geneticin, and resistant colonies were isolated, expanded, and then screened for baseline luciferase activity. Since the MK2−/− MEF were already resistant to geneticin, stable transfectants produced by co-transfection with pMEpuro were selected with puromycin, and resistant clones were isolated and expanded as above. The firefly luciferase activity was assayed using a kit from Promega (Madison, WI) according to the manufacturer's instructions. In brief, cells were lysed and the substrate (beetle luciferin) was added to the lysate. Next, chemiluminescence was measured using a luminometer.
Actin Cytoskeleton Examination
MEF were seeded on sterile glass coverslips. At various degrees of confluence, cells were rinsed twice with phosphate-buffered saline (PBS), and fixed for 10 min with 4% formaldehyde. Next the coverslips were washed twice with PBS, and then the cells were permeabilized for 10 min with 0.4% triton X-100 in PBS. The cells were stained with rhodamine–phalloidin (Molecular Probes/Invitrogen, Carlsbad, CA) for 20 min. The coverslips were then washed with PBS, mounted with 10% glycerol and examined using a Zeiss fluorescence microscope. For immunocytochemistry, after rhodamine–phalloidin staining the coverslips were washed with PBS then incubated overnight with mouse anti smα antibody (ASM-1, Chemicon, Temecula, CA). Next day, the coverslips were washed and the cells were incubated for 1 h with FITC-conjugated goat anti-mouse antibody (Jackson, Immunoresearch, West Grove, PA), washed, mounted with 10% glycerol and examined.
SDS–PAGE and Immunoblotting
Aliquots from the cell lysates prepared as described above were assayed for protein using the Bradford protein assay [Bradford, 1976] and then diluted with 2× Laemmli loading buffer for SDS–PAGE [Laemmli, 1970]. Equal amounts of protein were then loaded in each well of 4−20% Tris/glycine gels. After electrophoresis for 90 min at 125 V constant voltage, the gel was blotted onto an Immobilon-P membrane by electrophoretic transfer at 25 V constant voltage overnight. The membrane was then washed, blocked with 5% milk, and probed with antibodies against MK2 (Cell Signaling, Beverly, MA) smα (1A4, Sigma) or pan-actin (C-2, Santa Cruz, Santa Cruz, CA). The immunoreactive bands were visualized using a secondary antibody conjugated to horseradish peroxidase (Pierce, Rockford, IL), and a chemiluminescent substrate according to the manufacturer's instructions (SuperSignal, Pierce). The intensity of the bands was quantified using UnScanIt software (Orem, UT).
Quantitation of smα, β-actin and Procollagen RNA
To measure mRNA in MEF we used semiquantitative RT-PCR. In brief, RNA was isolated from treated or untreated MEF using the RNeasy kit (Qiagen, Valencia, CA) according to manufacturer's instructions. The amount of RNA was determined spectrophotometrically, and equal amounts were used to generate cDNA by reverse transcription using the Superscript III system (Invitrogen) according to manufacturer's instructions. Next an equal volume of the cDNA was added to a master mix containing the following primers. For smα, the primers 5′-GCATCCACGAAACCACCTA-3′ and 5′-CACGAGTAACAAATCAAAGC-3′ were used to generate a 418 bp PCR product from mouse smooth muscle α-actin cDNA [Arakawa et al., 2000]. For β-actin, the primers 5′-TGGAATCCTGTGGCATCCATGAAAC-3′ and 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′ were used to generate 348 bp product from mouse β-actin cDNA [Reynolds et al., 2004]. The primers 5′ -GAGCGGAGAGTACTGGATCG-3′ and 5′-GTTCGGGCTGATGTACCAGT-3′ were used to generate a 142 bp product from mouse procollagen type Iα1 cDNA [Yamazaki et al., 2005]. The PCR reaction was carried out as using a Bio-Rad I-cycler thermal cycler (Hercules, CA). The PCR products were then resolved on a 2% NuSieve/1% SeaKem agarose gel. After staining with ethidium bromide, photographs of the gels were taken, scanned, and the intensity of the bands was measured using UnScanIt software.
Statistical Analysis
Data are presented as means ± standard deviation (SD). Statistical differences were determined by either Student's t-test for comparison of two sample means or analysis of variance (ANOVA) for comparison of more than two sample means followed by Bonferroni posthoc testing for multiple comparisons between two sample means (P < 0.05 was considered statistically significant).
RESULTS
MK2−/− MEF Contain Less Actin Stress Fibers Than WT MEF
Myofibroblasts are known to contain more actin stress fibers than normal fibroblasts. We have previously reported that p38 activation leads to stress fiber formation in hypoxic endothelial cells and demonstrated that it is mediated by MK2 and its substrate HSP27 [Kayyali et al., 2002]. Thus, we predicted that cells from MK2−/− mice would contain less stress fibers than WT cells because the p38-MK2-HSP27 pathway is disrupted in these cells. After verifying that MK2 is not expressed in MK2−/− MEF at basal conditions or after TGFβ (1 ng/ml) treatment (Fig. 1), these cells and WT MEF were grown on coverslips and then fixed and stained for filamentous actin using rhodamine–phalloidin as we described earlier [Kayyali et al., 2002]. As shown in Figure 2, MEF from WT mice contain a significant amount of parallel actin stress fibers. MEF from MK2−/− mice, on the other hand, contain significantly less stress fibers and filamentous actin is organized in web like form rather than in parallel fibers.
Fig. 1.

MK2−/− MEF do not express the MK2 protein. Cell lysates from wild-type or MK2−/− MEF were immunoblotted with an anti-MK2 antibody. While significant bands were observed in wild-type MEF, MK2 protein was not detectable in the MK2−/− cells, either at baseline or after treatment with TGFβ (1 ng/ml for 24 h).
Fig. 2.
