

Abstract
Although it is well-established that G protein-coupled receptor signaling systems can network with those of tyrosine kinase receptors by several mechanisms, the point(s) of convergence of the two pathways remains largely undelineated, particularly for opioids. Here we demonstrate that opioid agonists modulate the activity of the extracellular signal-regulated protein kinase (ERK) in African green monkey kidney COS-7 cells transiently cotransfected with μ-, δ-, or κ-opioid receptors and ERK1- or ERK2-containing plasmids. Recombinant proteins in transfected cells were characterized by binding assay or immunoblotting. On treatment with corresponding μ- ([d-Ala2,Me-Phe4,Gly-ol5]enkephalin)-, δ-([d-Pen2,d-Pen5]enkephalin)-, or κ- (U69593)-selective opioid agonists, a dose-dependent, rapid stimulation of ERK1 and ERK2 activity was observed. This activation was inhibited by specific antagonists, suggesting the involvement of opioid receptors. Pretreatment of cells with pertussis toxin abolished ERK1 and ERK2 activation by agonists. Cotransfection of cells with dominant negative mutant N17-Ras or with a βγ scavenger, CD8-β-adrenergic receptor kinase-C, suppressed opioid stimulation of ERK1 and ERK2. When epidermal growth factor was used to activate ERK1, chronic (>2-h) opioid agonist treatment resulted in attenuation of the stimulation by the growth factor. This inhibition was blocked by the corresponding antagonists and CD8-β-adrenergic receptor kinase-C cotransfection. These results suggest a mechanism involving Ras and βγ subunits of Gi/o proteins in opioid agonist activation of ERK1 and ERK2, as well as opioid modulation of epidermal growth factor-induced ERK activity.
Keywords: Opioid receptor, Mitogen-activated protein kinase, Extracellular signal-regulated protein kinase, GTP binding regulatory protein, Ras, Epidermal growth factor
Mitogen-activated protein (MAP) kinases are a family of serine/threonine kinases, which include extracellular signal-regulated protein kinases (ERKs), JNKs, and p38, that can be activated by a wide range of extracellular signals (Johnson and Vaillancourt, 1994; Robbins et al., 1994; Cobb and Goldsmith, 1995; Hawes et al., 1995; Luttrell et al., 1997). MAP kinases are central intermediates in signal transduction from the cell surface to the cytoplasm and nucleus (Pelech and Sanghera, 1992; Seth et al., 1992; Avruch et al., 1994; Guan, 1994; Davis, 1995; Waskiewicz and Cooper, 1995). The ERK phosphorylation cascade can be evoked by growth factors acting through tyrosine kinase receptors and a series of protein-protein interactions that include the mediation of the GTP binding protein Ras (Dent et al., 1992; Howe et al., 1992; Lange-Carter et al., 1993; Creedon et al., 1996).
Several distinct mechanisms of activation of the ERK pathway by GTP binding regulatory protein (G protein)-coupled receptors have recently been discovered. There is evidence to suggest that the different pathways used to modulate ERK activity may depend on the type of the G protein (Gi, Gs, or Gq) that is coupled to the activated receptor as well as the type of signaling systems present in a given cell population (Crespo et al., 1994; Hawes et al., 1995; Van Biesen et al., 1996). Gi- and Gq-coupled receptors stimulate ERK1 and ERK2 via distinct signaling pathways and different G protein subunits (Hawes et al., 1995). The βγ dimers released from Gi on agonist stimulation activate Ras, possibly through phosphatidylinositol 3-kinase γ in some cells as recently suggested (Lopez-Ilasaca et al., 1997; Luttrell et al., 1997). Activated Ras stimulates the serine threonine kinase Raf, which then phosphorylates ERK kinase (MEK), leading to the phosphorylation and activation of both ERK1 (44 kDa) and ERK2 (42 kDa). On the other hand, Gαq acts on phospholipase Cβ resulting in increases in intracellular Ca2+ concentration and protein kinase C (PKC) activation, which in turn network with the ERK pathway, at different points of interaction (Ahn et al., 1992; De Vivo and Iyengar, 1994; Crespo et al., 1995; Hawes et al., 1995). Moreover, cell types appear to play a role in determining the mechanism of interaction (Van Biesen et al., 1996). In Chinese hamster ovary (CHO) cells, platelet-activating factor receptors and ml muscarinic receptors activate ERK through pertussis toxin (PTX)-sensitive, endogenously available Go, and this process is PKC-dependent but Ras-independent. In contrast, in African green monkey kidney (COS-7) cells, α1-adrenergic and ml receptors activate ERK via a PTX-insensitive mechanism. Once activated, a fraction of phosphorylated ERK is translocated into the nucleus where it regulates the activity of various transcription factors (Johnson and Vaillancourt, 1994; Luttrell et al., 1997). In addition, many other proteins can be phosphorylated by activated ERK, including other protein kinases, cytoskeletal proteins, and other enzymes.
Because of their pleiotropic potential, ERK activities are tightly controlled by both positive and negative mechanisms (Robbins et al., 1993; Cobb and Goldsmith, 1995). In mammals, the highest levels of ERK mRNA were found in brain and spinal cord (Boulton et al., 1991). Recently, the regional distributions of ERK and MEK were reported in brain (Ortiz et al., 1995). A growing body of evidence suggests that ERK is also modulated by opioids. Chronic but not acute administration of morphine to rats increased ERK1 catalytic activity specifically in the ventral tegmental area of the brain (Berhow et al., 1996). ERK2 was stimulated in a μ-opioid receptor (μ-OR) stably transfected CHO cell line by different opioid agonists (Li and Chang, 1996). Our understanding of the mechanism by which opioid agonists regulate ERK activity is rudimentary. In earlier studies on opioid superactivation of adenylyl cyclase, COS-7 cells were also transiently cotransfected with μ-OR and hemagglutinin (HA)-tagged ERK2. Both acute (5-min) and chronic (4-h) treatment with [d-Ala2,Me-Phe4,Gly-ol5]enkephalin (DAMGE) stimulated the activity of HA-ERK2, and this activation was completely abolished by an N17-Ras dominant negative mutant (Avidor-Reiss et al., 1996). Fukuda et al. (1996) reported that μ, δ, and κ receptors are functionally coupled to ERK2 in stably cotransfected CHO cell lines. The PTX-sensitive activation of ERK2 in their system was proposed to involve PKC and a tyrosine protein kinase. Burt et al. (1996) demonstrated that recombinant δ-OR in Rat-1 fibroblasts was able to initiate activation of ERK1 and ERK2 in a Gi-dependent manner.
Of the MAP kinases, the ERK isozymes have been implicated in cell proliferation. In prior studies on fetal brain cell aggregates and C6 glioma cell proliferation, activation of μ- and κ-ORs were shown to modulate DNA synthesis by PTX-insensitive and -sensitive mechanisms, respectively (Barg et al., 1992, 1993a, 1994). Both κ-opioid stimulation and inhibition of DNA synthesis were observed depending on the time of culture of fetal brain cell aggregates (Barg et al., 1993a). Chronic morphine treatment results in the inhibition of C6 cell proliferation. In spinal cord-dorsal root ganglion cocultures κ-opioid agonists increased thymidine incorporation into DNA (Barg et al., 1993b). Because ERKs have been implicated in cell proliferation, the possibility exists that ERK1 and ERK2 may play a role in opioid modulation of DNA synthesis.
Here we use COS-7 cells transiently cotransfected with κ-, δ-, or μ-OR and ERK1- or HA-ERK2-containing plasmids. The effects of receptor type-specific agonists on ERK1 or HA-ERK2 activity were examined in a time- and concentration-dependent manner. Evidence for the involvement of PTX-sensitive G protein and a Ras/Gβγ -dependent mechanism of opioid activation of ERK1 and ERK2 is presented. In addition, chronic opioid treatment inhibited epidermal growth factor (EGF)-induced ERK1 activity.
