

Abstract
Androgen receptor (AR) recruitment of transcriptional corepressors NCoR and SMRT can be enhanced by antagonists such as mifepristone. This study shows that enhanced NCoR binding to the mifepristone liganded AR is mediated by the NCoR C-terminal N1 CoRNR box, and that this selectivity is due to charged residues unique to the C-terminal CoRNR boxes of NCoR and SMRT. Significantly, these residues are on a helical face adjacent to oppositely charged residues in helix 4 of the AR ligand-binding domain (LBD). Mutagenesis of these AR residues in helix 4, as well as mutation of lysine 720 in helix 3 (predicted to interact with the CoRNR box), markedly impaired AR recruitment of NCoR, indicating that N1 CoRNR box binding is being stabilized by these ionic interactions in the AR LBD coactivator/corepressor binding site. Finally, results using a helix 12 deleted AR indicate that mifepristone induces allosteric changes in addition to helix 12 displacement that are critical for NCoR binding. These findings demonstrate that AR antagonists can enhance corepressor recruitment by stabilizing a distinct antagonist conformation of the AR coactivator/corepressor binding site, and support the development of additional antagonists that may be able to further enhance AR recruitment of corepressors.
Keywords: androgen receptor, prostate cancer, corepressor, antagonist, mifepristone
Introduction
The androgen receptor (AR) is a member of the steroid receptor/nuclear receptor family that plays a major role in normal prostate growth and in the development and progression of prostate cancer (PCa). Similarly to other nuclear receptors, binding of agonist ligands causes a shift in the position of helix 12 in the AR ligand binding domain (LBD) towards helices 3–5, which both stabilizes ligand binding and generates a hydrophobic cleft for binding of coactivator proteins via leucine-X-X-leucine-leucine (LXXLL) motifs (1, 2). A unique feature of AR is that an LXXLL-like motif in the N-terminus (amino acids 23–27, FQNLF) binds to this hydrophobic cleft, which further stabilizes helix 12 and ligand binding (3–10). In the absence of ligand, nonsteroidal nuclear receptors such as thyroid and retinoid receptors repress transcription by recruiting the corepressor proteins NCoR and SMRT, which are associated with histone deacetylases (11–15). Corepressor binding is mediated by extended LXXLL-like motifs (L-X-X-I/H-I-X-X-X-L/I), termed corepressor nuclear receptor boxes (CoRNR boxes), which are located in the C-terminal half of NCoR and SMRT (16–23). The positioning of helix 12 away from helices 3–5 in unliganded (or antagonist liganded) nuclear receptors opens the LXXLL site to accommodate these larger CoRNR boxes, with three helical turns (versus two turns for LXXLL motifs) (24).
While the nonsteroidal nuclear receptors bind DNA in the absence of ligand and actively repress transcription via NCoR and SMRT recruitment, DNA binding by steroid receptors is generally ligand dependent and mediated physiologically by agonist ligands. Nonetheless, certain steroid hormone receptor antagonists (or partial agonists) can stimulate DNA binding and recruitment of NCoR or SMRT. This appears to be the mechanism of action for the estrogen receptor α (ERα) antagonists tamoxifen and raloxifene. Crystal structures of the tamoxifen and raloxifene liganded ERα show that the side groups of these drugs force alternative nonagonist positions for helix 12 (25, 26). This repositioning of helix 12 allows for corepressor binding, and the tissue selective activities of these drugs (antagonists in breast cancer and agonists in bone and other tissues) appear to reflect the relative levels of transcriptional coactivators versus corepressors in the respective tissues (27). Biochemical studies of mifepristone (RU486), an antagonist of the glucocorticoid receptor (GR) and progesterone receptor (PR), indicate that antagonist activity is similarly due to recruitment of NCoR or SMRT (28–30). The critical structural feature of mifepristone is a bulky phenyl-aminodimethyl group in the 11β position that is presumed to displace helix 12. Therefore, while the physiological roles of NCoR and SMRT in the normal functioning of ERα, PR, and GR are uncertain, drugs that stimulate recruitment of these corepressors can be developed and have important clinical activities.
In contrast to other steroid receptors, the agonist liganded AR interacts with NCoR and SMRT (31–33), and RNAi approaches have shown that NCoR and SMRT function at physiological levels as negative regulators of androgen (testosterone and dihydrotestosterone, DHT) stimulated AR transcriptional activity (34–36). Moreover, AR recruitment of NCoR, as assessed by chromatin immunoprecipitation (ChIP), is increased by AR antagonists or partial agonists (bicalutamide, cyproterone acetate, or mifepristone), and NCoR/SMRT downregulation can increase the agonist activity of drugs such as hydroxyflutamide that have partial agonist activity (34, 35, 37–40). Significantly, the interactions between AR and these corepressors is complex, as NCoR and SMRT appear to interact with both the AR N-terminal and ligand binding domains (32, 34, 41, 42).