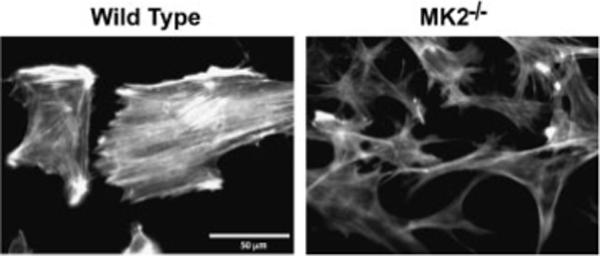
Disruption of MK2 expression reduces actin stress fibers. Wild-type and MK2−/− fibroblasts were grown on glass coverslips and then stained with rhodamine–phalloidin. MK2−/− MEF contained less filamentous actin than wild-type MEF in which actin was more organized into parallel stress fibers. Figure shows a representative area of several coverslips from several experiments.
Disruption of MK2 Expression Reduces smα Level in Embryonic Fibroblasts
Since increased actin stress fiber formation has been associated with differentiation of fibroblasts into myofibroblasts, we examined whether MK2−/− MEF differed from WT MEF in other fibroblast/myofibroblast characteristics. One hallmark of differentiation of fibroblasts into myofibroblasts is the increased expression of smα. To assess the level of smα expression in MK2−/− MEF versus WT MEF, both cell types were grown on coverslips then immunolabeled with an antibody against smα. As shown in Figure 3 only few MK2−/− MEF were immunolabeled with anti-smα antibody while the majority of WT MEF stained positive for smα. Furthermore, smα colocalized with rhodamine–phalloidin stained stress fibers in WT MEF (orange), while it did not colocalize with filamentous actin in the few smα-positive MK2 MEF (green).
Fig. 3.

Most MK2−/− fibroblasts do not express α-smooth muscle actin (smα). Wild-type and MK2−/− fibroblasts were grown on glass coverslips and then stained with rhodamine–phalloidin (red) and anti smα antibody (green). Most wild-type fibroblasts contained smα which colocalized with filamentous actin (yellow), while the majority of MK2 fibroblasts did not contain any smα.
These results were confirmed by immunoblotting such that when cell lysates were immunoblotted for smα the protein was barely detectable in MK2−/− MEF, compared to WT MEF which expressed much higher levels of smα (Fig. 4). On the other hand, levels of total actin were similar in the two types of MEF (Fig. 4). These results indicate that MK2−/− MEF are less differentiated into myofibroblasts than WT MEF.
Fig. 4.
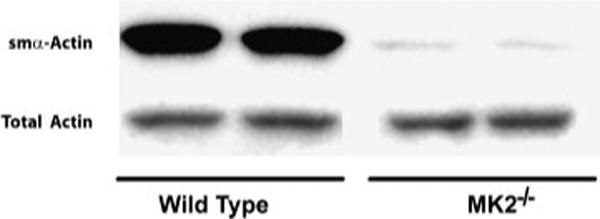
MK2−/− MEF contain less smα-actin than wild-type MEF. Immunoblotting cell lysates with a specific anti-smα antibody demonstrated that MK2−/− MEF express much less smα-actin than wild-type MEF. The level of total actin immunoblotted with anti-pan actin antibody is not different between the two cell types.
TGFβ Induces smα Expression in WT MEF but not in MK2−/− MEF
TGFβ has been implicated in the fibrotic response because of its ability to induce differentiation of fibroblasts into myofibroblasts. We tested whether TGFβ can induce myofibroblast differentiation in both WT MEF and MK2−/− MEF. Cells were growth arrested in serum free media for 24 h then exposed to TGFβ (1 ng/ml). As shown in Figure 5, TGFβ treatment causes WT MEF to produce more smα, but not MK2−/− MEF which produce even less smα in response to TGFβ.
Fig. 5.

TGFβ treatment increases smα-actin production in WT MEF but not in MK2−/− MEF. When WT MEF were treated with TGFβ (1 ng/ml for 24 h), smα-actin level increased consistent with the myofibroblast differentiating effect of TGFβ on fibroblasts. MK2−/− MEF on the other hand, did not respond to TGFβ by increasing smα-actin, but rather appeared to express less smα-actin in TGFβ-treated MK2−/− MEF than in vehicle-treated cells (Control). The MK2−/− film was exposed much longer than the WT film to reveal the difference due to TGFβ treatment, and does not reflect the difference between wild-type and MK2−/− smα-actin levels described earlier.
TGFβ Exerts Opposite Effects on the Level of smα mRNA in WT Versus MK2−/− MEF
To test if MK2 disruption affects the level of smα message as well as protein we compared the level of smα mRNA by semiquantitative RT-PCR. As shown in Figure 6, the level of smα message relative to the β-actin mRNA was lower in MK2−/− MEF than in WT MEF. TGFβ (1 ng/ml for 24 h) appeared to increase the smα message level in WT MEF. However, it significantly decreased the level of smα mRNA in MK2−/− MEF. This latter decrease in MK2−/− cells is consistent with the effect of TGFβ on smα protein level shown in Figure 5.
Fig. 6.

TGFβ produces opposite effects in MK2−/− MEF versus wild-type fibroblasts. The level of smα and β-actin mRNA were determined by RT-PCR on equal amounts of RNA isolated from WT or MK2−/− MEF treated with or without TGFβ (1 ng/ml) for 24 h. MK2−/− MEF contained less smα mRNA than wild-type MEF at baseline. TGFβ increased smα mRNA in wild-type MEF while it significantly reduced it in MK2−/− MEF.
The effect of MK2 suppression on the level of smα mRNA was specific in that the levels of collagen mRNA, another key protein produced by fibroblasts, were not different between MK2−/− MEF and WT MEF (Fig. 7). Furthermore, TGFβ treatment did not suppress the collagen mRNA level as it did for smα mRNA in MK2−/− MEF (Fig. 7).
Fig. 7.

Collagen message levels are not affected by MK2 suppression. The level of procollagen type I α1 mRNA were determined by RT-PCR on equal amounts of RNA isolated from wild-type or MK2−/− MEF treated with or without TGFβ (1 ng/ml) for 24 h. MK2−/− MEF contained comparable levels of message as wild-type MEF at baseline. TGFβ did not appear to alter collagen mRNA significantly in either cell types after 24 h of exposure.