Experimental Procedures
Chemicals
Chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.) with the exception of DAMGE, [d-Pen2,d-Pen5]enkephalin (DPDPE), and [3H] DAMGE (37 Ci/mmol) from Multiple Peptide Systems (San Diego, CA, U.S.A.), [3H]U69593 (56 Ci/mmol) and [γ-32P]ATP (3,000 Ci/mmol) from Amersham (Arlington Heights, IL, U.S.A.), anti-phosphorylated ERK antibody (anti-Active MAPK pAb) from Promega (Madison, WI, U.S.A.), 12CA5 monoclonal antibody from Babco (Berkeley, CA, U.S.A.), EGF (human, recombinant) from GibcoBRL, Life Technologies (Grand Island, NY, U.S.A.), norbinalthorphimine (nor-BNI) from RBI (Natick, MA, U.S.A.), and unlabeled U69593, CTAP (d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Peh-Thr-NH2) and naltrindole from NIDA Drug Supply (Research Triangle, NC, U.S.A.). We thank Drs. H. Akil (University of Michigan) for μ- and κ-OR plasmids, J. Baldassare (St. Louis University) for ERK1 in pcDNA-3 vector, C. Evans (UCLA) for a δ-OR vector, S. Gutkind (National Institutes of Health) for HA-ERK2 in pPcDNAI AMP, dominant negative mutant N17-Ras in pPcDNAIII, CD8 in pPcDNAI AMP, and CD8-β-adrenergic receptor kinase-C (CD8-βARK-C) in pPcDNAIII, and G. Toth (Hungarian Academy of Science) for [3H]naltrindole (44.6 Ci/mmol).
Cell culture growth and transfection
COS-7 cells were grown at 37°C in a humidified CO2 (5%) incubator in Dulbecco's modified Eagle's medium and Ham's nutrient mixture F12 containing 10% heat-inactivated calf serum. Cells were grown in 10-cm-diameter Petri dishes and routinely seeded 24 h before transfection to achieve 50–60% confluency on the day of transfection. Transfections were performed with rat μ- or κ-OR cDNA (pCMV-neo expression vector) or mouse δ-OR cDNA (pCI-neo expression vector) (3–5 μg per dish) in the presence or in the absence of ERK1 cDNA (1–2 μg per dish) in pcDNA-3 vector using the DEAE-dextran method (Avidor-Reiss et al., 1996). In some experiments, COS-7 cells were cotransfected with either the dominant negative mutant N17-Ras (2 μg per dish) or a cDNA chimera, designated pcDNA-CD8– βARK-C (2 μg per dish), which expresses the extracellular and transmembrane domains of CD8 fused to an intracellular domain containing the carboxyl terminus of βARK (the βγ binding portion) (Crespo et al., 1995). For ERK2 experiments COS-7 cells were cotransfected with cDNA for δ-OR and an expression plasmid containing an amino-terminal HA-tagged murine ERK2 (HA-ERK2) as previously described (Avidor-Reiss et al., 1996).
Binding experiments
Binding studies were performed as described (Belcheva et al., 1993). Cells were harvested 48 h after transfection and homogenized by gentle disruption in a “cell cracker” (Belcheva et al., 1993). A membrane fraction (P20) was prepared from cell homogenates by sedimenting a 1,000-g supernatant at 20,000 g. A cocktail containing 10 μg/ml leupeptin, 2 μg/ml pepstatin A, 200 μg/ml bacitracin, and 1 mM phenylmethylsulfonyl fluoride was added to the buffers used for preparation of this membrane fraction.
ERK assays
ERK1 activity was analyzed using the “in-gel” kinase method of Kameshita and Fujisawa (1989) with some modifications. In a 24-48-h culture, OR/ERK1 cotransfected cells from each dish were serum-starved overnight. Cell cultures were then exposed to opioids (agonist with or without antagonist) in the presence or absence of PTX under conditions described in the figure legends. In certain experiments, cultures were treated with EGF (100 ng/ml) for 5 min. Then cells were washed with cold phosphate-buffered saline and lysed with buffer containing 20 mM HEPES, 10 mM EGTA, 40 mM β-glycerophosphate, 2.5 mM MgCl2, 2 mM sodium vanadate, 1% Nonidet-40, 1 mM phenylmethylsulfonyl fluoride, 20 μg/ml aprotinin, and 20 μg/ml leupeptin as described by Crespo et al. (1995). Lysates were spun at 14,000 g for 20 min at 4°C, and protein concentration of the supernatants was determined before enzyme assay. Cell lysates (60–100 μg of protein per lane) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The resolving gels contained 0.5 mg/ml myelin basic protein (MBP). To remove sodium dodecyl sulfate after electrophoresis, gels were washed twice with 100 ml of 50 mM Tris-HC1 (pH 8.0) containing 20% isopropyl alcohol for 30 min. Then a 1-h wash with 50 mM Tris-HCl (pH 8.0) and 5 mM β-mercaptoethanol was followed by a protein denaturation step with 100 ml of 6 M guanidine chloride (twice, 30 min each). Finally, the proteins in the gels were renatured with buffer containing 50 mM Tris-HCl (pH 8.0), 5 mM β-mercaptoethanol, and 0.04% Tween 40 for 16 h at 4°C with at least four or five changes of buffer. Gels were incubated with 40 mM HEPES buffer (pH 8.0) containing 2 mM dithiothreitol and 10 mM MgCl2 at room temperature for 1 h. The kinase assay was performed by placing the gels in 40 mM HEPES buffer (pH 8.0) containing 0.5 mM EGTA, 10 mM MgCl2, 2 μM protein kinase inhibitor, 40 μM unlabeled ATP, and 50 μCi of [γ-32P]ATP for 3 h at room temperature. Then gels were rinsed with a solution of 5% trichloroacetic acid and 1% sodium pyrophosphate to remove noncovalently bound 32P. After drying, gels were exposed to Kodak BIOMAX MS film for 1–4 h at −70°C.
ERK2 activity was determined using HA-ERK2 immunoprecipitates of cotransfected COS-7 cells as described (Avidor-Reiss et al., 1996). COS-7 cells were cotransfected with the cDNA for δ-OR and an expression plasmid containing HA-ERK2 cDNA (Her et al., 1993; Crespo et al., 1994). Determination of ERK2 activation by opioid treatment was performed 48 h after transfection. Overnight serum-starved cells were exposed to opioids for the times indicated, the cells were lysed, and HA-ERK2 was immunoprecipitated using a specific monoclonal antibody (12CA5) against the HA moiety, followed by application of protein G-Sepharose beads. The beads were washed three times and incubated for 30 min at 30°C with 45 μg of MBP and 1 μCi of [γ-32P] ATP in a total volume of 30 μl of kinase reaction buffer (Crespo et al., 1995). The reaction was terminated by addition of 5× Laemmli buffer, and the mixture was boiled and separated by polyacrylamide gel electrophoresis. Phosphorylated MBP was visualized by autoradiography. For details, see Crespo et al. (1994, 1995) and Avidor-Reiss et al. (1996).
Kinase activity, as expressed by the degree of phosphorylation of MBP, was quantified using the Phosphorlmager (Molecular Dynamics) and Image Quant software. Background values were subtracted from the values determined for each band. Observed density changes are expressed as percentages of control values, which were taken as 100%.
Immunoblot analysis
COS-7 cells cotransfected with ERK1 and μ- or κ-receptor cDNA were lysed, and lysate proteins were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (4 μg protein per lane). Proteins were blotted on an ImmobilonP membrane, and immediately after transfer, the membrane was incubated with 3% H2O2 for 15 min. Nonspecific sites were blocked with 1% casein in Tris-buffered saline plus 0.2% Tween-20. The blot was then washed three times with Tris-buffered saline plus 0.2% Tween-20 and incubated with anti-phosphorylated ERK antibody diluted 1:20,000 in Tris-buffered saline plus 0.2% Tween-20 for 1 h at room temperature. After five washes with Tris-buffered saline plus 0.2% Tween-20 blots were incubated with 1:5,000 diluted goat anti-rabbit horseradish peroxidase-conjugated IgG for 1 h. Bands were visualized with a chemi-luminescence detection system.
Protein assays
Protein concentrations were determined by the method of Bradford (1976) with bovine serum albumin as the standard.
Statistical analysis
Data analyses were performed using ANOVA, Bartlett's test for homogeneity of variance, and/or post hoc Tukey-Kramer multiple comparisons test.
Results
Binding and ERK1 activity assays in OR/ERK1 transfected COS-7 cells
Cultures were cotransfected with μ-, δ-, or κ-OR and ERK1 cDNA-containing plasmids. High-affinity μ (DAMGE; KD = 1.5 ± 0.3 nM; Bmax = 854–1,070 fmol/mg of protein), δ (naltrindole; KD = 0.7 ± 0.4 nM; Bmax = 1,275–2,313 fmol/mg of protein), or κ (U69593; KD = 3.3 ± 0.3 nM; Bmax = 823–2,341 fmol/mg of protein) binding was detected in the corresponding transfected lines. Binding was not detected in the parental line. 125I-β-Endorphin cross-linking experiments with all three types of recombinant receptors in COS-7 cells corroborated the binding data (Belcheva et al., 1996 and manuscript in preparation).
