

Abstract
Hereditary hemochromatosis (HH) encompasses several inherited disorders of iron homeostasis characterized by increased gastrointestinal iron absorption and tissue iron deposition. The most common form of this disorder is HFE-related HH, nearly always caused by homozygosity for the C282Y mutation. A substantial proportion of C282Y homozygotes do not develop clinically significant iron overload, suggesting roles for environmental factors and modifier genes in determining the phenotype. Recent studies have demonstrated that the pathogenesis of nearly all forms of HH involves inappropriately decreased expression of the iron-regulatory hormone hepcidin. Hepcidin serves to decrease the export of iron from reticuloendothelial cells and absorptive enterocytes. Thus, HH patients demonstrate increased iron release from these cell types, elevated circulating iron, and iron deposition in vulnerable tissues. The mechanism by which HFE influences hepcidin expression is an area of current investigation and may offer insights into the phenotypic variability observed in persons with mutations in HFE.
Keywords: Hemochromatosis, iron, HFE, hepcidin, ferroportin
Hemochromatosis may be broadly defined as an abnormal increase in body iron stores, with consequent (or impending) tissue pathology. The term has usually been reserved for chronic rather than acute (for example, iron poisoning) situations. The term “hereditary hemochromatosis” (HH) is restricted to those causes that are considered primary, that is, due to mutations in genes that participate in iron homeostasis (Table 1). Secondary causes of hemochromatosis include parenteral iron loading from repeated erythrocyte transfusions and the excessive enteral iron absorption consequent to certain hematologic conditions (that is, those in which erythrocyte survival is shortened or erythropoiesis is ineffective). While many of the secondary causes of hemochromatosis are due to genetic defects, the term HH is restricted to heritable primary iron-overload disorders.
Table 1. Genetic Classification of Hereditary Hemochromatosis.
| Inheritance | Hepcidin | Iron Deposition | |
|---|---|---|---|
| HFE-related | AR | Low | Hepatocellular > RE |
| C282Y/C282Y | |||
| C282Y/H63D | |||
| Other HFE mutations | |||
| Non-HFE-Related | |||
| Transferrin receptor 2 | AR | Low | Hepatocellular > RE |
| Hemojuvelin | AR | Low | Hepatocellular > RE |
| Hepcidin | AR | Absent | Hepatocellular > RE |
| Ferroportin | |||
| Type I | AD | High | RE > hepatocellular |
| Type II | AD | High | Hepatocellular > RE |
AR, autosomal recessive; RE, reticuloendothelial; AD, autosomal dominant.
By far the most common form of HH, often designated classical HH, is an autosomal recessive disorder most commonly manifested in adults of northern European ancestry.1 The gene responsible, designated HFE, was found to encode a major histocompatibility complex (MHC) class I-like molecule2 without metal transport properties. The mechanisms by which this enigmatic molecule exerts its effects on iron metabolism have only begun to be dissected. The identification and characterization of gene mutations which cause non-HFE-related HH have greatly assisted the understanding of the regulation of iron homeostasis. This article will describe the current understanding of the pathophysiology of HH and present models to explain the underlying dysregulated signaling between body iron status and intestinal iron absorption.
HFE GENE AND PROTEIN
While the autosomal recessive inheritance pattern of classical HH had long been recognized, identification of the responsible gene proved elusive for many years. An important breakthrough in identifying the gene responsible for classical HH occurred in 1976 when HH was found to be closely linked to the MHC.3 Positional cloning identified the responsible gene, termed HFE, which encodes protein with structural similarities to other MHC class I proteins. A single nucleotide change, resulting in the substitution of tyrosine for cysteine at amino acid 282 of the unprocessed protein (C282Y), was identified in nearly all patients with MHC-linked HH. A second mutation in HFE was also identified, which results in the substitution of aspartate for histidine at amino acid 63 (H63D). Proof that HFE is the gene defective in classical HH was provided when knockout of the mouse Hfe gene resulted in iron overload.4-6
It was clear from the primary structure of the HFE protein that it was unlikely to have metal transporter characteristics.2 HFE, like other MHC class I molecules, is physically associated with β2-microglobulin (β2 M).7 The C282Y mutation leads to disruption of an intramolecular disulfide bond necessary for the interaction of mutant HFE protein with β2 M.7,8 As a result, the C282Y mutant protein is abnormally processed, leading to accelerated degradation and reduced cell surface expression.9 The HH-like phenotype of β2M-knockout mice provides independent evidence of the importance of the association between β2M and HFE protein for normal HFE function.10,11
The first potential link between HFE and cellular iron metabolism was made with the discovery that HFE protein forms a complex with transferrin receptor 1 (TfR1). This observation has led to a large number of investigations on the effect of HFE on cellular TfR1-mediated iron transport. While overexpression of HFE in cultured cells has been demonstrated to affect transferrin (Tf)-mediated iron transport and cellular iron status, the magnitude and direction of this effect has varied depending upon the cell type utilized.12,13 Moreover, HFE is capable of changing cellular iron status of cells which do not express TfR1 at all, suggesting that HFE can influence cellular transport of nontransferrin-bound iron.13 Possibly the interaction of HFE with TfR1 serves to regulate the bioavailability of HFE to influence this nontransferrin-bound cellular iron transport. Because HFE and diferric Tf have overlapping binding sites on TfR1,14 the bioavailability of HFE might be influenced by concentrations of both TfR1 and diferric Tf.