TGFβ Activates Serum Response Elements in WT MEF in a p38 Dependent Manner
Since smα induction by several agents including TGFβ has been reported to occur through activation of SRE in the smα promoter, we tested whether this activation occurs in WT MEF. First, we generated stable MEF transfectants that overexpress a construct of two copies of SRE.L driving the expression of firefly luciferase. Several clones were screened for baseline and TGFβ-induced activity. As shown in Figure 8A, TGFβ (1 ng/ml) caused activation of the SRE-luciferase, which increased with time up to 6 h and returned to baseline by 24 h. An inhibitor of p38 MAP kinase, SB-203580 (1 μM), partially blocked the activation of SRE-luciferase by TGFβ suggesting that p38 pathway might be involved in SRE promoter activation (Fig. 8B).
Fig. 8.
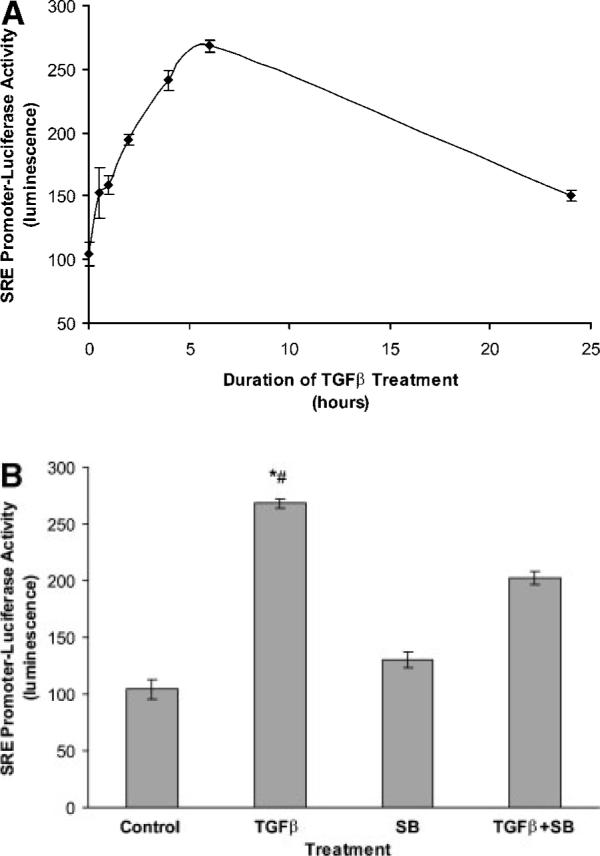
TGFβ treatment increases SRE-luciferase promoter activity in wild-type MEF. When wild-type MEF were treated with TGFβ (1 ng/ml for shown duration), SRE promoter activity increased with the greatest activation observed after 6 h of treatment (A). SB203580, a p38 inhibitor, partially blocked TGFβ activation of SRE promoter measured at 6 h (B). *Indicates statistically significant difference from Control mean, #indicates statistically significant difference from TGFβ + SB mean; P < 0.05.
MK2 Suppression Does not Abolish TGFβ-Induced Activation of SRE
Since MK2 has been reported to phosphorylate SRF [Heidenreich et al., 1999], we tested whether the effect of MK2 suppression on smα expression is due to its effect on the SRE promoter. Thus we generated stably transfected MK2−/− MEF to overexpress a construct of two copies of SRE.L driving the expression of firefly luciferase. As we did with WT MEF we screened several clones for baseline and TGFβ-induced activity. As shown in Figure 9A, TGFβ (1 ng/ml) caused activation of the SRE-luciferase in MK2−/− MEF, increasing with time up to 6 h and returning to baseline by 24 h. The extent of promoter activation and its time-course were similar to those observed in WT MEF suggesting that MK2 disruption has no effect on TGFβ-induced SRE promoter activity. An inhibitor of p38 MAP kinase, SB-203580 (1 μM), completely blocked the activation of SRE-luciferase by TGFβ suggesting that p38 pathway might be involved in SRE promoter activation (Fig. 9B). The difference in inhibition level might reflect a lower level of p38 in MK2−/− MEF as previously reported [Kotlyarov et al., 2002].
Fig. 9.

TGFβ treatment increases SRE-luciferase promoter activity in MK2−/− MEF. When MK2−/− MEF were treated with TGFβ (1 ng/ml for shown duration), SRE promoter activity increased similarly to WT MEF with the greatest activation observed after 6 h of treatment (A). SB203580, a p38 inhibitor, completely blocked TGFβ activation of SRE promoter measured at 6 h (B). *Indicates statistically significant difference from Control mean, #indicates statistically significant difference from TGFβ + SB mean; P < 0.05.
smα mRNA is Less Stable in MK2−/− MEF Than in WT MEF
Since MK2 has been described to regulate the expression of some proteins, such as IL-6 and TNF through altering their message stability [Neininger et al., 2002; Hitti et al., 2006], we measured smα mRNA and normalized it to β-actin mRNA in MK2−/− and WT MEF. Cells were incubated with 2.4 μM actinomycin D to inhibit transcription, and relative smα mRNA was assayed by semi quantitative RT-PCR at different time points. As shown in Figure 10, smα mRNA was degraded at a faster (8-fold) rate in MK2−/− compared to WT MEF. The half-life of smα mRNA was 23 h in MK2−/− MEF while it was 187 h in WT MEF. These results suggest that the reason MK2−/− MEF express less smα than WT MEF might be due to reduced smα message stability in these cells.
Fig. 10.

smα mRNA is less stable in MK2−/− MEF than in WT MEF. MEF were incubated with actinomycin D (2.4 μM) for 0, 2, 4, 6, 8, 16, and 24 h; then smα mRNA levels relative to β-actin mRNA were determined. Single-phase exponential decay regression lines are derived from three samples for each exposure group. The solid line and open circles represent WT MEF, and the dashed line and closed diamonds represent MK2−/− MEF. Regression curves were obtained from the exponential decay of mRNA using the formula y = ae−bx, where a is the smα mRNA value at time 0 (100%) and b the decay rate constant for each condition. smα mRNA half-life for each curve was calculated as t½ = [lna–ln50]/b.