We have screened candidate compounds for drugs that can enhance the AR-NCoR interaction, and may therefore function as novel and more potent AR antagonists. Mammalian one and two-hybrid protein interaction studies indicate that physiological weak androgens and AR antagonists currently used in PCa patients do not substantially increase AR binding of NCoR (34). In contrast, mifepristone, a progesterone and glucocorticoid receptor antagonist with weak partial agonist activity on the AR (43), can strongly enhance AR-NCoR and AR-SMRT binding (34, 39, 44). Previous results indicate that the interaction between NCoR and the mifepristone liganded AR is dependent on a region encompassing the C-terminal CoRNR box (N1), and that both the AR N-terminal and ligand binding domains are required to mediate the interaction (34). One interpretation of these data is that mifepristone alters the structure of the LXXLL coactivator-binding site in the AR LBD to allow for CoRNR box binding. However, there are no available data on antagonist conformations of AR, and the molecular basis for AR interaction with corepressor proteins remains unclear. Therefore, the objectives of this study were to determine the basis for NCoR binding to the mifepristone liganded AR and develop a model that can be used for the development of further more active AR antagonists.
Materials and Methods
Plasmids and Reagents
Expression vectors for AR (pSVARo), VP16-AR, VP16-TRβ1, VP16-NCoR fusions, Gal4-NCoR fusions, full length NCoR (PKCR2-NCoR), and Gal4-RARα have been described previously (22, 31, 34, 45). All amino acid residue numbering for NCoR is based on the human NCoR sequence. Mutant pSVARo, NCoR, Gal4-NCoR, and VP16-NCoR constructs were created by site-directed mutagenesis using the Quick-Change Site Directed Mutagenesis kit (Stratagene, La Jolla, CA). The androgen receptor constructs with N-terminal deletions (del 1–11 and del 1–37) and the control for these mutants (KNHA-AR) have an N-terminal HA-tag and were kindly provided by Dr Michael Lu (46). The helix 12-deleted AR was derived from pSVARo by inserting a stop codon in place of methionine at residue 886. The reporter construct ARE4-luciferase, containing four tandem copies of a synthetic ARE has been described (31). pG5-luciferase, regulated by five tandem Gal4 binding sites, and pRL-CMV, a CMV promoter regulated Renilla control, were from Promega (Madison, WI). DHT and mifepristone were from Sigma (St. Louis, MO) and were used as 1:1000 stock solutions in ethanol.
Cell culture and transfection
CV1 and COS7 cells were maintained in DMEM supplemented with 10% fetal bovine serum (Hyclone, Logan, UT). Cells in 48-well tissue culture plates in DMEM containing 10% charcoal dextran-stripped fetal bovine serum (CDS-FBS, Hyclone) were cotransfected using Lipofectamine 2000 (Invitrogen). Cells were transfected with 100 ng of reporter and expression vectors except where indicated per well, and 1.25 ng of pRL-CMV Renilla vector for normalisation. After 24 hours medium was replaced with fresh DMEM containing 10% CDS-FBS with hormones or drugs at the indicated final concentrations. Following a further 24 hours, firefly and Renilla luciferase activities were assayed with the dual-luciferase assay system as per the supplier’s instructions (Promega). All samples were in triplicate and firefly luciferase activities were normalised for cotransfected Renilla activity.
Results
NCoR interaction with the AR N-terminal domain (NTD) is mediated by a region flanking the N2 CoRNR box
Previous studies have shown that mifepristone can enhance AR binding to NCoR, and that both the AR NTD and the LBD are required for this mifepristone enhanced NCoR-AR interaction (34). We have further shown that this NCoR interaction with the mifepristone liganded AR is mediated by a C-terminal fragment of NCoR containing the N2 and N1 CoRNR boxes (see diagram in figure 1A) (34). To determine whether this region of NCoR interacts with the AR NTD independently of the LBD, we assessed coactivation of an AR fragment containing only the NTD and DBD (AR-NTD-DBD) (Fig. 1A). Consistent with previous studies, the AR-NTD-DBD was constitutively active in the absence of ligand when assayed on an androgen responsive element regulated luciferase reporter gene (ARE4-luciferase) (Fig. 1B). However, this activity could be increased by cotransfection of the NCoR(2005–2440) fragment (containing the N2 and N1 CoRNR boxes) fused to the VP16 transactivation domain, VP16-NCoR(2005–2440). This coactivation was decreased by deletion of the region immediately N-terminal to the N2 CoRNR box in VP16-NCoR(2043–2440) (Fig. 1C). Further deletion of the N2 CoRNR box in VP16-NCoR(2065–2440) completely abrogated coactivation. These findings indicate that a region encompassing the N2 CoRNR box (residues 2005–2065) mediates an interaction with the AR NTD that is independent of the LBD.
Figure 1.

Region encompassing NCoR N2 CoRNR box interacts with AR N-terminal domain. A, outlines of NCoR and AR. B, CV1 cells were cotransfected with plasmids encoding AR N-terminal and DNA binding domains (AR-NTD-DBD), VP16-NCoR(2005–2440), ARE4-Luc luciferase reporter, and a CMV regulated Renilla luciferase plasmid (pRL-CMV). Luciferase versus Renilla luciferase activities were determined from triplicate samples after 24 hours, and the data are expressed as RLU ± S.E. C, CV1 cells were transfected as above with AR-NTD-DBD, pRL-CMV, and A RE4-Luc luciferase reporter, in conjunction with the indicated VP16-NCoR plasmids.