ERK1 activity was measured by “in-gel” kinase analyses using MBP as a substrate, and several protein kinases were detected in COS-7 cell lysates (Fig. 1A). Assignment of the 44-kDa band to ERK1 is consistent with previous molecular size data (Robbins et al., 1993; Cobb and Goldsmith, 1995) and was confirmed by immunoblotting (Fig. 1B). Activated ERK was detected by a polyclonal antibody that interacts preferentially with the phosphorylated form of this enzyme. The presence of significantly higher ERK1 activity in DAMGE (1 μM)-stimulated μ-OR/ERKl cotransfected cells is demonstrated in Fig. 1A. Nontransfected cultures or cells transfected only with the OR or ERK1 cDNA alone served as negative controls in this experiment. DAMGE treatment of the cells did not affect the activity of the other four kinases detected in the gel (Fig. 1A).
FIG. 1.
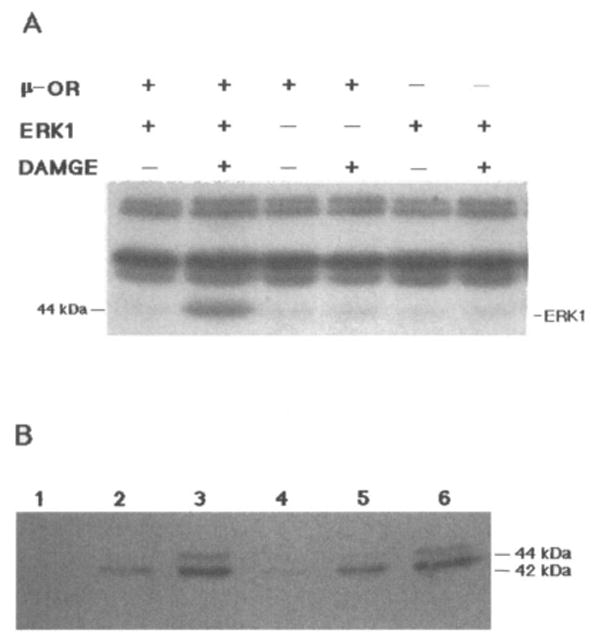
A: Stimulation of ERK1 activity by μ-opioid agonist. COS-7 cells transiently cotransfected with μ-OR with or without ERK1 cDNA were treated with 1 μM DAMGE for 10 min, and cell lysates were tested for ERK1 activity using the “in gel” assay as described in Experimental Procedures. A representative autoradiogram is shown with a band corresponding to ERK1 (44 kDa) and higher-molecular-mass kinases. B: Immunoblot analysis of activated ERK. In lanes 1–3 COS-7 cells were cotransfected with μ-OR and ERK1 cDNA: lane 1, nonstimulated cells; lane 2, cells treated with 1 μM DAMGE; and lane 3, serum-stimulated cells. In lanes 4–6 COS-7 cells were cotransfected with κ-OR and ERK1 cDNA: lane 4, nonstimulated cells; lane 5, cells treated with 1 μM U69593; and lane 6, serum-stimulated cells. These experiments were repeated twice.
Time- and concentration-dependent stimulation of ERK1 and ERK2 activity by opioid agonists
Time course studies for ERK1 activation were performed with DAMGE in μ-OR transfected cells or with U69593 in κ-OR transfected cells. Similarly, ERK2 activity was determined after different intervals of DPDPE exposure in δ-OR transfected COS-7 cells. Agonist-induced ERK1 and ERK2 stimulation was time-dependent, with a maximal effect observed at 5–10 min, which persisted for 30–60 min and decreased during the next 4–48 h of exposure (Fig. 2).
FIG. 2.
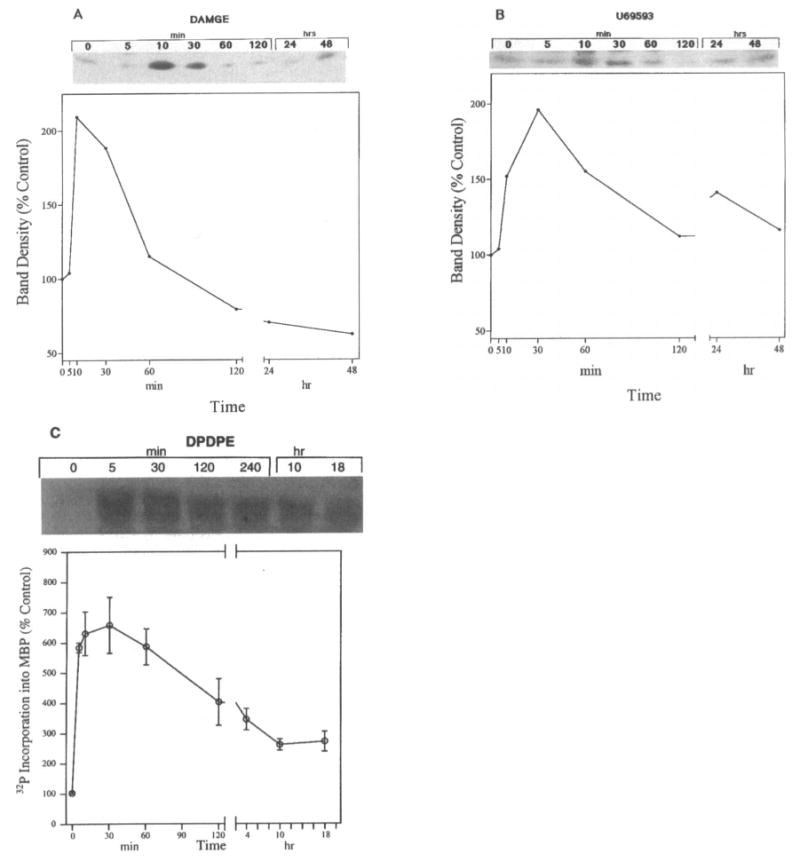
Time-dependent stimulation of ERK1 and ERK2 activity by opioid agonists. COS-7 cells were cotransfected with μ- (A) or κ-OR (B) and ERK1 cDNA or δ-OR (C) and HA-ERK2 and treated with either 1 μM DAMGE (μ), 1 μM U69593 (κ), or 0.1 μM DPDPE (δ) for different intervals. Top: Representative autoradiograms show the phosphorylated MBP bands. Bottom: Quantification of ERK1 or HA-ERK2 activity. These experiments were repeated three times.
Based on the time course study, all subsequent experiments testing the effects of opioid agonists on ERK1 activity were performed by incubating the transfected cell cultures for 5–10 min with increasing concentrations of either DAMGE or U69593. Concentration-dependent stimulation of ERK1 activity is shown for μ- (Fig. 3A) and κ-OR (Fig. 3B). At concentrations as low as 10 nM opioid agonist, an appreciable enhancement of enzyme activity was observed. At the two highest concentrations tested (5–10 μM), the DAMGE induction plateaued at fourfold augmentation, whereas at the same concentrations, U69593 reached a maximal threefold induction of ERK1 activity (Fig. 3A and B). The δ-selective agonist DPDPE induced dose-dependent activation of HA-ERK2 as measured by the immunoprecipitation method (Fig. 3C). DPDPE stimulated HA-ERK2 activity at concentrations as low as 0.1 nM, and a fivefold maximal induction (using 0.1 and 1 μM DPDPE) was observed in these experiments.
FIG. 3.