DISCUSSION
Differentiation of fibroblasts into myofibroblasts has been a subject of interest because of the role this step is believed to play in wound healing and fibrosis. Several signaling pathways have been implicated in this process. Our results indicate that MK2 plays an important role in myofibroblast differentiation as defined by the expression of smα. In particular we have demonstrated that embryonic fibroblasts derived from MK2-deficient mice lack stress fibers and smα. In addition, unlike their WT counterparts the cells do not respond to TGFβ by increasing smα production but by suppressing it. These findings might implicate MK2 in wound healing and the pathogenesis of fibrotic disease.
Fibrosis occurs in different organs in response to different types of injury, or it can arise without a clear stimulus as in the case of IPF [Cherniack et al., 1991; ATS and ERS, 2000]. Current thinking on the mechanism by which PF might arise includes a model of abnormal wound healing. For example, in response to injury or some unknown stimulus, inflammation occurs leading to the recruitment of fibroblasts. These fibroblasts proliferate and differentiate into myofibroblasts, and when unresolved lead to fibrotic foci which are the hallmarks of fibrosis. Thus, one of the important cells in PF is the myofibroblast, which occurs in areas of active fibrosis and is responsible for production and deposition of extracellular matrix proteins [Vyalov et al., 1993]. The factors that determine the path to resolution versus fibrosis are not fully understood, however, considerable focus has been placed on signals that cause fibroblasts to differentiate into myofibroblasts such as those generated by TGFβ [Desmouliere et al., 1993; Ronnov-Jessen and Petersen, 1993; Yokozeki et al., 1997; Roy et al., 2001]. TGFβ has been reported to be elevated in the lungs of IPF patients [Khalil et al., 1991]. This growth factor belongs to a large family of proteins, which include members such as bone morphogenetic proteins, and that are important in a variety of diseases, such as, cancer and pulmonary hypertension. TGFβ has been shown to stimulate a pathway that proceeds from cell surface receptors to the nucleus through Smad proteins (for review, see Attisano and Wrana [2002]). Yet TGFβ can signal independently of Smad proteins by activating Ras and Erk [Hartsough and Mulder, 1995; Hartsough et al., 1996], Rho GTPase and JNK [Atfi et al., 1997], as well as p38 MAP kinase [Hanafusa et al., 1999]. Indeed TGFβ-induced myofibroblast differentiation has been suggested to be mediated by p38, and inhibition of p38 reduced pulmonary [Underwood et al., 2000; Matsuoka et al., 2002] and renal [Stambe et al., 2004] fibrosis in animal models.
p38 is a stress-activated kinase that is activated in response to stimuli such as ultraviolet radiation and hyperosmolarity. Once p38 is activated, it phosphorylates a variety of substrates, including the kinase MK2. Upon its phosphorylation by p38, MK2 becomes activated, and in turn, phosphorylates additional substrates, such as the small heat shock protein HSP27. When phosphorylated, HSP27 no longer blocks the polymerization of actin, thus resulting in the stabilization of actin fibers [Benndorf et al., 1994]. Recent studies both in vivo and in vitro [Yoshida et al., 2002; Stambe et al., 2004] have suggested that p38 plays an important role in the pathogenesis of fibrosis, even though some researchers reported no effect of inhibiting p38 on differentiation of lung fibroblasts into myofibroblasts [Hashimoto et al., 2001]. We launched the experiments described in this report to examine if signaling downstream of the p38 pathway plays a role in fibroblast differentiation into myofibroblasts.
Our laboratory has already published that the p38-MK2-HSP27 pathway mediates cytoskeletal changes in endothelial cells exposed to hypoxia, which lead to increased actin stress fiber formation [Kayyali et al., 2002; An et al., 2005]. Since similar actin reorganization is believed to occur when fibroblasts switch to myofibroblasts, we examined whether embryonic fibroblasts from MK2−/− knockout mice differed in their filamentous actin distribution when compared to embryonic fibroblasts from WT mice. Our results demonstrated that the MK2−/− fibroblasts indeed contained less actin stress fibers than WT cells (Fig. 2), consistent with what we observed in endothelial cells in which the p38-MK2-HSP27 pathway was disrupted [Kayyali et al., 2002]. Not only did the MK2−/− fibroblasts contain much less actin stress fibers than WT fibroblasts, but they also lacked smα, which is the hallmark for myofibroblast differentiation (Fig. 3). These results were significant and were further confirmed through immunoblotting (Fig. 4) and RT-PCR (Fig. 6). MK2−/− MEF also did not express smα in response to TGFβ which increased smα in WT cells (Figs. 5 and 6). In fact TGFβ treatment reduced the amount of smα in MK2−/− MEF. The significance of this reduction has yet to be determined and might reflect tipping the balance between enhancers and repressors that target TGFβ responsive elements discussed below. Since smα expression has been shown to be regulated, in particular by TGFβ, at the transcriptional level, we explored the mechanisms that might explain the different levels of smα in MK2−/− versus WT MEF.
The regulation of smα expression in fibroblasts and other cell types is quite complex and at the transcriptional level alone involves several interacting factors, enhancers and suppressors. The promoter region of the smα gene contains elements (SRE) that are responsive to SRF. The CArG box, which is a core component of the SRE has been shown to be responsive to serum in the mouse [Foster et al., 1992; Stoflet et al., 1992], as well as, the human [Kim et al., 1993] smα promoter in fibroblasts. However, while mutation of the CArG sites in the promoter abolished its activity in smooth muscle cells in vitro and in vivo [Mack and Owens, 1999], it had no effect in endothelial cells suggesting that regulation varies in different cell types [Shimizu et al., 1995]. In addition to the CArG, a TGFβ responsive element (TCE) has been reported that enhanced SRF binding and appeared to be necessary for TGFβ induction of smα in smooth muscle cells [Hautmann et al., 1997] and fibroblasts [Hautmann et al., 1999]. One study also suggested that CArG is less important for induction of smα in fibroblasts treated with TGFβ than TCE [Roy et al., 2001]. Recent studies suggested that the TCE site in the smα promoter mediates both activation and repression of the gene [Liu et al., 2003]. In addition, Smad3 which mediates some of TGFβ effects has been reported to bind to Smad3-binding elements in the smα promoter and activate it [Hu et al., 2003]. Several other studies suggested that repressor proteins such as, the Pur family [Subramanian et al., 2004], MYS1 [Kelm et al., 1999, 2003] and its human analog YB-1 [Zhang et al., 2005] also contributed to the complex tissue specific expression of smα possibly through interacting with SRF, SP1, and Smad2/3. Since TGFβ has been described to activate p38 in fibroblasts and since SRF activation has been shown to be p38 dependent [Deaton et al., 2005], we chose to focus on SRF activation as a mechanism that might explain the different levels of smα expression in WT versus MK2−/− fibroblasts. Indeed one report demonstrating that MK2 directly phosphorylates and activates SRF [Heidenreich et al., 1999] suggested a direct link between p38 activation and smα expression. Thus, we hypothesized that the reason MK2−/− fibroblasts lack smα is due to their inability to activate SRE in the promoter region of the smα gene since SRF in these cells is not being phosphorylated. We could not identify differences in the phosphorylation level of SRF between MK2−/− versus WT fibroblasts either under normal conditions or in response to TGFβ (data not shown). Still we tested whether MK2 level might affect the SRE indirectly by expressing SRE-luciferase in WT and TGFβ fibroblasts.