The N1 CoRNR box is critical for NCoR binding to the mifepristone liganded AR
We showed previously that removal of a region encompassing the N1 CoRNR box abrogated NCoR binding to the mifepristone liganded AR (34). To further assess whether the N1 CoRNR box mediates the interaction with the mifepristone liganded AR LBD, we mutated a double isoleucine in N1 to alanines in the NCoR(1806–2440) fragment, which contains all three CoRNR boxes. We also mutated a double isoleucine in the N2 CoRNR box to alanines. These fragments were fused to the Gal4 DBD, and assessed for interaction with VP16-AR using a pG5-luciferase reporter. There was a strong interaction between the mifepristone liganded VP16-AR and Gal4DBD-NCoR(1806–2440), and this interaction was not impaired by the N2(AA) mutation (Fig. 2A). However, the interaction was abrogated by the N1(AA) mutation, indicating that the N1 CoRNR box was critical for binding. As a further control to confirm that the N1(AA) mutation was not non-specifically altering the structure or expression of the protein, we determined whether the interaction with unliganded TRβ (which is mediated by the N3 CoRNR box) was intact. As shown in figure 2B, the wild-type, N1(AA) and N2(AA) Gal4-NCoRc interacted with the VP16-TRβ.
Figure 2.

Isoleucines in NCoR N1 CoRNR are required for recruitment of NCoR C-terminal to mifepristone liganded AR. A, CV1 cells were cotransfected with Gal4-NCoR(1806–2440) wild type (WT) or with double isoleucine to alanine substitutions in the N1 (N1AA) or N2(N2AA) CoRNR boxes, VP16-AR, pG5-luciferase reporter vector, and Renilla luciferase control (pRL-CMV). Cells in this experiment and below were treated with the indicated ligands for 24 hrs in steroid depleted medium and luciferase versus Renilla luciferase activities were determined from triplicate samples. B, CV1 cells were transfected with an expression vector for full-length VP16-TRβ1, pG5-luciferase reporter, and pRL-CMV control in the absence of ligand. C, cells were transfected with full-length AR (pSVARo), ARE4-luciferase reporter, pRL-CMV control, VP16-NCoR(1806–2440) wild type or N1AA mutant. D, CV1 cells were transfected with full-length AR (pSVARo), VP16-NCoR(2005–2440) wild type or N1AA mutant, ARE4-luciferase reporter, and pRL-CMV control.
To confirm that the N1 CoRNR box was critical for binding to the intact AR bound to an androgen responsive element, we cloned the N1(AA) mutation into the VP16-NCoR(1806–2440) vector. As shown in figure 3C, the N1(AA) mutation abrogated NCoR recruitment by the mifepristone liganded wild-type AR. The N1(AA) mutation cloned into the VP16-NCoR(2005–2440) vector (containing the N2 and N1 CoRNR boxes) similarly abrogated recruitment (Fig. 4D). To control for non-specific effects of the mutation in the NCoR(2005–2440) fragment, we showed that the mutation did not impair recruitment by the unliganded RARα fused to the Gal4 DNA binding domain (data not shown).
Figure 3.
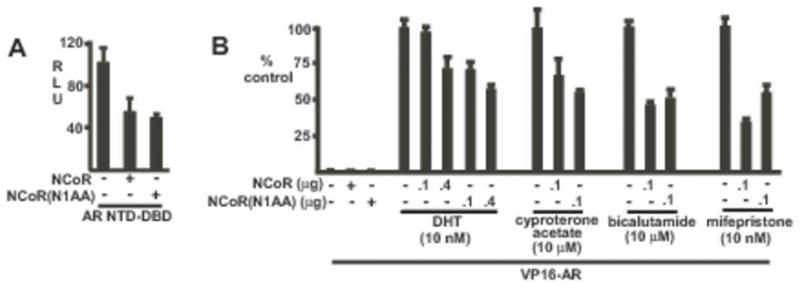
NCoR N1 CoRNR box enhances recruitment of full length NCoR by the mifepristone liganded AR. A, CV1 cells were transfected with an AR N-terminus-DBD expression vector (AR NTD-DBD), full length NCoR wild type or N1AA mutant (NCoR(N1AA), ARE4-luciferase reporter, and pRL-CMV control. B, CV1 cells were transfected with VP16-AR in the presence of full length NCoR wild type or N1AA mutant, ARE4-luciferase and pRL-CMV. Cells were treated for 24 hrs with the indicated ligands and assayed in triplicate for luciferase activity.
Figure 4.

Conserved charged residues in the NCoR and SMRT N1 CoRNR boxes are required for N1 CoRNR box binding to the mifepristone liganded AR. A, sequence alignment for NCoR and SMRT CoRNR boxes. B, CV1 cells were transfected with wild type AR, VP16-NCoR(2005–2440) (wild type, E2264A or R2285A mutants), ARE4-luciferase and the pRL-CMV control. Cells were treated with the indicated ligands for 24 hrs and luciferase versus Renilla luciferase activities were determined from triplicate samples.