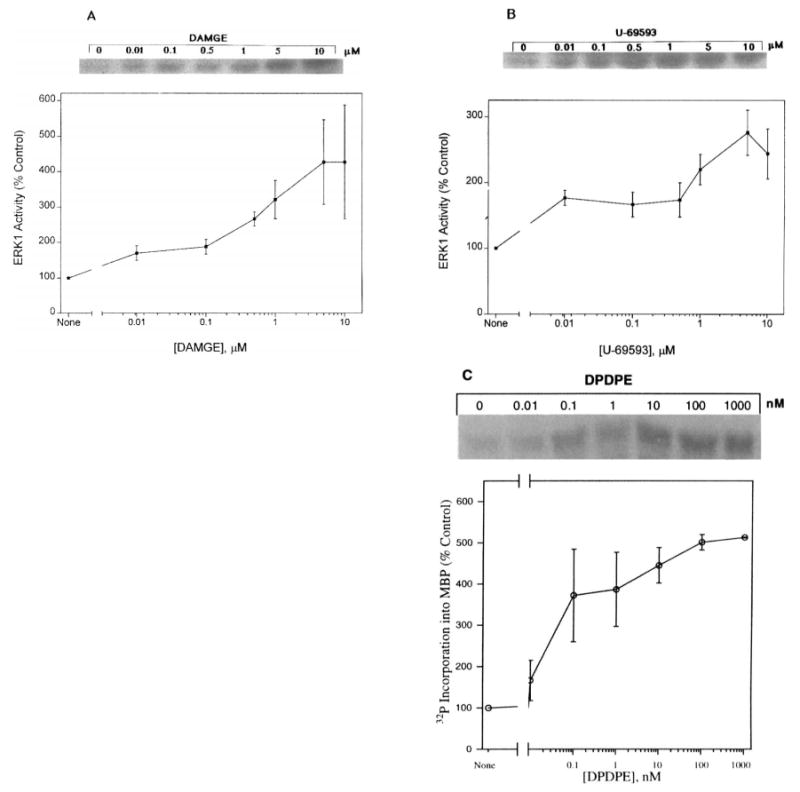
Concentration-dependent stimulation of ERK1 and ERK2 activity by opioid agonists. COS-7 cells were cotransfected with μ-, κ-, or δ-OR and ERK1 or ERK2 cDNA and treated with different concentrations of (A) DAMGE (μ) or (B) U69593 (κ) for 10 min and (C) DPDPE (δ) for 5 min. Top: Representative autoradiograms show the phosphorylated MBP bands. Bottom: Quantification of ERK1 and ERK2 activity. Data are mean ± SEM (bars) values from three or four experiments. Basal levels are different from all agonist-treated cell values: p < 0.05.
PTX and receptor type-specific opioid antagonists abolish ERK1 and ERK2 stimulation by opioid agonists
When OR/ERK1 cotransfected cells were preincubated with a μ- (CTAP), κ- (nor-BNI), or δ- (naltrindole) selective antagonist before treatment with the corresponding agonists, a significant reduction in the stimulation of ERK1 activity was observed (Fig. 4). Similar antagonism was observed for δ-opioid activation of HA-ERK2 (Fig. 5). These results, along with the evidence for discrete OR transfection by binding assay, confirm the mediation of ERK1 and ERK2 stimulation by μ-, δ-, or κ-OR. Activation of ERK1 by DPDPE (Fig. 4C) was comparable to the effect of this (δ-opioid agonist on ERK2 as described above (Fig. 3C).
FIG. 4.
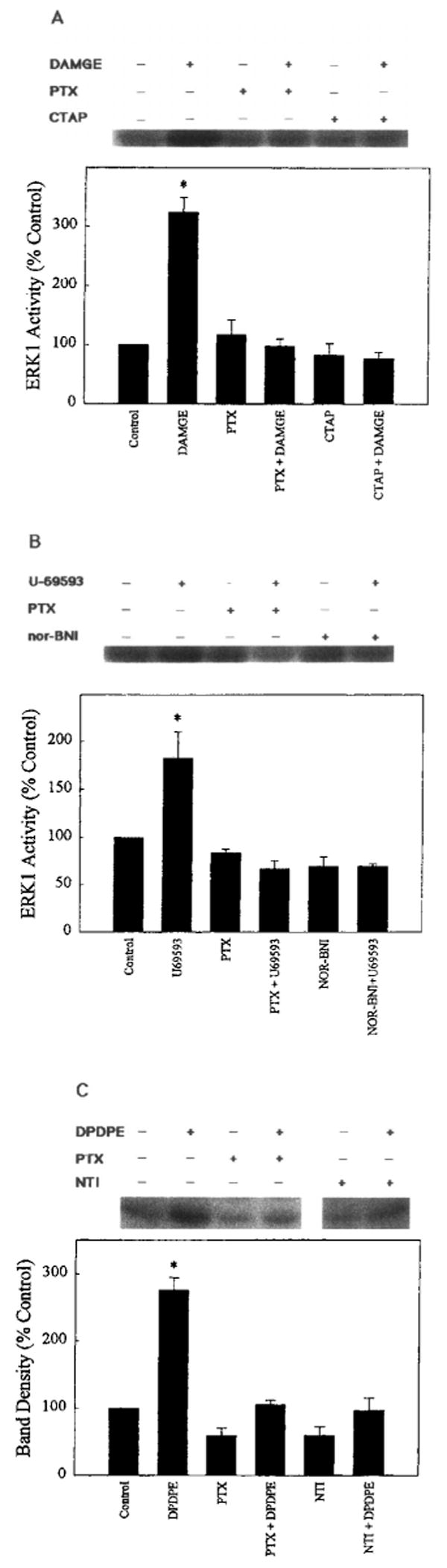
Inhibition of opioid agonist-induced ERK1 activity by PTX or antagonist. COS-7 cells were cotransfected with μ- or κ-OR and ERK1 cDNA and pretreated for 8 h with PTX (100 ng/ml) or for 1 h with (A) 1 μM CTAP (μ) before enzyme stimulation with DAMGE (100 nM), (B) 1 μM nor-BNI (κ) before enzyme stimulation with U69593 (100 nM), or (C) 1 μM naltrindole (NTI; δ) before enzyme stimulation with DPDPE (100 nM). ERK1 was stimulated with agonist for 10 min. Top: Representative autoradiograms show the phosphorylated MBP bands. Bottom: Quantification of ERK1 activity. Data are mean ± SEM (bars) values from two or three experiments. Significantly different from control and all other values: *p < 0.06.
FIG. 5.
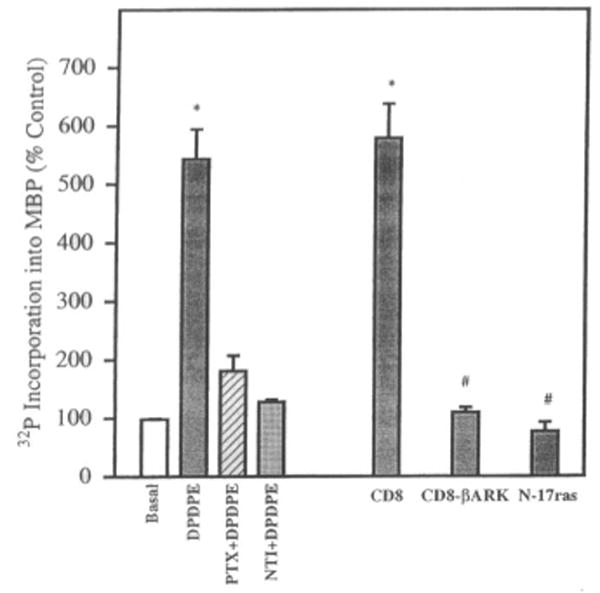
HA-ERK2 activation by a δ-agonist is PTX-sensitive and is abolished by a βγ scavenger and a Ras dominant negative mutant. COS-7 cells were cotransfected with cDNAs encoding HA-ERK2 and δ-OR and as indicated with CD8, CD8-βARK-C, and the dominant negative mutant N17-Ras. Cells were exposed to 1 μM DPDPE for 5 min. PTX, cells were pretreated for 18 h with 100 ng/ml PTX; NTI, the antagonist naltrindole (1 μM) was added 2 min before DPDPE. Data are mean ± SEM (bars) values of three experiments. Significantly different from control (not treated with DPDPE): *p < 0.001. Significantly different from DPDPE-stimulated CD8 cotransfected cells: *p < 0.001.
To investigate the role of G proteins in the activation of the ERK pathway by opioids, cotransfected cultures were pretreated with 100 ng/ml PTX in serum-free medium at least 8 h before exposure to opioid agonists. This concentration of PTX effectively ADP-ribosylates and inactivates Gi and Go proteins in COS-7 cells (Crespo et al., 1994). On PTX treatment, μ-, δ-, and κ-opioid agonist-stimulated ERK1 and δ-induced HA-ERK2 activities were reduced to control levels (Figs. 4 and 5). This finding implies the involvement of G proteins, presumably Gi/o, in the enzyme activation process.
Effect of a Gβγ scavenger and dominant negative mutant N17-Ras on ERK1 and ERK2 activity
To determine the role of Ras in opioid agonist-induced ERK activation, OR/ERK transfected cells were cotransfected with cDNA encoding N17-Ras, which is a dominant inhibitory mutant. The results show that opioid agonist-induced ERK1 and ERK2 activation was significantly attenuated by N17-Ras (Figs. 5 and 6), suggesting that opioid agonists acting via their receptors modulate ERK1 and ERK2 by a Ras-dependent pathway. As a positive control, we tested EGF stimulation of ERK1 activity in the absence and presence of N17-Ras (Fig. 6).