Most studies on the cell biology of HFE have utilized cultured cell lines. While HFE is expressed at relatively low levels in most human tissues,2 its role in iron homeostasis is thought to be related to its expression in hepatocytes,15,16 Kupffer cells,17 and/or duodenal crypt cells.18 Proposed mechanisms by which HFE exerts its effects in each of these cell types will be presented below.
HFE Mutations
The HFE mutation found in the majority of classical HH patients is the substitution of tyrosine for cysteine at amino acid 282 of the unprocessed protein (C282Y).2 Approximately 80 to 90% of Caucasian subjects of northern European ancestry with clinical HH are homozygous for C282Y.19 In other ethnic populations, the C282Y mutation is less common. Population studies suggest that the C282Y mutation occurred on an ancestral (possibly Celtic) haplotype ∼2000 years ago.20 It has been speculated that the C282Y mutation, by causing increased iron absorption and augmented body iron stores, provided a selective advantage to a population with limited dietary iron availability.
The frequency of the C282Y mutation in Caucasian populations is quite high (10 to 15% carriers).21 However, it is now clear from population studies that many C282Y homozygotes do not develop clinically significant iron overload,22-24 indicating that there is incomplete penetrance of the C282Y mutation. For example, about one third of C282Y homozygotes have serum ferritin values within the normal range.23-26 Only a small percentage have signs and symptoms of HFE-related HH.22 While factors such as blood loss (for example, menstruation, blood donation) and diet may contribute to the lack of phenotypic expression, they cannot explain most cases. Thus, incomplete penetrance of the C282Y mutation appears largely due to genetic modifiers.
The H63D mutation in HFE is even more common than C282Y and is found in 15 to 40% of Caucasians.27 Unlike C282Y, this mutation appears to have arisen multiple times in different ethnic populations.20 Homozygosity for H63D slightly increases body iron status (Tf saturation, serum ferritin) but does not result in clinically significant iron overload.28 However, compound heterozygosity for the H63D mutation with C282Y is found with an increased frequency in patients with iron overload than predicted for the general population.29 The risk for significant iron loading in the C282Y/H63D compound heterozygote is increased, but is estimated to be nearly 200-fold lower than in the C282Y homozygote.30 Rarely, patients with HH have been found to have HFE mutations other than C282Y or H63D.31,32 However, the relative contribution of mutations other than C282Y or H63D to the overall incidence of HFE-related HH appears to be quite small.
MOLECULAR PARTICIPANTS IN DUODENAL IRON ABSORPTION
An increase in intestinal iron absorption is a pathogenic characteristic of classical HH. Understanding the pathogenesis of HH, therefore, requires a review of the molecular determinants of duodenal iron absorption (see Fig. 1). Because there are no significant physiological mechanisms to regulate iron loss, iron homeostasis is dependent upon a tight linkage between body iron requirements and intestinal iron absorption. Nearly all absorption of dietary iron occurs in the duodenum, where iron may be taken up either as ionic iron or as heme.33 The absorption of both forms of iron is increased in patients with HH. Absorption of ionic iron across the enterocyte occurs in two stages: uptake across the apical membrane and transfer across the basolateral membrane. Prior to uptake, ionic iron requires reduction from the ferric to the ferrous state. This is accomplished by ferric reductase(s) (for example, duodenal cytochrome b related ferric reductase (Dcytb)) on the luminal surface of duodenal enterocytes.34 The ferrous iron crosses the apical membrane via the divalent metal transporter 1 (DMT1).35,36 Iron taken up by the enterocyte may be stored as ferritin (and excreted in the feces when the senescent enterocyte is sloughed) or transferred across the basolateral membrane to the plasma. This latter process occurs via the transporter ferroportin.37-39 The basolateral transfer of iron requires oxidation of iron to the ferric state by the molecule hephaestin.40
Figure 1.