Finally, we cloned the N1(AA) mutation into the intact full length NCoR. As expected, both the wild-type and mutant NCoR could suppress the constitutive activity of the AR NTD (Fig. 3A). We then assessed inhibition of the DHT versus mifepristone liganded AR. As the latter does not have substantial transcriptional activity, we carried out these cotransfections with a VP16-AR fusion protein. Significantly, both the wild-type and N1(AA) mutant NCoR suppressed the DHT liganded VP16-AR, with the N1(AA) mutant being more effective (Fig. 3B). The N1AA was also more effective at repressing VP16-AR transactivation by a partial agonist (cyproterone acetate), while its effect on a pure antagonist (bicalutamide) was comparable to the wild-type NCoR. In contrast, the N1(AA) mutant was less active at repressing the mifepristone liganded VP16-AR, consistent with the N1 CoRNR box enhancing recruitment of the full length NCoR. It should be noted that while the N1(AA) mutation abrogates AR interaction with NCoR C-terminal fragments, it does not do so in the context of full length NCoR. This likely reflects additional contacts between NCoR and the AR NTD mediated by N-terminal regions of NCoR (42).
Charged residues common to N1 CoRNR boxes in NCoR and SMRT are critical for binding
A previous study found that the interaction between unliganded AR and SMRT was dependent on the C-terminal (N1) CoRNR box (33). An alignment of the SMRT and NCoR CoRNR boxes shows that the N1 CoRNR boxes are almost identical (Fig. 4A). Moreover, they are distinct from the other CoRNR boxes in having a charged residue (arginine) at position 6. They also share a glutamate at position 2, which is aspartate in the N3 CoRNR box of NCoR and alanine in the other CoRNR boxes. Therefore, we carried out further mutagenesis to determine whether these charged residues common to the NCoR and SMRT C-terminal CoRNR boxes contribute to NCoR binding to the mifepristone liganded AR. Significantly, mutations at either site markedly impaired NCoR recruitment by the mifepristone liganded AR (Fig. 4B).
N1 is interacting with the AR coactivator binding site
To further assess the molecular basis for N1 CoRNR box binding to the AR, and in particular to test the hypothesis the N1 CoRNR box was binding to the LXXLL coactivator site, we compared the available crystal structures of the agonist liganded AR binding to a FQNLF peptide and the antagonist liganded PPARγ binding to a SMRT N1 CoRNR box peptide (47). A conserved lysine at the C-terminal end of helix 3 in AR and PPARγ anchors both peptides by forming hydrogen bonds with the C-terminal phenylalanine or leucine residues, respectively (Fig. 5A). The FQNLF peptide forms 2 helical turns and is anchored at its N-terminus by helix 12 in the AR. In contrast, displacement of this helix in the antagonist conformation of PPARγ allows the site to accommodate a third helical turn in the CoRNR box, with leucines at position 1 and 9, and isoleucine at position 5, forming a hydrophobic face that binds to helix 3. Another face of the CoRNR box helix is formed by glutamic acid at position 2 and arginine at position 5, which interact with K310 and N303 in helix 4 of PPARγ. Significantly, these residues in PPARγ correspond to Q738 and D731 in helix 4 of the AR, suggesting that strong interactions between these acidic and basic residues may stabilize CoRNR box binding to the mifepristone liganded AR (Fig. 5A). A recent crystal structure has been reported for the PR complexed with a mifepristone related antagonist (asoprisnil) and an NCoR N1 CoRNR box (48). Significantly, this structure shows a similar positioning of the CoRNR box peptide and a bond between glutamic acid at position 2 and a glutamine in PR that is equivalent to Q738 in AR (Fig. 5B).
Figure 5.

Charged residues in helix 4 of AR coactivator binding site mediate NCoR binding to mifepristone liganded AR. A, outline showing proposed interaction between N1 CoRNR box and AR helix 4. B, detail of asoprisnil liganded progesterone receptor LBD (helix 4) cocrystalized with NCoR N1 CoRNR box. Numbering of the aspartate and glutamine in helix 4 are based on the AR sequence. C and D, CV1 cells were cotransfected with wild type AR (pSVARo) or mutant ARs as indicated, VP16-NCoR(2005–2440), ARE4-luciferase reporter, and pRL-CMV control. Cells were then treated with 10 nM DHT or mifepristone for 24 hrs and assayed for firefly versus Renilla luciferase activity.
To determine whether K720, D731, and Q738 in AR do contribute to NCoR binding by the mifepristone liganded AR, we next examined site directed mutants. A K720A mutation in the AR only moderately decreased DHT stimulated activity, possibly due to strong hydrophobic interactions mediated by the phenylalanines in the FQNLF peptide (Fig. 5C). In contrast, the K720A mutation markedly impaired recruitment of VP16-NCoR(2005–2440) in response to mifepristone. The mutations in helix 4 (D731A and Q738A) had substantial effects on DHT stimulated AR activity, but again more markedly impaired the response to mifepristone (Fig. 5D). Taken together, the NCoR mutagenesis data above and these AR mutagenesis data strongly support the conclusion that the N1 CoRNR box is binding to the coactivator/corepressor binding site in the AR LBD.