FIG. 6.
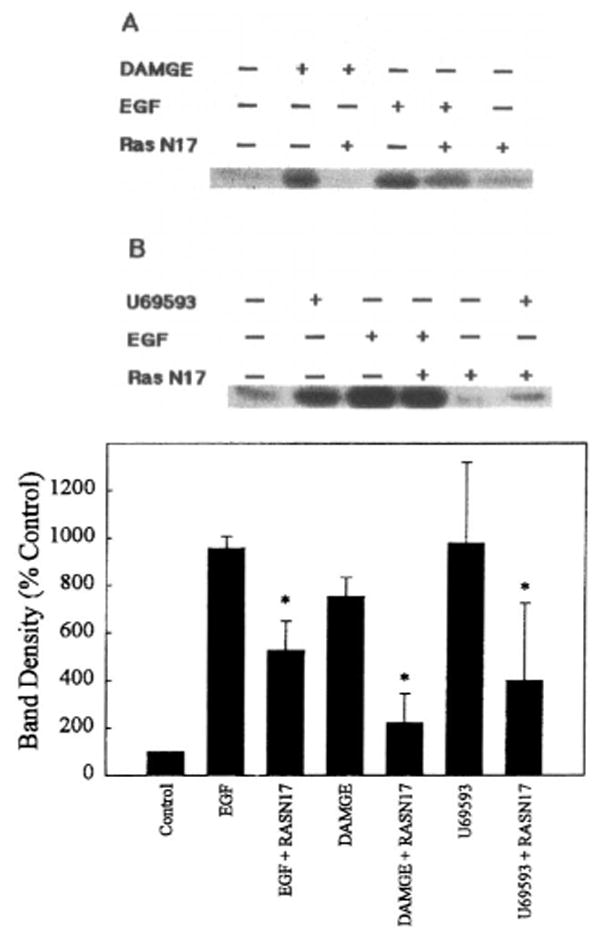
ERK1 activation by opioid agonists is attenuated by a Ras dominant negative mutant. COS-7 cells were cotransfected with cDNAs encoding ERK1, μ- or κ-OR, and the dominant negative mutant N17-Ras. Cells were exposed either to EGF (100 ng/ml) for 5 min or to the corresponding opioid ligand at 1 μM for 10 min. Top: Representative autoradiograms show the phosphorylated MBP bands for (A) μ- or (B) κ-opioid ligands. Bottom: Quantification of ERK1 activity. Data are mean ± SEM (bars) values from three to five experiments. Significantly different from ERK activation in the absence of Ras: *p < 0.05.
The involvement of Gβγ in ERK1 and ERK2 stimulation was studied with cultures cotransfected with the cDNA of CD8– βARK-C, a fusion plasmid that carries the carboxyl terminus of βARK (the βγ binding portion) and the amino terminus of CD8 (which allows anchoring to the membrane). The presence of this scavenger of βγ dimers abolished ERK activity induced by the corresponding opioid agonists, whereas the CD8 fragment by itself had no effect (Figs. 5 and 7). Previous immunofluorescence analysis of intact transfected COS-7 cells revealed that both CD8 and the CD8– βARK-C chimera were efficiently expressed and that CD8– βARK-C abolished the activation of MAP kinase by β-adrenergic and muscarinic receptor agonists (Crespo et al., 1994, 1995; Coso et al., 1996).
FIG. 7.
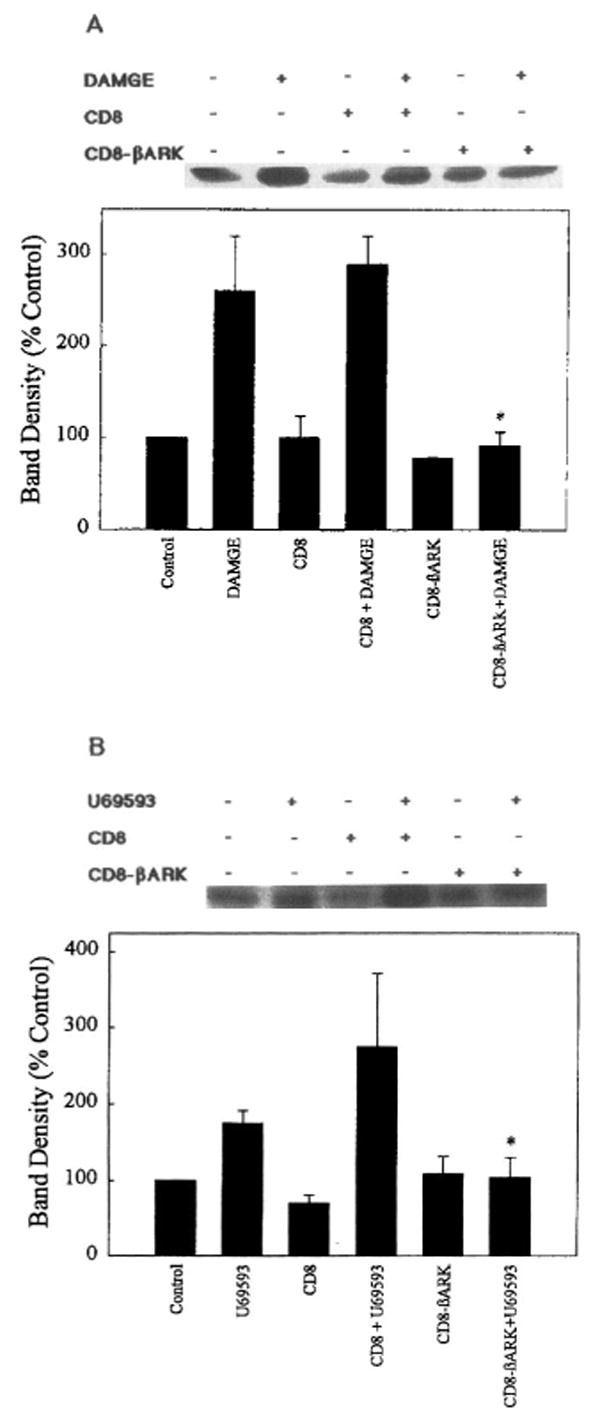
Inhibition of κ- and μ-agonist-stimulated ERK1 activity by Gβγ sequestration. COS-7 cells were cotransfected with cDNA encoding (A) μ- or (B) κ-OR, ERK1, and CD8 or CD8– βARK-C. Cells were then treated with either 0.1 μM DAMGE (μ) or 0.1 μM U69593 (κ) for 10 min before determination of ERK1 activity. Top: Representative autoradiograms showing phosphorylated MBP bands. Bottom: Quantification of ERK1 activity. Data are mean ± SEM (bars) values from two or three experiments. Significantly different from ERK activation by agonist with or without CD8: *p < 0.05.
Opioids inhibit EGF stimulation of ERK1 activity by a mechanism that requires the presence of Gβγ
In previous studies of primary brain cultures and C6 glioma cells, chronic opioid treatment resulted in the attenuation of mitogen-mediated DNA synthesis and cell proliferation (Barg et al., 1992, 1993a, 1994). As seen in Fig. 2, chronic (>60-min) opioid treatment of transfected COS-7 cells leads to a reduction of ERK activation when compared with the acute (10-min) opioid effect. To investigate the mechanism further, we studied the effect of opioid agonists on EGF activation of ERK1. For this purpose, μ- or κ-OR and ERK1 transfected COS-7 cells were treated with 1 μM DAMGE or U69593, respectively, for different intervals (0, 2, 24, and 48 h), and then the cultures were exposed to EGF (100 ng/ml) for 5 min. As shown in Fig. 8, the two opioid agonists induced a significant inhibition of EGF-stimulated ERK1 activity at the 2- and 24-h time points. The 2-h opioid agonist effects were reversed by the antagonists CTAP for μ and nor-BNI for κ (data not shown).
FIG. 8.