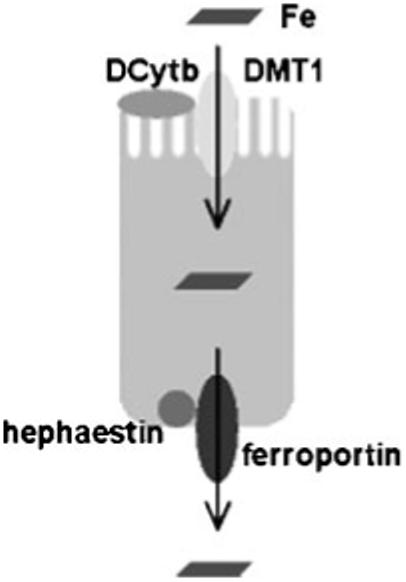
Iron absorption by duodenal enterocytes. Duodenal villus cells are the major site of iron absorption from the diet. Prior to uptake, dietary ionic iron requires reduction from the ferric to the ferrous state. This is accomplished by ferric reductase(s) (for example, Dcytb) expressed on the luminal surface of villus enterocytes. Ferrous iron is taken up by the apical transporter, DMT1. Iron may be stored within the cell as ferritin and lost with the sloughed senescent enterocyte or transferred across the basolateral membrane to the plasma. This latter process occurs via the transporter ferroportin and is facilitated by oxidation of iron to the ferric state by the molecule hephaestin. Dcytb, duodenal cytochrome b related ferric reductase; DMT1, divalent metal transporter 1.
In addition to increased uptake of iron from the diet, patients with HFE-related HH demonstrate increased basolateral transfer of iron from the enterocytes to the plasma. Data suggest that the increased basolateral transfer of iron may be the driving force behind the increased intestinal iron absorption observed in HFE-related HH.41 This may induce secondary changes in the expression of the genes participating in enterocyte iron uptake (Dcytb and DMT1). Indeed, some studies on patients with HFE-related HH42-44 and Hfe-knockout mice45,46 have demonstrated increased expression of mRNAs encoding these transporters.
Several biologic processes influence the rate of dietary iron absorption. Reduction in body iron stores, increased marrow erythropoietic activity, decreased blood hemoglobin content, and decreased blood oxygen saturation all increase the absorption of iron from the diet. In contrast, the presence of systemic inflammation decreases dietary iron absorption. The discovery of hepcidin provides a unifying model to explain the regulation of dietary iron absorption by each of these biologic processes (see below).
IRON UPTAKE BY LIVER AND RETICULOENDOTHELIAL CELLS
Absorbed iron is bound to Tf and delivered via the portal system to the liver, which is the major site of iron storage. Hepatocytes take up Tf-bound iron via TfR1 and possibly by the more highly expressed homologous protein, transferrin receptor 2 (TfR2).47,48 Hepatocytes can also take up Tf-bound iron by nonreceptor-mediated mechanisms.49 Non-Tf bound iron, found in the circulation when Tf becomes highly saturated, may be taken up by hepatocytes as well.50 This latter mechanism appears to contribute to the hepatic iron deposition in HH.51 It is not clear whether the liver is a passive recipient of the excess iron presented to it by the intestine, or whether hepatocellular iron uptake is upregulated with HH.52
Reticuloendothelial macrophages acquire iron primarily from phagocytosis of senescent erythrocytes.53 However, these cells also express TfR1, and this is a potential additional mechanism for iron uptake. The iron is either retained (stored as ferritin) or released into the plasma via the iron export protein ferroportin. The released iron is oxidized to the ferric state in the plasma by ceruloplasmin and is bound to circulating Tf. Patients with HFE-related HH have paradoxical sparing of iron in the reticuloendothelial system,54 probably due to increased release of iron from these cells.55
ROLE OF LIVER IN REGULATION OF IRON ABSORPTION
The role of the liver in iron homeostasis has received increased attention in recent years. Hepatocytes have long been recognized as a storage reservoir for iron, taking up dietary iron from the portal circulation and, at times of increased demand, releasing iron into the hepatic circulation. Recently, several genes expressed in the liver have been identified that, when mutated, cause HH. These include hepcidin,56 TfR2,57 and hemojuvelin.58-60 The finding that each of these genes has high hepatic expression has focused attention on the potential role of the liver in the regulation of intestinal iron absorption.
HEPCIDIN
Hepcidin is a 25-amino acid peptide first identified in urine and plasma as an antimicrobial peptide. However, its role in influencing systemic iron status has become paramount and it is now considered to be the master iron regulatory hormone.61,62 The first evidence that hepcidin is involved in iron homeostasis came from the observation that liver hepcidin mRNA expression is increased in mice with dietary iron loading.63 The fortuitous discovery that knockout of the hepcidin gene in the mouse led to a hemochromatosis-like phenotype established the critical role of hepcidin as a negative regulator of intestinal iron absorption.64 It was later discovered that hepcidin mutations are responsible for one form of juvenile hemochromatosis in several human pedigrees.56 Moreover, transgenic overexpression of hepcidin in mouse hepatocytes leads to a severe form of iron-deficiency anemia that could be overcome by parenteral but not enteral iron.65 Most important, factors regulating intestinal iron absorption (iron stores, erythropoietic activity, hemoglobin, oxygen content, and inflammation) also regulate liver hepcidin expression. In each of these situations, intestinal iron absorption varies inversely with liver hepcidin expression. Specifically, animals subjected to hypoxia or hemolytic anemia show decreased hepatic hepcidin mRNA concentrations,66 while animals with dietary iron overload67 or systemic inflammation66 have increased hepatic mRNA concentrations. While technical problems have prevented the measurement of the mature hepcidin peptide in serum, urinary hepcidin concentrations can be measured. As expected, urinary hepcidin levels are increased in human subjects with inflammation but decreased in HH patients.68-71
A temporal association exists between a decrease in liver hepcidin mRNA levels and an increase in intestinal iron transporter gene expression.72,73 Hepcidin decreases the functional activity of the iron exporter ferroportin by causing its internalization and degradation.74 In the enterocyte, this would lead to decreased basolateral iron transfer and thus decreased dietary iron absorption. The increased intracellular iron might then cause secondary (but perhaps physiologically less relevant) decreases in expression of genes involved in apical enterocyte iron uptake, that is, Dcytb and DMT1. These secondary effects are likely of less relevance because iron taken up, but retained, by the enterocytes is excreted with enterocyte sloughing.