Both WT and MK2−/− fibroblasts expressed significant activity of the SRE promoter when stable transfectants were generated (Figs. 8 and 9). Moreover, we did not observe a significant difference in the extent or time-course of activation of the smα promoter by TGFβ in either cell type (Figs. 8 and 9). These observations lead us to conclude that MK2 affects smα by mechanisms that are independent of the SRE or SRF. The fact that a p38 inhibitor was able to block SRE promoter activation (more so in the MK2−/− MEF than in WT) led us to postulate that the p38-mediated activation of SRF/SRE by TGFβ remained functional after disrupting MK2 expression. Hence, MK2 might affect smα expression through actions on other transcription enhancers or repressors.
MK2 might regulate smα expression by posttranscriptional mechanisms. For example MK2 has been shown to increase IL-6 and TNF message stability, as well as TNF translation upon induction by lipopolysaccharide [Kotlyarov and Gaestel, 2002; Neininger et al., 2002; Hitti et al., 2006]. Some of these actions are believed to be mediated through direct phosphorylation of 3′UTR-binding proteins by MK2, which inhibits their mRNA degrading activity. Indeed the data described in Figure 10 indicate that smα mRNA is less stable in MK2−/− versus WT fibroblasts. Experiments are underway to identify the potential role of 3′ UTR-binding proteins in regulating smα mRNA level in response to MK2 modulation. Finally, MK2 might regulate smα expression at the posttranslational level since the difference in smα mRNA levels at baseline between MK2−/− and WT MEF was not as great as the difference in protein levels. Nevertheless, the RT-PCR we employed is only semiquantitative and we cannot draw definite conclusions about the relative amount of message.
As to the physiological significance of our findings, MK2 might turn out to be a key participant in the differentiation of fibroblasts into myofibroblasts, and hence in fibrosis. Yet MK2−/− fibroblasts continue to produce collagen (Fig. 7). Recently it has been suggested that collagen deposition in an animal model of fibrosis may be derived from bone marrow cells rather than lung fibroblasts or myofibroblasts [Dunsmore and Shapiro, 2004; Hashimoto et al., 2004]. Whether this finding indicates that myofibroblast differentiation is beneficial rather than detrimental remains to be determined. We are conducting experiments to determine if MK2−/− fibroblasts are likely to be less or more fibrotic in vivo using MK2−/− knockout mice. In conclusion, our findings indicate that MK2 plays an important role in development of the myofibroblast phenotype, and implicate the kinase and its targets in the processes of wound healing and fibrosis.
ACKNOWLEDGMENTS
The authors are grateful to Dr. Kozo Kaibuchi (Nagoya University) for providing the SRE.L construct, to Dr. Howard Surks (Tufts-New England Medical Center), and Dr. Kazuo Maruyama (Tokyo Medical and Dental University) for providing the pMEpuro vector.
Grant sponsor: NIH; Grant number: HL079320; Grant sponsor: National Heart, Lung and Blood Institute.
REFERENCES
- An SS, Pennella CM, Gonnabathula A, Chen J, Wang N, Gaestel M, Hassoun PM, Fredberg JJ, Kayyali US. Hypoxia alters biophysical properties of endothelial cells via p38 MAPK- and Rho kinase-dependent pathways. Am J Physiol Cell Physiol. 2005;289:C521–C530. doi: 10.1152/ajpcell.00429.2004. [DOI] [PubMed] [Google Scholar]
- Ando M, Miyazaki E, Fukami T, Kumamoto T, Tsuda T. Interleukin-4-producing cells in idiopathic pulmonary fibrosis: An immunohistochemical study. Respirology. 1999;4:383–391. doi: 10.1046/j.1440-1843.1999.00209.x. [DOI] [PubMed] [Google Scholar]
- Arakawa E, Hasegawa K, Yanai N, Obinata M, Matsuda Y. A mouse bone marrow stromal cell line, TBR-B, shows inducible expression of smooth muscle-specific genes. FEBS Lett. 2000;481:193–196. doi: 10.1016/s0014-5793(00)01995-5. [DOI] [PubMed] [Google Scholar]
- Atfi A, Djelloul S, Chastre E, Davis R, Gespach C. Evidence for a Role of Rho-like GTPases and stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) in transforming growth factor beta-mediated signaling. J Biol Chem. 1997;272:1429–1432. doi: 10.1074/jbc.272.3.1429. [DOI] [PubMed] [Google Scholar]
- ATS, ERS Idiopathic pulmonary fibrosis: Diagnosis and treatment. International consensus statement. Am J Respir Crit Care Med. 2000;161:646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- Attisano L, Wrana JL. Signal Transduction by the TGF-beta superfamily. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- Benndorf R, Hayess K, Ryazantsev S, Wieske M, Behlke J, Lutsch G. Phosphorylation and supramolecular organization of murine small heat shock protein HSP25 abolish its actin polymerization-inhibiting activity. J Biol Chem. 1994;269:20780–20784. [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Carini R, De Cesaris MG, Splendore R, Vay D, Domenicotti C, Nitti MP, Paola D, Pronzato MA, Albano E. Signal pathway involved in the development of hypoxic preconditioning in rat hepatocytes. Hepatology. 2001;33:131–139. doi: 10.1053/jhep.2001.21050. [DOI] [PubMed] [Google Scholar]