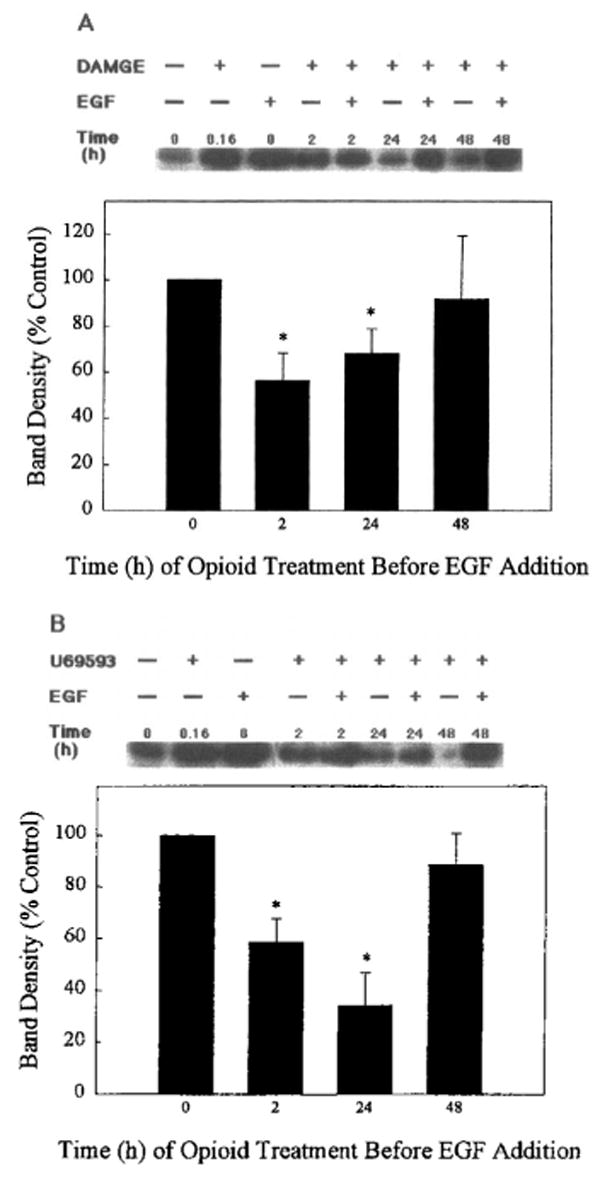
Time-dependent inhibition of EGF activation of ERK1 by chronic opioid agonist treatment. COS-7 cells were cotransfected with (A) μ- or (B) κ-OR and ERK1 cDNA and treated with either 1 μM DAMGE (μ) or 1 μM U69593 (κ) for different intervals, and then cells were exposed to EGF (100 ng/ml) for 5 min. Top: Representative autoradiograms show the phosphorylated MBP bands. Time signifies the interval of opioid exposure. Bottom: Quantification of ERK1 activity after pretreatment of cells with opioids for different intervals and addition of EGF for 5 min. Data are mean ± SEM (bars) values from three to five experiments. Significantly different from EGF alone: *p < 0.05.
The inhibitory effect of chronic (2-h) κ- but not μ-opioid exposure was sensitive to the presence of PTX (Fig. 9). This finding again implies the involvement of G proteins, presumably Gi/o, at least for the κ-opioid action. However, reversal of both μ- and κ-agonist actions by CD8– βARK-C (Fig. 9) suggests that the presence of βγ subunits is required as seen for μ- and κ-opioid-mediated activation of ERK (Fig. 7). In addition as a negative control, we show that EGF stimulation of ERK1 activity is not affected by the presence of CD8– βARK-C (Fig. 9). These results demonstrate cross-talk between the Gi/o-coupled ORs and EGF tyrosine kinase signaling pathways.
FIG. 9.
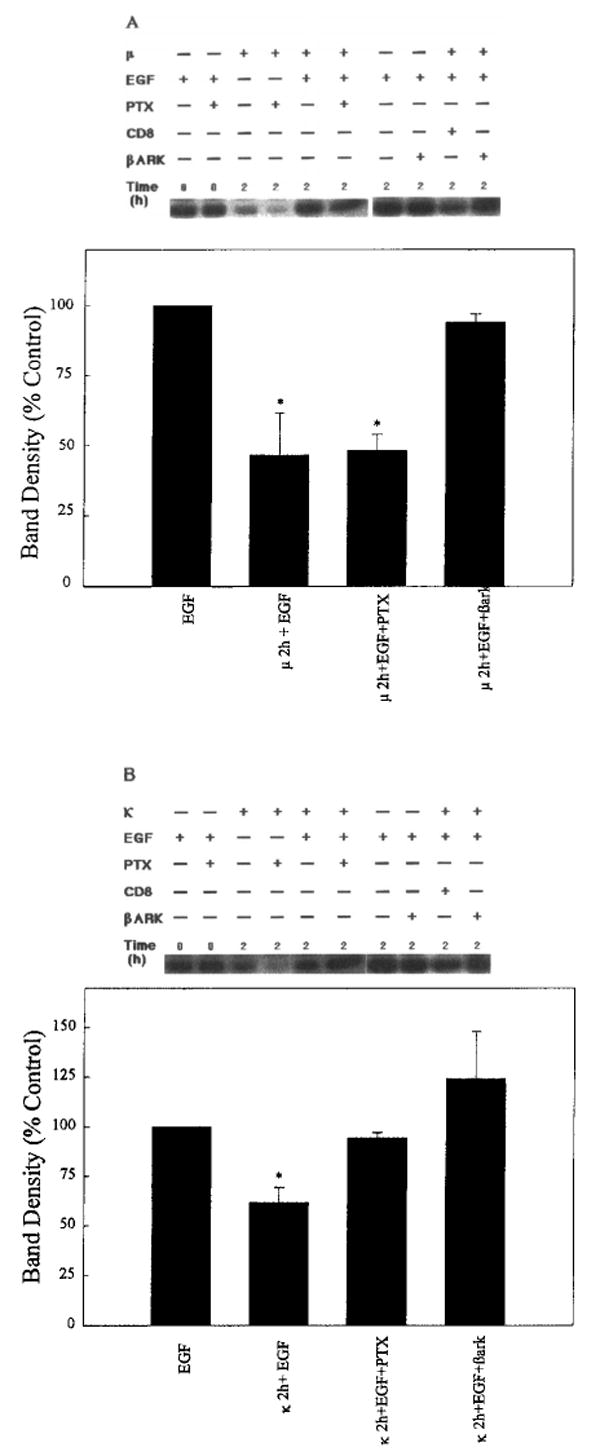
Opioid inhibition of EGF-induced ERK1 activity requires the presence of Gβγ. COS-7 cells were cotransfected with (A) μ- or (B) κ-OR and ERK1 cDNA in the presence or absence of CD8 or CD8– βARK-C cDNA. Some cultures were grown in the presence of PTX (100 ng/ml) for at least 15 h before treatment with 1 μM DAMGE (μ) or 1 μM U69593 (κ) for 2 h. ERK1 activity was stimulated with EGF (100 ng/ml) for 5 min. Top: Representative autoradiograms show the phosphorylated MBP bands. Time signifies the interval of opioid exposure. Bottom: Quantification of ERK1 activity after opioid and EGF treatment. Data are mean ± SEM (bars) values from two or three experiments. Significantly different from EGF alone: *p < 0.05.
Discussion
Here we examined the mechanism of opioid regulation of ERK1 and ERK2 in COS-7 cells and demonstrated that these members of the MAP kinase family are downstream components of the signal transduction cascade of μ-, δ-, and κ-ORs. Although opioid activation of ERK isozyme activity has been detected both in vivo and in vitro, previous in vitro investigations have focused on opioid activation of ERK2 activity (Ortiz et al., 1995; Avidor-Reiss et al., 1996; Berhow et al., 1996; Burt et al., 1996; Fukuda et al., 1996; Li and Chang, 1996). Nevertheless, ERK1 differs from ERK2 in some properties (Robbins et al., 1994; Cobb and Goldsmith, 1995). Moreover, we have now provided evidence to implicate Gβγ in opioid regulation of ERK1 and ERK2 activity and demonstrated that chronic opioid exposure modulates ERK activation by EGF.
The results obtained here show that the μ-, δ-, and κ-agonists DAMGE, DPDPE, and U69593, respectively, can rapidly stimulate ERK1 and ERK2 activity in μ-, δ-, or κ-OR (respectively) cotransfected COS-7 cells. This activation is ligand concentration-dependent and is reduced to control levels by preincubation of the cells with receptor type-specific antagonists, suggesting an OR-mediated process. Furthermore, opioid agonists stimulate ERK1 and ERK2 activity via ORs that are coupled to PTX-sensitive G proteins. There is evidence to suggest that βγ subunits are involved in the stimulation of ERK by other Gi-coupled receptors (Crespo et al., 1994; Koch et al., 1994). The results of this study with CD8– βARK-C and the Ras dominant negative mutant N17-Ras suggest that opioid stimulation of ERK1 and ERK2 activity is mediated by βγ subunits of PTX-sensitive heterotrimeric G proteins and is Ras dependent. Although it has been shown that Ras controls the activity of ERKs, recent data support a role for Ras and Racl in the regulation of JNK activity as well (Coso et al., 1996). Therefore, it can be concluded that βγ heterodimers provide a link between cell surface receptors coupled to PTX-sensitive heterotrimeric G proteins and small GTP-binding proteins.