Displacement of helix 12 is not sufficient to obtain NCoR recruitment
A structural feature of mifepristone that contributes to its antagonist activity is its bulky phenyl-aminodimethyl group in the 11β position, which is presumed to displace helix 12. However, it was not clear whether helix 12 displacement was sufficient for NCoR binding, or whether NCoR binding is enhanced by further ligand induced allosteric changes in the AR ligand binding domain. Therefore, to determine whether mifepristone enhances NCoR recruitment by mechanisms in addition to displacement of helix 12, we generated a mutant AR that was truncated after helix 11. As expected, this mutation markedly impaired transactivation in response to DHT, and mifepristone was similarly inactive (Fig. 6A). Significantly, the truncated AR was not coactivated by VP16-NCoR(2005–2440) in the absence of ligand or in response to DHT, demonstrating that removal of helix 12 alone is not sufficient. In contrast, the truncated mifepristone liganded AR was strongly coactivated by VP16-NCoR(2005–2440), indicating that NCoR binding was dependent on further ligand induced conformational changes in the AR ligand binding domain.
Figure 6.

Removal of AR helix 12 is insufficient to allow unliganded AR recruitment of NCoR, but has little impact on mifepristone liganded AR-NCoR interaction. A, COS7 cells were cotransfected with wild type AR (AR WT) or truncated AR lacking helix 12 of the LBD (AR M886X), VP16-NCoR(2005–2440), ARE4-Luc luciferase reporter, and pRL-CMV control. Cells were treated with the indicated ligands for 24 hrs and luciferase versus Renilla luciferase activities were determined. Results are representative of three independent experiments. B, model indicating that the NCoR interaction with the agonist (DHT) liganded AR is mediated primarily through the AR NTD, and may be enhanced by TAB2. In contrast, mifepristone (MIF) alters the structure of the AR LBD so that it no longer binds to the AR N-terminal FXXLF peptide or to LXXLL peptides in p160 coactivators, and instead accommodates an NCoR CoRNR box (CoRNR box 1) to provide a second stabilizing interaction in addition to the AR NTD interaction.
Discussion
Structural studies of glucocorticoid, estrogen, and progesterone receptors have shown that binding of certain antagonists causes displacement of helix 12, and this appears to allow for CoRNR box mediated recruitment of NCoR and SMRT. Recent studies have shown that NCoR and SMRT interact with both the agonist and antagonist liganded AR, and that this interaction can be enhanced by certain antagonists, particularly by mifepristone. However, there are no available crystal structures of AR in an antagonist conformation, and the basis for NCoR/SMRT recruitment by the agonist versus antagonist liganded AR has been unclear. This study first confirms that there is a ligand independent interaction between NCoR and the AR NTD. Using site directed mutagenesis, we then establish that NCoR binding to the mifepristone liganded AR LBD is mediated by the C-terminal N1 CoRNR box. Moreover, we demonstrate that the basis for this selectivity is unique charged residues in the NCoR and SMRT N1 CoRNR boxes, which are predicted to be positioned on a helical face adjacent to oppositely charged residues in helix 4 of the AR LBD. Mutagenesis of these residues in helix 4, in conjunction with mutation of K720 in helix 3, further support the conclusion that the N1 CoRNR box is positioned in the coactivator binding site and is being stabilized by charge interactions. Finally, results using a helix 12 deleted AR indicate that mifepristone induces further conformational changes in the LBD (in addition to helix 12 displacement) that are critical for NCoR binding.
Based on the data in this study, a model for AR-NCoR binding to the agonist versus antagonist liganded AR is outlined in figure 6B. We suggest that NCoR interaction with the agonist liganded AR is mediated primarily by the AR N-terminal domain. Significantly, multiple regions of NCoR may mediate this AR N-terminal domain binding, including a region flanking the N2 CoRNR box identified in this study, and a domain in the middle of NCoR that encompassed the third repressor domain (RD3) (42). Moreover, the N-terminal interaction may be both direct and indirect through TAB2 (40). SMRT has also been shown to interact with the AR N-terminal domain, and this interaction may inhibit the agonist liganded AR by competing for binding of p160 coactivators and impairing the AR N-C terminal interaction, as well as by recruiting histone deacetylases. Mifepristone can stabilize this weak binding to the AR N-terminal domain by mediating a further interaction between the C-terminal N1 CoRNR box of NCoR and the coactivator binding site in the AR LBD. However, it should be emphasized that while mifepristone enhances CoRNR box binding to the AR LBD, this interaction is still quite weak and not readily detectable in the absence of the AR N-terminal domain interactions.