- Cherniack RM, Crystal RG, Kalica AR. NHLBI workshop summary. Current concepts in idiopathic pulmonary fibrosis: A road map for the future. Am Rev Respir Dis. 1991;143:680–683. doi: 10.1164/ajrccm/143.3.680. [DOI] [PubMed] [Google Scholar]
- Chihara K, Amano M, Nakamura N, Yano T, Shibata M, Tokui T, Ichikawa H, Ikebe R, Ikebe M, Kaibuchi K. Cytoskeletal rearrangements and transcriptional activation of c-fos serum response element by Rho-kinase. J Biol Chem. 1997;272:25121–25127. doi: 10.1074/jbc.272.40.25121. [DOI] [PubMed] [Google Scholar]
- Conrad PW, Rust RT, Han J, Millhorn DE, Beitner-Johnson D. Selective activation of p38alpha and p38gamma by hypoxia. Role in regulation of cyclin D1 by hypoxia in PC12 cells. J Biol Chem. 1999;274:23570–23576. doi: 10.1074/jbc.274.33.23570. [DOI] [PubMed] [Google Scholar]
- Conrad PW, Millhorn DE, Beitner-Johnson D. Novel regulation of p38gamma by dopamine D2 receptors during hypoxia. Cell Signal. 2000;12:463–467. doi: 10.1016/s0898-6568(00)00091-7. [DOI] [PubMed] [Google Scholar]
- Das M, Bouchey DM, Moore MJ, Hopkins DC, Nemenoff RA, Stenmark KR. Hypoxia-induced proliferative response of vascular adventitial fibroblasts is dependent on g protein-mediated activation of mitogen-activated protein kinases. J Biol Chem. 2001;276:15631–15640. doi: 10.1074/jbc.M010690200. [DOI] [PubMed] [Google Scholar]
- Deaton RA, Su C, Valencia TG, Grant SR. Transforming growth factor-{beta}1-induced expression of smooth muscle marker genes involves activation of PKN and p38 MAPK. J Biol Chem. 2005;280:31172–31181. doi: 10.1074/jbc.M504774200. [DOI] [PubMed] [Google Scholar]
- Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103–111. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunsmore SE, Shapiro SD. The bone marrow leaves its scar: New concepts in pulmonary fibrosis. J Clin Invest. 2004;113:180–182. doi: 10.1172/JCI20782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DN, Min B, Foster LK, Stoflet ES, Sun S, Getz MJ, Strauch AR. Positive and negative cis-acting regulatory elements mediate expression of the mouse vascular smooth muscle alpha-actin gene. J Biol Chem. 1992;267:11995. [PubMed] [Google Scholar]
- Hanafusa H, Ninomiya-Tsuji J, Masuyama N, Nishita M, Fujisawa J, Shibuya H, Matsumoto K, Nishida E. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. J Biol Chem. 1999;274:27161–27167. doi: 10.1074/jbc.274.38.27161. [DOI] [PubMed] [Google Scholar]
- Hartsough MT, Mulder KM. Transforming growth factor beta activation of p44[IMAGE] in proliferating cultures of epithelial cells. J Biol Chem. 1995;270:7117–7124. doi: 10.1074/jbc.270.13.7117. [DOI] [PubMed] [Google Scholar]
- Hartsough MT, Frey RS, Zipfel PA, Buard A, Cook SJ, McCormick F, Mulder KM. Altered transforming growth factor signaling in epithelial cells when ras activation is blocked. J Biol Chem. 1996;271:22368–22375. doi: 10.1074/jbc.271.37.22368. [DOI] [PubMed] [Google Scholar]
- Hashimoto S, Gon Y, Takeshita I, Matsumoto K, Maruoka S, Horie T. Transforming growth factor-beta1 induces phenotypic modulation of human lung fibroblasts to myofibroblast through a c-Jun-NH2-terminal kinase-dependent pathway. Am J Respir Crit Care Med. 2001;163:152–157. doi: 10.1164/ajrccm.163.1.2005069. [DOI] [PubMed] [Google Scholar]
- Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113:243–252. doi: 10.1172/JCI18847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hautmann MB, Madsen CS, Owens GK. A transforming growth factor? (TGF?) control element drives TGF?- induced stimulation of smooth muscle?-actin gene expression in concert with two CArG elements. J Biol Chem. 1997;272:10948. doi: 10.1074/jbc.272.16.10948. [DOI] [PubMed] [Google Scholar]
- Hautmann MB, Adam PJ, Owens GK. Similarities and differences in smooth muscle [alpha]-actin induction by TGF- [beta] in smooth muscle versus non-smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:2049. doi: 10.1161/01.atv.19.9.2049. [DOI] [PubMed] [Google Scholar]
- Heidenreich O, Neininger A, Schratt G, Zinck R, Cahill MA, Engel K, Kotlyarov A, Kraft R, Kostka S, Gaestel M, Nordheim A. MAPKAP kinase 2 phosphorylates serum response factor in vitro and in vivo. J Biol Chem. 1999;274:14434–14443. doi: 10.1074/jbc.274.20.14434. [DOI] [PubMed] [Google Scholar]
- Hinz B, Gabbiani G. Mechanisms of force generation and transmission by myofibroblasts. Curr Opin Biotechnol. 2003;14:538–546. doi: 10.1016/j.copbio.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Hirano S, Rees RS, Gilmont RR. MAP kinase pathways involving Hsp27 regulate fibroblast-mediated wound contraction. J Surg Res. 2002;102:77. doi: 10.1006/jsre.2001.6315. [DOI] [PubMed] [Google Scholar]
- Hitti E, Iakovleva T, Brook M, Deppenmeier S, Gruber AD, Radzioch D, Clark AR, Blackshear PJ, Kotlyarov A, Gaestel M. Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol Cell Biol. 2006;26:2399–2407. doi: 10.1128/MCB.26.6.2399-2407.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Wu Z, Phan SH. Smad3 mediates transforming growth factor-{beta}-induced {alpha}-smooth muscle actin expression. Am J Respir Cell Mol Biol. 2003;29:397–404. doi: 10.1165/rcmb.2003-0063OC. [DOI] [PubMed] [Google Scholar]