The mechanism by which liver hepcidin expression is modulated by iron status is conjectural. However, a potential hepatocyte iron “sensor” has been proposed to be TfR2.48,75 Mutation of TfR2 causes a rare form of HH in humans.57 Likewise, TfR2-mutant mice have an HH phenotype.76 Despite hepatic iron loading, hepcidin expression is decreased in patients with TfR2-related HH77 and in TfR2-mutant mice.78 This suggests that TfR2 is necessary for the appropriate transduction of the signal between body iron status and hepcidin expression.
ROLE OF HEPCIDIN IN HFE-RELATED HH
Despite excess hepatic iron stores, patients with HFE-related HH (and Hfe-knockout mice) have decreased hepatic expression of hepcidin.79-81 The reason functional loss of HFE leads to inappropriate underexpression of hepcidin is uncertain. One possibility is that HFE is a participant in the hepatocellular sensing of body iron status (see Fig. 2). In this model, hepatocytes modulate hepcidin expression by sensing the circulating levels of diferric Tf. The concentration of diferric Tf in the portal venous system reflects the rate of iron absorption. The binding of diferric Tf to hepatocellular TfR2 might transduce a signal that modulates expression of hepcidin.
Figure 2.
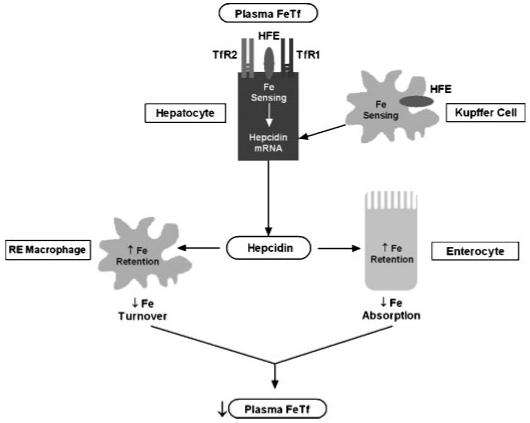
Role of hepcidin in the pathogenesis of hereditary hemochromatosis. In this model, diferric Tf in the portal circulation serves as the ligand for hepatocyte TfRs (TfR1 and/or TfR2). This interaction transduces a signal that increases hepatocellular expression of hepcidin. Alternatively, Kupffer cells may sense iron stores and send an as yet unknown signal to the hepatocytes to increase hepcidin expression. Circulating hepcidin binds ferroportin in target cells (duodenal enterocytes and reticuloendothelial macrophages) to decrease ferroportin-mediated iron export from these cells. In duodenal enterocytes, this leads to a decrease in the amount of dietary iron absorbed into the circulation. Thus, circulating levels of diferric Tf are normalized and homeostasis is maintained. In HFE-related HH, loss of functional HFE protein (in hepatocytes and/or Kupffer cells) leads to aberrant hepatocellular sensing of plasma iron, inappropriately low levels of hepcidin, decreased reticuloendothelial iron stores, and increased duodenal iron absorption. Tf, transferrin; TfR1, transferrin receptor 1; TfR2, transferrin receptor 2.
HFE might influence the sensitivity of the hepatocellular iron sensor in such a way as to increase the amount of hepcidin expressed at any given circulating concentration of differic Tf. In HFE-related HH, loss of hepatocellular cell surface expression of HFE leads to a relative decrease in the expression of hepcidin. Decreased circulating hepcidin would cause increased ferroportin-mediated efflux of iron from certain cell types, including reticuloendothelial cells and duodenal enterocytes.