Significantly, displacement of helix 12 alone is not sufficient to obtain N1 CoRNR box binding, which indicates that further mifepristone induced conformational changes are required to enhance binding. Our site directed mutagenesis results indicate that one effect of mifepristone may be to reposition helix 4 in the AR LBD so that charged residues on one face of this helix (D731 and Q738) can bond with oppositely charged residues (arginine and glutamate) that are unique to the N1 CoRNR boxes of NCoR and SMRT. In addition, it is possible that the phenyl-aminodimethyl group in the 11β position makes direct contact with the CoRNR box. This mechanism has been suggested for the PR antagonist asoprisnil, which is structurally related to mifepristone and enhances NCoR binding to the PR (48). In any case, the important conclusions from these data are that antagonists can alter the structure of the AR coactivator binding site to enhance CoRNR box binding, and that this enhancement requires both displacement of helix 12 and additional antagonist mediated changes in the coactivator binding site.
AR antagonists currently in use (flutamide and bicalutamide) are effective when used as single agents or in combination with castration (androgen deprivation therapy) to suppress AR activity in PCa. However, patients invariably relapse with tumors that have been termed hormone refractory, androgen independent, or castration resistant PCa. Significantly, AR transcriptional activity is reactivated in these recurrent tumors through unclear mechanisms, but appear to include increased tumor synthesis of androgens and other adaptations that enhance AR activation in response to low levels of androgens (49). Unfortunately, AR antagonists are no longer effective at suppressing AR activity in these recurrent tumors. The basis for this loss of activity is unclear, but may include alterations in AR or associated proteins that further decrease AR affinity for these antagonists (relative to intracellular testosterone and DHT), or enhance their partial agonist activity. To determine whether mifepristone has activity in these recurrent tumors, we recently completed a small trial of mifepristone in 19 patients with advanced castration resistant PCa (50). Interpretation of the results was confounded by substantial increases in adrenal androgen levels in response to GR blockade by mifepristone, but only 6 patients had evidence of a response based on stable levels of serum PSA and no patients had significant declines in their PSA.
As noted above, while mifepristone has provided a proof of principle that ligand induced conformational changes in the AR LBD can enhance NCoR binding, the interaction between the mifepristone liganded AR and the N1 CoRNR box is still relatively weak and may limit the efficacy of this drug in advanced castration resistant PCa. Nonetheless, based on results in this study we would predict that additional antagonists can be developed that will further enhance AR recruitment of NCoR and SMRT. Such drugs may be effective in recurrent PCa, and may also have novel tissue selective activities based on relative levels of coactivators versus corepressors.
Acknowledgments
This work was supported by NIH grants CA111803 (S.P.B), DK56123 (A.N.H), and T32 CA081156 (H.C.S), and by Department of Defense grant PC040246 (S.P.B.).
Abbreviations List
- AR
androgen receptor
- CDS-FBS
charcoal/dextran stripped fetal bovine serum
- CoRNR
corepressor nuclear receptor
- DBD
DNA binding domain
- DHT
dihydrotestosterone
- ER
estrogen receptor
- GR
glucocorticoid receptor
- LBD
ligand binding domain
- NTD
N-terminal domain
- PCa
prostate cancer
- PR
progesterone receptor
References
- 1.Matias PM, Donner P, Coelho R, et al. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J Biol Chem. 2000;275:26164–71. doi: 10.1074/jbc.M004571200. [DOI] [PubMed] [Google Scholar]
- 2.Sack JS, Kish KF, Wang C, et al. Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc Natl Acad Sci U S A. 2001;98:4904–9. doi: 10.1073/pnas.081565498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong CI, Zhou ZX, Sar M, Wilson EM. Steroid requirement for androgen receptor dimerization and DNA binding. Modulation by intramolecular interactions between the NH2-terminal and steroid-binding domains. J Biol Chem. 1993;268:19004–12. [PubMed] [Google Scholar]
- 4.Berrevoets CA, Doesburg P, Steketee K, Trapman J, Brinkmann AO. Functional interactions of the AF-2 activation domain core region of the human androgen receptor with the amino-terminal domain and with the transcriptional coactivator TIF2 (transcriptional intermediary factor2) Mol Endocrinol. 1998;12:1172–83. doi: 10.1210/mend.12.8.0153. [DOI] [PubMed] [Google Scholar]
- 5.Doesburg P, Kuil CW, Berrevoets CA, et al. Functional in vivo interaction between the amino-terminal, transactivation domain and the ligand binding domain of the androgen receptor. Biochemistry. 1997;36:1052–64. doi: 10.1021/bi961775g. [DOI] [PubMed] [Google Scholar]