- Kacimi R, Chentoufi J, Honbo N, Long CS, Karliner JS. Hypoxia differentially regulates stress proteins in cultured cardiomyocytes: Role of the p38 stress-activated kinase signaling cascade, and relation to cytoprotection. Cardiovasc Res. 2000;46:139–150. doi: 10.1016/s0008-6363(00)00007-9. [DOI] [PubMed] [Google Scholar]
- Kayyali US, Donaldson C, Huang H, Abdelnour R, Hassoun PM. Phosphorylation of xanthine dehydrogenase/oxidase in hypoxia. J Biol Chem. 2001;276:14359–14365. doi: 10.1074/jbc.M010100200. [DOI] [PubMed] [Google Scholar]
- Kayyali US, Pennella CM, Trujillo C, Villa O, Gaestel M, Hassoun PM. Cytoskeletal changes in hypoxic pulmonary endothelial cells are dependent on the MAP kinase-associated protein kinase MK2. J Biol Chem. 2002;28:28. doi: 10.1074/jbc.M205863200. [DOI] [PubMed] [Google Scholar]
- Kelm RJ, Jr., Cogan JG, Elder PK, Strauch AR, Getz MJ. Molecular interactions between single-stranded DNA-binding proteins associated with an essential MCAT element in the mouse smooth muscle alpha—actin promoter. J Biol Chem. 1999;274:14238–14245. doi: 10.1074/jbc.274.20.14238. [DOI] [PubMed] [Google Scholar]
- Kelm RJ, Jr., Wang S-X, Polikandriotis JA, Strauch AR. Structure/function analysis of mouse Pur{beta}, a single-stranded DNA-binding repressor of vascular smooth muscle {alpha}-actin gene transcription. J Biol Chem. 2003;278:38749–38757. doi: 10.1074/jbc.M306163200. [DOI] [PubMed] [Google Scholar]
- Khalil N, O'Connor RN, Unruh HW, Warren PW, Flanders KC, Kemp A, Bereznay OH, Greenberg AH. Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 1991;5:155–162. doi: 10.1165/ajrcmb/5.2.155. [DOI] [PubMed] [Google Scholar]
- Kim JH, Bushel PR, Kumar CC. Smooth muscle [alpha]-actin promoter activity is induced by serum stimulation of fibroblast cells. Biochem Biophys Res Commun. 1993;190:1115. doi: 10.1006/bbrc.1993.1165. [DOI] [PubMed] [Google Scholar]
- Kotlyarov A, Gaestel M. Is MK2 (mitogen-activated protein kinase-activated protein kinase 2) the key for understanding post-transcriptional regulation of gene expression? Biochem Soc Trans. 2002;30:959–963. doi: 10.1042/bst0300959. [DOI] [PubMed] [Google Scholar]
- Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk HD, Gaestel M. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol. 1999;1:94–97. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- Kotlyarov A, Yannoni Y, Fritz S, Laass K, Telliez JB, Pitman D, Lin LL, Gaestel M. Distinct cellular functions of MK2. Mol Cell Biol. 2002;22:4827–4835. doi: 10.1128/MCB.22.13.4827-4835.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sinha S, Owens G. A transforming growth factor-{beta} control element required for SM {alpha}-actin expression in vivo also partially mediates GKLF-dependent transcriptional repression. J Biol Chem. 2003;278:48004–48011. doi: 10.1074/jbc.M301902200. [DOI] [PubMed] [Google Scholar]
- Mack CP, Owens GK. Regulation of smooth muscle {alpha}-actin expression in vivo is dependent on CArG elements within the 5′ and first intron promoter regions. Circ Res. 1999;84:852–861. doi: 10.1161/01.res.84.7.852. [DOI] [PubMed] [Google Scholar]
- Marais E, Genade S, Huisamen B, Strijdom JG, Moolman JA, Lochner A. Activation of p38 MAPK induced by a multi-cycle ischaemic preconditioning protocol is associated with attenuated p38 MAPK activity during sustained ischaemia and reperfusion. J Mol Cell Cardiol. 2001;33:769–778. doi: 10.1006/jmcc.2001.1347. [DOI] [PubMed] [Google Scholar]
- Matsuoka H, Arai T, Mori M, Goya S, Kida H, Morishita H, Fujiwara H, Tachibana I, Osaki T, Hayashi S. A p38 MAPK inhibitor, FR-167653, ameliorates murine bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2002;283:L103–L112. doi: 10.1152/ajplung.00187.2001. [DOI] [PubMed] [Google Scholar]
- Neininger A, Kontoyiannis D, Kotlyarov A, Winzen R, Eckert R, Volk H-D, Holtmann H, Kollias G, Gaestel M. MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem. 2002;277:3065–3068. doi: 10.1074/jbc.C100685200. [DOI] [PubMed] [Google Scholar]
- Pan LH, Ohtani H, Yamauchi K, Nagura H. Coexpression of TNF alpha and IL-1 beta in human acute pulmonary fibrotic diseases: An immunohistochemical analysis. Pathol Int. 1996;46:91–99. doi: 10.1111/j.1440-1827.1996.tb03584.x. [DOI] [PubMed] [Google Scholar]
- Piguet PF, Ribaux C, Karpuz V, Grau GE, Kapanci Y. Expression and localization of tumor necrosis factor-alpha and its mRNA in idiopathic pulmonary fibrosis. Am J Pathol. 1993;143:651–655. [PMC free article] [PubMed] [Google Scholar]
- Reynolds PR, Mucenski ML, Le Cras TD, Nichols WC, Whitsett JA. Midkine is regulated by hypoxia and causes pulmonary vascular remodeling. J Biol Chem. 2004;279:37124–37132. doi: 10.1074/jbc.M405254200. [DOI] [PubMed] [Google Scholar]
- Ronnov-Jessen L, Petersen OW. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab Invest. 1993;68:696–707. [PubMed] [Google Scholar]