The model described above identifies the hepatocyte as the cell type requiring HFE for a normal hepcidin response to iron status. Some evidence, however, suggests that HFE expression by Kupffer cells may be important in the regulation of hepcidin expression. Hepatocytes in cell culture do not demonstrate a change in hepcidin expression in response to changes in iron content,82 raising the possibility that another cell type is needed for functional iron-sensing. Moreover, Hfe-knockout mice demonstrate improved iron status upon repopulating the reticuloendothelial cells with transplanted bone marrow from wild-type mice.83
While it is clear that dysregulation of hepcidin is central to the pathogenesis of HFE-related HH, HFE might be able to influence body iron homeostasis independently of hepcidin. As outlined previously, transfection of HFE into cultured cells directly influences their iron status. Possibly, loss of HFE in certain cell types (for example, duodenal crypt cells) directly contributes to the iron homeostasis abnormalities observed in HFE-related HH. Duodenal crypt cells express HFE and have been proposed to act as a sensor of body iron status via uptake of plasma diferric Tf.84 The duodenal uptake of plasma iron is indeed impaired in Hfe-knockout mice.85 This observation supports a model in which functional loss of HFE may decrease the iron pool in duodenal crypt cells, resulting in a relatively iron-deficient state in these cells. This could result in the increased expression of iron transport genes in daughter villus enterocytes and lead to increased dietary iron absorption. While this “crypt cell hypothesis” has been posed as an explanation for the excess dietary iron absorption in HH, the effect of HFE on liver hepcidin expression appears to be paramount.86,87
NON-HFE-RELATED HH
While HFE mutations account for the vast majority of HH, other forms have been recognized and are generally grouped together under the term “non-HFE-related HH”88 (Table 1). Mutations of the TfR2 gene produce an autosomal recessive form of HH57,89 that is clinically very similar to classical HH. It is not yet known how these uncommon mutations of the TfR2 gene result in iron overload, but it is possible that they cause abnormal iron sensing by hepatocytes, the predominant site of TfR2 expression.
Mutations in two different genes have been shown to cause forms of juvenile HH. The more common of these occurs in the hemojuvelin gene.59 Hemojuvelin is expressed in hepatocytes and may be involved in regulating the hepcidin pathway, as hepcidin levels are low in this condition. As mentioned earlier, mutations in the hepcidin gene also produce a form of juvenile HH.57
Two distinct types of ferroportin mutations cause autosomal dominant HH.90,91 The first type results in ferroportin inactivation; the second type interferes with the interaction between ferroportin and hepcidin but retains iron export capability. Inactivating ferroportin mutations cause a cellular distribution of iron loading which differs from classical HH, as iron is retained primarily in RE cells rather than hepatocytes. Moreover, serum iron levels tend to be lower than in HFE-related HH. Individuals with the second type of ferroportin mutation fail to respond to hepcidin and thus demonstrate a more classical HH phenotype. In both forms of ferroportin-related HH (unlike other forms of HH discussed in this article), hepcidin expression is elevated rather than decreased (see Table 1).
African iron overload is a form of HH exacerbated by dietary iron loading that occurs primarily in sub-Saharan Africa.92 The distribution of accumulated iron in African iron overload is different than in HFE-related HH, in that iron loading appears more prominent in reticuloendothelial cells relative to hepatocytes (as seen with inactivating ferroportin mutations). Indeed, a particular mutation in ferroportin has been associated with some cases of African iron overload.93 This mutation might account for elevated ferritin levels observed in some African-Americans as well.93-95
SUMMARY
The identification of HFE and other genes involved in iron metabolism has greatly expanded our understanding of many aspects of HH. The introduction of a commercially available genetic test for the C282Y and H63D mutations of HFE allows presymptomatic diagnosis and adds precision to studies of the population genetics of HFE-related HH. It is now recognized that a substantial proportion of C282Y homozygotes do not develop clinically significant iron overload, and modifier genes may be involved in this phenomenon. Mouse models of HH and cell culture studies have increased our understanding of the regulation of iron homeostasis. The pathogenesis of nearly all forms of HH involves an inappropriately low expression of hepcidin, an iron-regulatory hormone that acts to decrease the export of iron from duodenal enterocytes and reticuloendothelial cells. As a consequence of this low hepcidin expression, HH patients with phenotypic expression have increased absorption of dietary iron and elevated Tf saturations. Future studies will refine our knowledge of the mechanisms of action of HFE protein, hepcidin, and other iron-related proteins, and further define the roles of these molecules in the pathogenesis of HH.
ACKNOWLEDGMENTS
This work was supported in part by NIH grants HL66225 (R.E.F.), DK53405 (W.S.S.), and DK41816 (B.R.B.).
ABBREVIATIONS
- AD
autosomal dominant
- AR
autosomal recessive
- B2M
B2 microglobulin
- Dcytb
duodenal cytochrome b related ferric reductase
- DMT1
divalent metal transporter 1
- HH
hereditary hemochromatosis
- MHC
major histocompatability complex
- RE
reticuloendotheliasl
- Tf
transferrin
- TfR1
transferrin receptor 1
- TfR2
transferrin receptor 2
Footnotes
Objectives: Upon completion of this article, the reader should be able to (1) identify the key processes regulating intestinal iron absorption, (2) understand the role of hepcidin in regulating the amount of iron released from duodenal enterocytes and reticuloendothelial cells, (3) understand the pathogenesis of HH in terms of decreased expression of hepcidin, and (4) be familiar with genes responsible for non-HFE-related HH.