Previously we established that chronic opioid treatment affects cell proliferation in various types of primary neural cultures. DNA synthesis in both 14-day brain cell aggregates (Barg et al., 1993a) and spinal cord–dorsal root ganglion cocultures (Barg et al., 1993b) was stimulated by opioid agonists via a PTX-sensitive mechanism. Because ERK1 and ERK2 are known to mediate the mitogenic actions of growth factors (Robbins et al., 1994), it is possible that these MAP kinases are involved in this pathway. Although the results presented here were performed with OR and ERK transfected COS-7 cells, they are relevant to the above-mentioned primary neural cells that normally express ORs.
In this study, conditions under which chronic opioid treatment attenuated mitogen-mediated cell proliferation and DNA synthesis, as observed in early stages of fetal brain cell aggregate and C6 glioma cells (Barg et al., 1992, 1993a, 1994), were also investigated. As COS-7 cells are known to respond to EGF, we investigated the effect of chronic opioid treatment on the activation of ERK 1 by this growth factor. Preincubation of μ- / κ-OR and ERK1 transfected cell cultures with corresponding agonists for 2 and 24 h inhibited EGF stimulation of ERK-1 activity. Moreover, this chronic treatment with μ- and κ-opioids inhibits EGF-induced ERK1 activation via βγ subunits akin to the mechanism of acute opioid stimulatory action on ERK in this system. The unique PTX insensitivity of the chronic μ-opioid action seen here is reminiscent of that observed for μ but not κ agonists in fetal brain cell aggregates (Barg et al., 1992) and in rat C6 glioma cells (Barg et al., 1994). Antagonist blockade indicates the involvement of μ- and κ-ORs in this inhibitory effect.
These results complement findings showing that the EGF receptor is rapidly phosphorylated on treatment of Rat-1 cells with one of several different agonists of G protein-coupled receptors (Daub et al., 1996). In COS-7 cells, this process was shown to be Src-dependent and to involve Shc (Luttrell et al., 1997). Taken together, the data suggest a role of tyrosine kinase receptors and associated nonreceptor tyrosine kinases (Src) as downstream mediators of G protein-coupled mitogenic signaling via βγ subunits. Interactions with both acute and chronic opioid systems then may elicit a ligand-independent tyrosine kinase receptor activation that positively affects the MAP kinase cascade. Alternatively, chronic opioid actions may elicit a desensitization of a tyrosine kinase receptor or nonreceptor tyrosine kinase that attenuates ligand-dependent stimulation (Daub et al., 1996; Luttrell et al., 1997). Because the MAP kinase phosphorylation cascade has the potential to be feedback-regulated, yet another possibility is that ERK itself can become overactivated and phosphorylate an upstream element to attenuate the signal (Davis, 1995). Opioid activation of ERK in the absence of EGF also declines after 2 h (see autoradiograms in Fig. 8), suggesting possible desensitization of the G protein-coupled receptor system as well. In fact, opioid receptor levels may be down-regulated under these conditions. Subsequently (24–48 h), an increase in ERK levels may ensue to explain the rebound seen (Fig. 8). Elucidation of the PTX-sensitive and -insensitive mechanisms of convergence of opioid and tyrosine kinase receptor pathways will allow the development of new strategies to modulate cell proliferation.
Acknowledgments
This work was supported in part by grants DA05412 (to C.J.C.) and DA06265 (to Z.V.) from the National Institute on Drug Abuse and funds from the Forschheimer Center for Molecular Genetics (to Z.V.) and the Henry and Anne Reich Research Fund for Molecular Research (to Z.V.). We thank Dr. David Ford for sharing his expertise in conducting “in gel” ERK assays and Mirya Kim for technical assistance.
Abbreviations used
- βARK
β-adrenergic receptor kinase
- CHO
Chinese hamster ovary
- COS
African green monkey kidney
- DAMGE
[d-Ala2,Me-Phe4,Gly-ol5]enkephalin
- DPDPE
[d-Pen2,d-Pen5]enkephalin
- EGF
epidermal growth factor
- ERK
extracellular signal-regulated protein kinase
- G protein
GTP binding regulatory protein
- HA
hemagglutinin
- MAP
mitogen-activated protein
- MBP
myelin basic protein
- MEK
extracellular signal-regulated protein kinase kinase
- nor-BNI
norbinalthorphimine
- OR
opioid receptor
- PKC
protein kinase C
- PTX
pertussis toxin
Contributor Information
Mariana M. Belcheva, E. A. Doisy Department of Biochemistry and Molecular Biology, St. Louis University School of Medicine, St. Louis, Missouri, U.S.A.
Zvi Vogel, Department of Neurobiology, Weizmann Institute of Science, Rehovot.
Elena Ignatova, E. A. Doisy Department of Biochemistry and Molecular Biology, St. Louis University School of Medicine, St. Louis, Missouri, U.S.A..
Tomer Avidor-Reiss, Department of Neurobiology, Weizmann Institute of Science, Rehovot.
Renata Zippel, Department of Neurobiology, Weizmann Institute of Science, Rehovot.
Rivka Levy, Department of Neurobiology, Weizmann Institute of Science, Rehovot.
Eric C. Young, E. A. Doisy Department of Biochemistry and Molecular Biology, St. Louis University School of Medicine, St. Louis, Missouri, U.S.A.
Jacob Barg, Department of Neurobiology, Weizmann Institute of Science, Rehovot, and Israel Rammot Yehuda Therapeutic Community, Zoharim, Israel.
Carmine J. Coscia, E. A. Doisy Department of Biochemistry and Molecular Biology, St. Louis University School of Medicine, St. Louis, Missouri, U.S.A.
References
- Ahn NG, Robbins DJ, Haycock JW, Seger R, Cobb MH, Krebs EG. Identification of an activator of the microtubule-associated protein 2 kinases ERK1 and ERK2 in PC 12 cells stimulated with nerve growth factor or bradykinin. J Neurochem. 1992;59:147–156. doi: 10.1111/j.1471-4159.1992.tb08885.x. [DOI] [PubMed] [Google Scholar]
- Avidor-Reiss T, Nevo I, Levy R, Pfeuffer T, Vogel Z. Chronic opioid treatment induces adenylyl cyclase V superactivation. J Biol Chem. 1996;271:21309–21315. doi: 10.1074/jbc.271.35.21309. [DOI] [PubMed] [Google Scholar]
- Avruch J, Zhang XF, Kyriakis JM. Raf meets ras: completing the framework of a signal transduction pathway. Trends Biochem Sci. 1994;19:279–283. doi: 10.1016/0968-0004(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Barg J, Belcheva MM, Coscia CJ. Evidence for the implication of phosphoinositol signal transduction in μ-opioid inhibition of DNA synthesis. J Neurochem. 1992;59:1145–1152. doi: 10.1111/j.1471-4159.1992.tb08357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barg J, Belcheva MM, Rowinski J, Coscia CJ. κ opioid agonist modulation of 3H-thymidine incorporation into DNA; evidence for the involvement of pertussis toxin-sensitive G protein-coupled phosphoinositide turnover. J Neurochem. 1993a;60:1505–1511. doi: 10.1111/j.1471-4159.1993.tb03314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barg J, Nah SY, Levy R, Saya D, Vogel Z. Modulation of thymidine incorporation by kappa-opioid ligands in rat spinal cord-dorsal root ganglion co-cultures. Brain Res. 1993b;629:109–114. doi: 10.1016/0006-8993(93)90488-9. [DOI] [PubMed] [Google Scholar]