- Roy SG, Nozaki Y, Phan SH. Regulation of [alpha]-smooth muscle actin gene expression in myofibroblast differentiation from rat lung fibroblasts. Int J Biochem Cell Biol. 2001;33:723. doi: 10.1016/s1357-2725(01)00041-3. [DOI] [PubMed] [Google Scholar]
- Seko Y, Takahashi N, Tobe K, Kadowaki T, Yazaki Y. Hypoxia and hypoxia/reoxygenation activate p65PAK, p38 mitogen-activated protein kinase (MAPK), and stress-activated protein kinase (SAPK) in cultured rat cardiac myocytes. Biochem Biophys Res Commun. 1997;239:840–844. doi: 10.1006/bbrc.1997.7570. [DOI] [PubMed] [Google Scholar]
- Shimizu RT, Blank RS, Jervis R, Lawrenz-Smith SC, Owens GK. The smooth muscle alpha-actin gene promoter is differentially regulated in smooth muscle versus non-smooth muscle cells. J Biol Chem. 1995;270:7631–7643. doi: 10.1074/jbc.270.13.7631. [DOI] [PubMed] [Google Scholar]
- Short M, Nemenoff RA, Zawada WM, Stenmark KR, Das M. Hypoxia induces differentiation of pulmonary artery adventitial fibroblasts into myofibroblasts. Am J Physiol Cell Physiol. 2004;286:C416–C425. doi: 10.1152/ajpcell.00169.2003. Epub 2003 Oct 15. [DOI] [PubMed] [Google Scholar]
- Stambe C, Atkins RC, Tesch GH, Masaki T, Schreiner GF, Nikolic-Paterson DJ. The role of p38alpha mitogen-activated protein kinase activation in renal fibrosis. J Am Soc Nephrol. 2004;15:370–379. doi: 10.1097/01.asn.0000109669.23650.56. [DOI] [PubMed] [Google Scholar]
- Stenmark KR, Gerasimovskaya E, Nemenoff RA, Das M. Hypoxic activation of adventitial fibroblasts: Role in vascular remodeling. Chest. 2002;122:326S–334S. doi: 10.1378/chest.122.6_suppl.326s. [DOI] [PubMed] [Google Scholar]
- Stoflet ES, Schmidt LJ, Elder PK, Korf GM, Foster DN, Strauch AR, Getz MJ. Activation of a muscle-specific actin gene promoter in serum-stimulated fibroblasts. Mol Biol Cell. 1992;3:1073–1083. doi: 10.1091/mbc.3.10.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian SV, Polikandriotis JA, Kelm RJ, Jr., David JJ, Orosz CG, Strauch AR. Induction of vascular smooth muscle {alpha}-actin gene transcription in transforming growth factor {beta}1-activated myofibroblasts mediated by dynamic interplay between the pur repressor proteins and Sp1/Smad coactivators. Mol Biol Cell. 2004;15:4532–4543. doi: 10.1091/mbc.E04-04-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood DC, Osborn RR, Bochnowicz S, Webb EF, Rieman DJ, Lee JC, Romanic AM, Adams JL, Hay DW, Griswold DE. SB 239063, a p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lung. Am J Physiol Lung Cell Mol Physiol. 2000;279:L895–L902. doi: 10.1152/ajplung.2000.279.5.L895. [DOI] [PubMed] [Google Scholar]
- Vyalov SL, Gabbiani G, Kapanci Y. Rat alveolar myofibroblasts acquire alpha-smooth muscle actin expression during bleomycin-induced pulmonary fibrosis. Am J Pathol. 1993;143:1754–1765. [PMC free article] [PubMed] [Google Scholar]
- Wallace WA, Ramage EA, Lamb D, Howie SE. A type 2 (Th2-like) pattern of immune response predominates in the pulmonary interstitium of patients with cryptogenic fibrosing alveolitis (CFA). Clin Exp Immunol. 1995;101:436–441. doi: 10.1111/j.1365-2249.1995.tb03131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh DJ, Peacock AJ, MacLean M, Harnett M. Chronic hypoxia induces constitutive p38 mitogen-activated protein kinase activity that correlates with enhanced cellular proliferation in fibroblasts from rat pulmonary but not systemic arteries. Am J Respir Crit Care Med. 2001;164:282–289. doi: 10.1164/ajrccm.164.2.2008054. [DOI] [PubMed] [Google Scholar]
- Yamazaki K, Fukata H, Adachi T, Tainaka H, Kohda M, Yamazaki M, Kojima K, Chiba K, Mori C, Komiyama M. Association of increased type I collagen expression and relative stromal overgrowth in mouse epididymis neonatally exposed to diethylstilbestrol. Mol Reprod Dev. 2005;72:291–298. doi: 10.1002/mrd.20347. [DOI] [PubMed] [Google Scholar]
- Yokozeki M, Moriyama K, Shimokawa H, Kuroda T. Transforming growth factor-beta 1 modulates myofibroblastic phenotype of rat palatal fibroblasts in vitro. Exp Cell Res. 1997;231:328–336. doi: 10.1006/excr.1997.3473. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Kuwano K, Hagimoto N, Watanabe K, Matsuba T, Fujita M, Inoshima I, Hara N. MAP kinase activation and apoptosis in lung tissues from patients with idiopathic pulmonary fibrosis. J Pathol. 2002;198:388–396. doi: 10.1002/path.1208. [DOI] [PubMed] [Google Scholar]
- Zhang A, Liu X, Cogan JG, Fuerst MD, Polikandriotis JA, Kelm RJ, Jr., Strauch AR. YB-1 coordinates vascular smooth muscle {alpha}-actin gene activation by transforming growth factor {beta}1 and thrombin during differentiation of human pulmonary myofibroblasts. Mol Biol Cell. 2005;16:4931–4940. doi: 10.1091/mbc.E05-03-0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziesche R, Hofbauer E, Wittmann K, Petkov V, Block LH. A preliminary study of long-term treatment with interferon gamma-1b and low-dose prednisolone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 1999;341:1264–1269. doi: 10.1056/NEJM199910213411703. [DOI] [PubMed] [Google Scholar]