Accreditation: Tufts University School of Medicine (TUSM) is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Credit: TUSM designates this educational activity for a maximum of 1 category 1 credit toward the AMA Physicians Recognition Award. Each physician should claim only those credits that he/she actually spent in the activity.
REFERENCES
- 1.Pietrangelo A. Hereditary hemochromatosis—a new look at an old disease. N Engl J Med. 2004;350:2383–2397. doi: 10.1056/NEJMra031573. [DOI] [PubMed] [Google Scholar]
- 2.Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 3.Simon M, Bourel M, Fauchet R, et al. Association of HLA-A3 and HLA-B14 antigens with idiopathic haemochromatosis. Gut. 1976;17:332–334. doi: 10.1136/gut.17.5.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou XY, Tomatsu S, Fleming RE, et al. HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc Natl Acad Sci USA. 1998;95:2492–2497. doi: 10.1073/pnas.95.5.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bahram S, Gilfillan S, Kuhn LC, et al. Experimental hemochromatosis due to MHC class I HFE deficiency: immune status and iron metabolism. Proc Natl Acad Sci USA. 1999;96:13312–13317. doi: 10.1073/pnas.96.23.13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levy JE, Montross LK, Cohen DE, et al. The C282Y mutation causing hereditary hemochromatosis does not produce a null allele. Blood. 1999;94:9–11. [PubMed] [Google Scholar]
- 7.Feder JN, Tsuchihashi Z, Irrinki A, et al. The hemochromatosis founder mutation in HLA-H disrupts beta2-microglobulin interaction and cell surface expression. J Biol Chem. 1997;272:14025–14028. doi: 10.1074/jbc.272.22.14025. [DOI] [PubMed] [Google Scholar]
- 8.Waheed A, Parkkila S, Zhou XY, et al. Hereditary hemochromatosis: effects of C282Y and H63D mutations on association with beta2-microglobulin, intracellular processing, and cell surface expression of the HFE protein in COS-7 cells. Proc Natl Acad Sci USA. 1997;94:12384–12389. doi: 10.1073/pnas.94.23.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parkkila S, Parkkila AK, Waheed A, et al. Cell surface expression of HFE protein in epithelial cells, macrophages, and monocytes. Haematologica. 2000;85:340–345. [PubMed] [Google Scholar]
- 10.de Sousa M, Reimao R, Lacerda R, et al. Iron overload in beta 2-microglobulin-deficient mice. Immunol Lett. 1994;39:105–111. doi: 10.1016/0165-2478(94)90094-9. [DOI] [PubMed] [Google Scholar]
- 11.Santos M, Schilham MW, Rademakers LH, et al. Defective iron homeostasis in beta 2-microglobulin knockout mice recapitulates hereditary hemochromatosis in man. J Exp Med. 1996;184:1975–1985. doi: 10.1084/jem.184.5.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waheed A, Grubb JH, Zhou XY, et al. Regulation of transferrin-mediated iron uptake by HFE, the protein defective in hereditary hemochromatosis. Proc Natl Acad Sci USA. 2002;99:3117–3122. doi: 10.1073/pnas.042701499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlson H, Zhang AS, Fleming WH, et al. The hereditary hemochromatosis protein, HFE, lowers intracellular iron levels independently of transferrin receptor 1 in TRVb cells. Blood. 2005;105:2564–2570. doi: 10.1182/blood-2004-03-1204. [DOI] [PubMed] [Google Scholar]
- 14.Giannetti AM, Bjorkman PJ. HFE and transferrin directly compete for transferrin receptor in solution and at the cell surface. J Biol Chem. 2004;279:25866–25875. doi: 10.1074/jbc.M401467200. [DOI] [PubMed] [Google Scholar]
- 15.Holmstrom P, Dzikaite V, Hultcrantz R, et al. Structure and liver cell expression pattern of the HFE gene in the rat. J Hepatol. 2003;39:308–314. doi: 10.1016/s0168-8278(03)00293-9. [DOI] [PubMed] [Google Scholar]
- 16.Zhang AS, Xiong S, Tsukamoto H, et al. Localization of iron metabolism-related mRNAs in rat liver indicate that HFE is predominantly expressed in hepatocytes. Blood. 2004;103:1509–1514. doi: 10.1182/blood-2003-07-2378. [DOI] [PubMed] [Google Scholar]
- 17.Bastin JM, Jones M, O’Callaghan CA, et al. Kupffer cell staining by an HFE-specific monoclonal antibody: implications for hereditary haemochromatosis. Br J Haematol. 1998;103:931–941. doi: 10.1046/j.1365-2141.1998.01102.x. [DOI] [PubMed] [Google Scholar]