- Barg J, Belcheva MM, Levy R, Saya D, McHale RJ, Johnson FE, Coscia CJ, Vogel Z. Opioids inhibit endothelin-mediated DNA synthesis, phosphatidylinositol turnover and Ca2+ mobilization in rat C6 glioma cells. J Neurosci. 1994;14:5858–5864. doi: 10.1523/JNEUROSCI.14-10-05858.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcheva MM, Barg J, Clark WG, Rowinski J, Gloeckner CA, Ho AM, Gao XM, Chuang DM, Coscia CJ. Novel intracellular opioid binding sites associated with the nuclei of neurohybrid cells. J Neurosci. 1993;13:104–114. doi: 10.1523/JNEUROSCI.13-01-00104.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcheva MM, Ignatova EG, Young EC, Coscia CJ. Agonist-induced desensitization and down-regulation of δ opioid receptors alter the levels of their I-125-β-endorphin cross-linked products in subcellular fractions from NG108-15 cells. Biochemistry. 1996;35:14818–14824. doi: 10.1021/bi961579+. [DOI] [PubMed] [Google Scholar]
- Berhow MT, Hiroi N, Nestler EJ. Regulation of ERK (extracellular signal regulated kinase), part of the neurotrophin signal transduction cascade, in the rat mesolimbic dopamine system by chronic exposure to morphine or cocaine. J Neurosci. 1996;16:4707–4715. doi: 10.1523/JNEUROSCI.16-15-04707.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Burt AR, Carr IC, Mullaney I, Anderson NG, Milligan G. Agonist activation of p42 and p44 mitogen-activated protein kinases following expression of the mouse δ opioid receptor in Rat-1 fibroblasts: effects of receptor expression levels and comparisons with G-protein activation. Biochem J. 1996;320:227–235. doi: 10.1042/bj3200227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- Coso OA, Teramoto H, Simonds WF, Gutkind JS. Signaling from G protein-coupled receptors to c-Jun kinase involves βγ subunits of heterotrimeric G proteins acting on a Ras and Racl-dependent pathway. J Biol Chem. 1996;271:3963–3966. doi: 10.1074/jbc.271.8.3963. [DOI] [PubMed] [Google Scholar]
- Creedon DJ, Johnson EM, Jr, Lawrence JC., Jr Mitogen-activated protein kinase-independent pathways mediate the effects of nerve growth factor and cAMP on neuronal survival. J Biol Chem. 1996;271:20713–20718. doi: 10.1074/jbc.271.34.20713. [DOI] [PubMed] [Google Scholar]
- Crespo P, Xu N, Simonds WR, Gutkind JS. Ras-dependent activation of MAP kinase pathway mediated by G-protein βγ subunits. Nature. 1994;369:418–420. doi: 10.1038/369418a0. [DOI] [PubMed] [Google Scholar]
- Crespo P, Cachero TG, Xu N, Gutkind JS. Dual effect of β-adrenergic receptors on mitogen-activated protein kinase. J Biol Chem. 1995;270:25259–25264. doi: 10.1074/jbc.270.42.25259. [DOI] [PubMed] [Google Scholar]
- Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Transcriptional regulation by MAP kinases. Mol Reprod Dev. 1995;42:459–467. doi: 10.1002/mrd.1080420414. [DOI] [PubMed] [Google Scholar]
- Dent P, Haser W, Haystead TAJ, Vincent LA, Roberts TM, Sturgill TW. Activation of mitogen-activated protein kinase: kinase by v-Raf in NIH 3T3 cells and in vitro. Science. 1992;251:1404–1406. doi: 10.1126/science.1326789. [DOI] [PubMed] [Google Scholar]
- De Vivo M, Iyengar R. Activated Gq-alpha potentiates platelet-derived growth factor-stimulated mitogenesis in confluent cell cultures. J Biol Chem. 1994;269:19671–19674. [PubMed] [Google Scholar]
- Fukuda K, Kato S, Morikawa H, Shoda T, Mori K. Functional coupling of the δ-, μ-, and κ-opioid receptors to mitogen-activated protein kinase and arachidonate release in Chinese hamster ovary cells. J Neurochem. 1996;67:1309–1316. doi: 10.1046/j.1471-4159.1996.67031309.x. [DOI] [PubMed] [Google Scholar]
- Guan KL. The mitogen activated protein kinase signal transduction pathway: from the cell surface to the nucleus. Cell Signal. 1994;6:581–589. doi: 10.1016/0898-6568(94)90041-8. [DOI] [PubMed] [Google Scholar]
- Hawes BE, van Biesen T, Koch WJ, Luttrell LM, Lefkowitz RJ. Distinct pathways of Gi-mediated mitogen-activated protein kinase activation. J Biol Chem. 1995;270:17148–17153. doi: 10.1074/jbc.270.29.17148. [DOI] [PubMed] [Google Scholar]
- Her JH, Lakhani S, Zu K, Vila J, Dent P, Sturgill TW, Weber MJ. Dual phosphorylation and autophosphorylation in mitogen-activated protein (MAP) kinase activation. Biochem J. 1993;296:25–31. doi: 10.1042/bj2960025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe LR, Leevers SJ, Gomez N, Nakielny S, Cohen P, Marshall CJ. Activation of the MAP kinase pathway by the protein kinase raf. Cell. 1992;71:335–342. doi: 10.1016/0092-8674(92)90361-f. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Vaillancourt RR. Sequential protein kinase reactions controlling cell growth and differentiation. Curr Opin Cell Biol. 1994;6:230–238. doi: 10.1016/0955-0674(94)90141-4. [DOI] [PubMed] [Google Scholar]
- Kameshita I, Fujisawa H. A sensitive method for detection of calmodulin-dependent protein kinase II activity in sodium dodecyl sulfate-polyacrylamide gel. Anal Biochem. 1989;183:139–143. doi: 10.1016/0003-2697(89)90181-4. [DOI] [PubMed] [Google Scholar]
- Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ. Cellular expression of the carboxyl terminus of a G protein-coupled receptor kinase attenuates Gβγ- mediated signaling. J Biol Chem. 1994;269:6193–6197. [PubMed] [Google Scholar]
- Lange-Carter CA, Pleiman CM, Gardner AM, Blumer KJ, Johnson GL. A divergence in the MAP kinase regulatory network defined by MEK kinase and Raf. Science. 1993;260:315–319. doi: 10.1126/science.8385802. [DOI] [PubMed] [Google Scholar]
- Li LY, Chang KJ. The stimulatory effect of opioids on mitogen-activated protein kinase in Chinese hamster ovary cells transfected to express μ-opioid receptors. Mol Pharmacol. 1996;50:599–602. [PubMed] [Google Scholar]
- Lopez-Ilasaca M, Crespo P, Pellici PG, Gutkind JS, Wetzker R. Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase γ. Science. 1997;275:394–397. doi: 10.1126/science.275.5298.394. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Delia Rocca GJ, van Biesen T, Luttrell DK, Lefkowitz RJ. Gβγ subunits mediate Src-dependent phosphorylation of the epidermal growth factor receptor. J Biol Chem. 1997;270:4637–4644. doi: 10.1074/jbc.272.7.4637. [DOI] [PubMed] [Google Scholar]
- Ortiz J, Harris HW, Guitart X, Terwilliger RZ, Haycock JW, Nestler EJ. Extracellular signal-regulated protein kinases (ERKs) and ERK kinase (MEK) in brain: regional distribution and regulation by chronic morphine. J Neurosci. 1995;15:1285–1297. doi: 10.1523/JNEUROSCI.15-02-01285.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelech SL, Sanghera JS. Distinct pathways of Gi-mediated mitogen-activated protein kinase activation. Trends Biol Sci. 1992;17:233–238. doi: 10.1016/s0968-0004(00)80005-5. [DOI] [PubMed] [Google Scholar]
- Robbins DJ, Zhen E, Owaki H, Vanderbilt CA, Ebert D, Geppert TD, Cobb MH. Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J Biol Chem. 1993;268:5097–5106. [PubMed] [Google Scholar]
- Robbins DJ, Zhen E, Cheng M, Xu S, Ebert D, Cobb MH. Map kinases ERK1 and ERK2: pleiotropic enzymes in a ubiquitous signaling network. Adv Cancer Res. 1994;63:93–116. doi: 10.1016/s0065-230x(08)60399-1. [DOI] [PubMed] [Google Scholar]
- Seth A, Gonzalez FA, Gupta S, Raden DL, Davis RJ. Signal transduction within the nucleus by mitogen-activated protein kinase. J Biol Chem. 1992;267:24796–24804. [PubMed] [Google Scholar]
- Van Biesen T, Hawes BE, Raymond JR, Luttrell LM, Koch WJ, Lefkowitz RJ. Go-protein α-subunits activate mitogen-activated protein kinase via a novel protein kinase C-dependent mechanism. J Biol Chem. 1996;271:1266–1269. doi: 10.1074/jbc.271.3.1266. [DOI] [PubMed] [Google Scholar]
- Waskiewicz AJ, Cooper JA. Mitogen and stress response pathways: MAP kinase cascades and phosphatase regulation in mammals and yeast. Curr Opin Cell Biol. 1995;7:798–805. doi: 10.1016/0955-0674(95)80063-8. [DOI] [PubMed] [Google Scholar]