- 6.He B, Kemppainen JA, Wilson EM. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J Biol Chem. 2000;275:22986–94. doi: 10.1074/jbc.M002807200. [DOI] [PubMed] [Google Scholar]
- 7.Chang CY, McDonnell DP. Evaluation of ligand-dependent changes in AR structure using peptide probes. Mol Endocrinol. 2002;16:647–60. doi: 10.1210/mend.16.4.0818. [DOI] [PubMed] [Google Scholar]
- 8.He B, Wilson EM. Electrostatic modulation in steroid receptor recruitment of LXXLL and FXXLF motifs. Mol Cell Biol. 2003;23:2135–50. doi: 10.1128/MCB.23.6.2135-2150.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Estebanez-Perpina E, Moore JM, et al. The molecular mechanisms of coactivator utilization in ligand-dependent transactivation by the androgen receptor. J Biol Chem. 2005;280:8060–8. doi: 10.1074/jbc.M407046200. [DOI] [PubMed] [Google Scholar]
- 10.Hur E, Pfaff SJ, Payne ES, Gron H, Buehrer BM, Fletterick RJ. Recognition and accommodation at the androgen receptor coactivator binding interface. PLoS Biol. 2004;2:E274. doi: 10.1371/journal.pbio.0020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horlein AJ, Naar AM, Heinzel T, et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor [see comments] Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 12.Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–7. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 13.Alland L, Muhle R, Hou H, Jr, et al. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature. 1997;387:49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- 14.Heinzel T, Lavinsky RM, Mullen TM, et al. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387:43–8. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 15.Nagy L, Kao HY, Chakravarti D, et al. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89:373–80. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 16.Hu X, Lazar MA. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature. 1999;402:93–6. doi: 10.1038/47069. [DOI] [PubMed] [Google Scholar]
- 17.Perissi V, Staszewski LM, McInerney EM, et al. Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev. 1999;13:3198–208. doi: 10.1101/gad.13.24.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagy L, Kao HY, Love JD, et al. Mechanism of corepressor binding and release from nuclear hormone receptors. Genes Dev. 1999;13:3209–16. doi: 10.1101/gad.13.24.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Webb P, Anderson CM, Valentine C, et al. The nuclear receptor corepressor (N-CoR) contains three isoleucine motifs (I/LXXII) that serve as receptor interaction domains (IDs). [In Process Citation] Mol Endocrinol. 2000;14:1976–85. doi: 10.1210/mend.14.12.0566. [DOI] [PubMed] [Google Scholar]
- 20.Cohen RN, Wondisford FE, Hollenberg AN. Two separate NCoR (nuclear receptor corepressor) interaction domains mediate corepressor action on thyroid hormone response elements. Mol Endocrinol. 1998;12:1567–81. doi: 10.1210/mend.12.10.0188. [DOI] [PubMed] [Google Scholar]
- 21.Cohen RN, Putney A, Wondisford FE, Hollenberg AN. The nuclear corepressors recognize distinct nuclear receptor complexes. Mol Endocrinol. 2000;14:900–14. doi: 10.1210/mend.14.6.0474. [DOI] [PubMed] [Google Scholar]
- 22.Cohen RN, Brzostek S, Kim B, Chorev M, Wondisford FE, Hollenberg AN. The Specificity of Interactions between Nuclear Hormone Receptors and Corepressors Is Mediated by Distinct Amino Acid Sequences within the Interacting Domains. Mol Endocrinol. 2001;15:1049–61. doi: 10.1210/mend.15.7.0669. [DOI] [PubMed] [Google Scholar]
- 23.Makowski A, Brzostek S, Cohen RN, Hollenberg AN. Determination of nuclear receptor corepressor interactions with the thyroid hormone receptor. Mol Endocrinol. 2003;17:273–86. doi: 10.1210/me.2002-0310. [DOI] [PubMed] [Google Scholar]
- 24.Xu HE, Stanley TB, Montana VG, et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature. 2002;415:813–7. doi: 10.1038/415813a. [DOI] [PubMed] [Google Scholar]
- 25.Brzozowski AM, Pike AC, Dauter Z, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–8. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 26.Shiau AK, Barstad D, Loria PM, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–37. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 27.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–8. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 28.Jackson TA, Richer JK, Bain DL, Takimoto GS, Tung L, Horwitz KB. The partial agonist activity of antagonist-occupied steroid receptors is controlled by a novel hinge domain-binding coactivator L7/SPA and the corepressors N-CoR or SMRT. Mol Endocrinol. 1997;11:693–705. doi: 10.1210/mend.11.6.0004. [DOI] [PubMed] [Google Scholar]
- 29.Schulz M, Eggert M, Baniahmad A, Dostert A, Heinzel T, Renkawitz R. RU486-induced glucocorticoid receptor agonism is controlled by the receptor N terminus and by corepressor binding. J Biol Chem. 2002;277:26238–43. doi: 10.1074/jbc.M203268200. [DOI] [PubMed] [Google Scholar]