- 18.Parkkila S, Waheed A, Britton RS, et al. Immunohistochemistry of HLA-H, the protein defective in patients with hereditary hemochromatosis, reveals unique pattern of expression in gastrointestinal tract. Proc Natl Acad Sci USA. 1997;94:2534–2539. doi: 10.1073/pnas.94.6.2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Powell LW, Subramaniam VN, Yapp TR. Haemochromatosis in the new millennium. J Hepatol. 2000;32:48–62. doi: 10.1016/s0168-8278(00)80415-8. [DOI] [PubMed] [Google Scholar]
- 20.Rochette J, Pointon JJ, Fisher CA, et al. Multicentric origin of hemochromatosis gene (HFE) mutations. Am J Hum Genet. 1999;64:1056–1062. doi: 10.1086/302318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beckman LE, Saha N, Spitsyn V, et al. Ethnic differences in the HFE codon 282 (Cys/Tyr) polymorphism. Hum Hered. 1997;47:263–267. doi: 10.1159/000154422. [DOI] [PubMed] [Google Scholar]
- 22.Beutler E, Felitti VJ, Koziol JA, et al. Penetrance of 845G → A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet. 2002;359:211–218. doi: 10.1016/S0140-6736(02)07447-0. [DOI] [PubMed] [Google Scholar]
- 23.Adams PC, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352:1769–1778. doi: 10.1056/NEJMoa041534. [DOI] [PubMed] [Google Scholar]
- 24.Olynyk JK, Cullen DJ, Aquilia S, et al. A population-based study of the clinical expression of the hemochromatosis gene. N Engl J Med. 1999;341:718–724. doi: 10.1056/NEJM199909023411002. [DOI] [PubMed] [Google Scholar]
- 25.Burt MJ, George PM, Upton JD, et al. The significance of haemochromatosis gene mutations in the general population: implications for screening. Gut. 1998;43:830–836. doi: 10.1136/gut.43.6.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McDonnell SM, Hover A, Gloe D, et al. Population-based screening for hemochromatosis using phenotypic and DNA testing among employees of health maintenance organizations in Springfield, Missouri. Am J Med. 1999;107:30–37. doi: 10.1016/s0002-9343(99)00163-1. [DOI] [PubMed] [Google Scholar]
- 27.Steinberg KK, Cogswell ME, Chang JC, et al. Prevalence of C282Y and H63D mutations in the hemochromatosis (HFE) gene in the United States. JAMA. 2001;285:2216–2222. doi: 10.1001/jama.285.17.2216. [DOI] [PubMed] [Google Scholar]
- 28.Gochee PA, Powell LW, Cullen DJ, et al. A population-based study of the biochemical and clinical expression of the H63D hemochromatosis mutation. Gastroenterology. 2002;122:646–651. doi: 10.1016/s0016-5085(02)80116-0. [DOI] [PubMed] [Google Scholar]
- 29.Bacon BR. Hemochromatosis: diagnosis and management. Gastroenterology. 2001;120:718–725. doi: 10.1053/gast.2001.21913. [DOI] [PubMed] [Google Scholar]
- 30.Risch N. Haemochromatosis, HFE and genetic complexity [letter] Nat Genet. 1997;17:375–376. doi: 10.1038/ng1297-375. [DOI] [PubMed] [Google Scholar]
- 31.Pointon JJ, Wallace D, Merryweather-Clarke AT, et al. Uncommon mutations and polymorphisms in the hemochromatosis gene. Genet Test. 2000;4:151–161. doi: 10.1089/10906570050114867. [DOI] [PubMed] [Google Scholar]
- 32.Britton RS, Fleming RE, Parkkila S, et al. Pathogenesis of hereditary hemochromatosis: genetics and beyond. Semin Gastrointest Dis. 2002;13:68–79. [PubMed] [Google Scholar]
- 33.Andrews NC. Disorders of iron metabolism. N Engl J Med. 1999;341:1986–1995. doi: 10.1056/NEJM199912233412607. [DOI] [PubMed] [Google Scholar]
- 34.McKie AT, Barrow D, Latunde-Dada GO, et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. 2001;291:1755–1759. doi: 10.1126/science.1057206. [DOI] [PubMed] [Google Scholar]
- 35.Fleming MD, Trenor CC, III, Su MA, et al. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet. 1997;16:383–386. doi: 10.1038/ng0897-383. [DOI] [PubMed] [Google Scholar]
- 36.Gunshin H, Mackenzie B, Berger UV, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 37.Donovan A, Brownlie A, Zhou Y, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776–781. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 38.McKie AT, Marciani P, Rolfs A, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- 39.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275:19906–19912. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 40.Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21:195–199. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- 41.McLaren GD, Nathanson MH, Jacobs A, et al. Regulation of intestinal iron absorption and mucosal iron kinetics in hereditary hemochromatosis. J Lab Clin Med. 1991;117:390–401. [PubMed] [Google Scholar]