- 30.Wagner BL, Norris JD, Knotts TA, Weigel NL, McDonnell DP. The nuclear corepressors NCoR and SMRT are key regulators of both ligand- and 8-bromo-cyclic AMP-dependent transcriptional activity of the human progesterone receptor. Mol Cell Biol. 1998;18:1369–78. doi: 10.1128/mcb.18.3.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng S, Brzostek S, Lee SR, Hollenberg AN, Balk SP. Inhibition of the dihydrotestosterone-activated androgen receptor by nuclear receptor corepressor. Mol Endocrinol. 2002;16:1492–501. doi: 10.1210/mend.16.7.0870. [DOI] [PubMed] [Google Scholar]
- 32.Dotzlaw H, Moehren U, Mink S, Cato AC, Iniguez Lluhi JA, Baniahmad A. The amino terminus of the human AR is target for corepressor action and antihormone agonism. Mol Endocrinol. 2002;16:661–73. doi: 10.1210/mend.16.4.0798. [DOI] [PubMed] [Google Scholar]
- 33.Liao G, Chen LY, Zhang A, et al. Regulation of androgen receptor activity by the nuclear receptor corepressor SMRT. J Biol Chem. 2003;278:5052–61. doi: 10.1074/jbc.M206374200. [DOI] [PubMed] [Google Scholar]
- 34.Hodgson MC, Astapova I, Cheng S, et al. The androgen receptor recruits nuclear receptor CoRepressor (N-CoR) in the presence of mifepristone via its N and C termini revealing a novel molecular mechanism for androgen receptor antagonists. J Biol Chem. 2005;280:6511–9. doi: 10.1074/jbc.M408972200. [DOI] [PubMed] [Google Scholar]
- 35.Yoon HG, Wong J. The corepressors SMRT and N-CoR are involved in agonist- and antagonist-regulated transcription by androgen receptor. Mol Endocrinol. 2005;20:1048–60. doi: 10.1210/me.2005-0324. [DOI] [PubMed] [Google Scholar]
- 36.Hodgson MC, Astapova I, Hollenberg AN, Balk SP. Activity of Androgen Receptor Antagonist Bicalutamide in Prostate Cancer Cells Is Independent of NCoR and SMRT Corepressors. Cancer Res. 2007;67:8388–95. doi: 10.1158/0008-5472.CAN-07-0617. [DOI] [PubMed] [Google Scholar]
- 37.Shang Y, Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell. 2002;9:601–10. doi: 10.1016/s1097-2765(02)00471-9. [DOI] [PubMed] [Google Scholar]
- 38.Kang Z, Janne OA, Palvimo JJ. Coregulator recruitment and histone modifications in transcriptional regulation by the androgen receptor. Mol Endocrinol. 2004;18:2633–48. doi: 10.1210/me.2004-0245. [DOI] [PubMed] [Google Scholar]
- 39.Berrevoets CA, Umar A, Trapman J, Brinkmann AO. Differential modulation of androgen receptor transcriptional activity by the nuclear receptor corepressor (N-CoR) Biochem J. 2004;379:731–8. doi: 10.1042/BJ20031456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu P, Baek SH, Bourk EM, et al. Macrophage/Cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006;124:615–29. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 41.Dotzlaw H, Papaioannou M, Moehren U, Claessens F, Baniahmad A. Agonist-antagonist induced coactivator and corepressor interplay on the human androgen receptor. Mol Cell Endocrinol. 2003;213:79–85. doi: 10.1016/j.mce.2003.10.036. [DOI] [PubMed] [Google Scholar]
- 42.Wu Y, Kawate H, Ohnaka K, Nawata H, Takayanagi R. Nuclear compartmentalization of N-CoR and its interactions with steroid receptors. Mol Cell Biol. 2006;26:6633–55. doi: 10.1128/MCB.01534-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kemppainen JA, Lane MV, Sar M, Wilson EM. Androgen receptor phosphorylation, turnover, nuclear transport, and transcriptional activation. Specificity for steroids and antihormones. J Biol Chem. 1992;267:968–74. [PubMed] [Google Scholar]
- 44.Song LN, Coghlan M, Gelmann EP. Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor. Mol Endocrinol. 2004;18:70–85. doi: 10.1210/me.2003-0189. [DOI] [PubMed] [Google Scholar]
- 45.Masiello D, Chen SY, Xu Y, et al. Recruitment of beta-catenin by wild-type or mutant androgen receptors correlates with ligand-stimulated growth of prostate cancer cells. Mol Endocrinol. 2004;18:2388–401. doi: 10.1210/me.2003-0436. [DOI] [PubMed] [Google Scholar]
- 46.Lu ML, Schneider MC, Zheng Y, Zhang X, Richie JP. Caveolin-1 interacts with androgen receptor. A positive modulator of androgen receptor mediated transactivation. J Biol Chem. 2001;276:13442–51. doi: 10.1074/jbc.M006598200. [DOI] [PubMed] [Google Scholar]
- 47.Xu HE, Stanley TB, Montana VG, et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature. 2002;415:813–7. doi: 10.1038/415813a. [DOI] [PubMed] [Google Scholar]
- 48.Madauss KP, Grygielko ET, Deng SJ, et al. A structural and in vitro characterization of asoprisnil: a selective progesterone receptor modulator. Mol Endocrinol. 2007;21:1066–81. doi: 10.1210/me.2006-0524. [DOI] [PubMed] [Google Scholar]
- 49.Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 50.Taplin ME, Manola J, Oh WK, et al. A phase II study of mifepristone (RU-486) in castration-resistant prostate cancer, with a correlative assessment of androgen-related hormones. BJU Int. 2008;101:1084–9. doi: 10.1111/j.1464-410X.2008.07509.x. [DOI] [PubMed] [Google Scholar]