- 42.Zoller H, Pietrangelo A, Vogel W, et al. Duodenal metal-transporter (DMT-1, NRAMP-2) expression in patients with hereditary haemochromatosis. Lancet. 1999;353:2120–2123. doi: 10.1016/S0140-6736(98)11179-0. [DOI] [PubMed] [Google Scholar]
- 43.Zoller H, Koch RO, Theurl I, et al. Expression of the duodenal iron transporters divalent-metal transporter 1 and ferroportin 1 in iron deficiency and iron overload. Gastroenterology. 2001;120:1412–1419. doi: 10.1053/gast.2001.24033. [DOI] [PubMed] [Google Scholar]
- 44.Rolfs A, Bonkovsky HL, Kohlroser JG, et al. Intestinal expression of genes involved in iron absorption in humans. Am J Physiol Gastrointest Liver Physiol. 2002;282:G598–G607. doi: 10.1152/ajpgi.00371.2001. [DOI] [PubMed] [Google Scholar]
- 45.Fleming RE, Migas MC, Zhou X, et al. Mechanism of increased iron absorption in murine model of hereditary hemochromatosis: increased duodenal expression of the iron transporter DMT1. Proc Natl Acad Sci USA. 1999;96:3143–3148. doi: 10.1073/pnas.96.6.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dupic F, Fruchon S, Bensaid M, et al. Inactivation of the hemochromatosis gene differentially regulates duodenal expression of iron-related mRNAs between mouse strains. Gastroenterology. 2002;122:745–751. doi: 10.1053/gast.2002.31877. [DOI] [PubMed] [Google Scholar]
- 47.Kawabata H, Yang R, Hirama T, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem. 1999;274:20826–20832. doi: 10.1074/jbc.274.30.20826. [DOI] [PubMed] [Google Scholar]
- 48.Fleming RE, Migas MC, Holden CC, et al. Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc Natl Acad Sci USA. 2000;97:2214–2219. doi: 10.1073/pnas.040548097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonkovsky HL. Iron and the liver. Am J Med Sci. 1991;301:32–43. doi: 10.1097/00000441-199101000-00006. [DOI] [PubMed] [Google Scholar]
- 50.Brissot P, Wright TL, Ma WL, et al. Efficient clearance of non-transferrin-bound iron by rat liver. Implications for hepatic iron loading in iron overload states. J Clin Invest. 1985;76:1463–1470. doi: 10.1172/JCI112125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Batey RG, Lai Chung Fong P, Shamir S, et al. A non-transferrin-bound serum iron in idiopathic hemochromatosis. Dig Dis Sci. 1980;25:340–346. doi: 10.1007/BF01308057. [DOI] [PubMed] [Google Scholar]
- 52.Chua AC, Olynyk JK, Leedman PJ, et al. Nontransferrin-bound iron uptake by hepatocytes is increased in the Hfe knockout mouse model of hereditary hemochromatosis. Blood. 2004;104:1519–1525. doi: 10.1182/blood-2003-11-3872. [DOI] [PubMed] [Google Scholar]
- 53.Deiss A. Iron metabolism in reticuloendothelial cells. Semin Hematol. 1983;20:81–90. [PubMed] [Google Scholar]
- 54.McLaren GD. Reticuloendothelial iron stores and hereditary hemochromatosis: a paradox [editorial] J Lab Clin Med. 1989;113:137–138. [PubMed] [Google Scholar]
- 55.Moura E, Noordermeer MA, Verhoeven N, et al. Iron release from human monocytes after erythrophagocytosis in vitro: an investigation in normal subjects and hereditary hemochromatosis patients. Blood. 1998;92:2511–2519. [PubMed] [Google Scholar]
- 56.Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33:21–22. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 57.Roetto A, Totaro A, Piperno A, et al. New mutations inactivating transferrin receptor 2 in hemochromatosis type 3. Blood. 2001;97:2555–2560. doi: 10.1182/blood.v97.9.2555. [DOI] [PubMed] [Google Scholar]
- 58.Papanikolaou G, Samuels ME, Ludwig EH, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36:77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 59.Lanzara C, Roetto A, Daraio F, et al. Spectrum of hemojuvelin gene mutations in 1q-linked juvenile hemochromatosis. Blood. 2004;103:4317–4321. doi: 10.1182/blood-2004-01-0192. [DOI] [PubMed] [Google Scholar]
- 60.Lee PL, Beutler E, Rao SV, et al. Genetic abnormalities and juvenile hemochromatosis: mutations of the HJV gene encoding hemojuvelin. Blood. 2004;103:4669–4671. doi: 10.1182/blood-2004-01-0072. [DOI] [PubMed] [Google Scholar]
- 61.Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood. 2003;102:783–788. doi: 10.1182/blood-2003-03-0672. [DOI] [PubMed] [Google Scholar]
- 62.Nicolas G, Viatte L, Bennoun M, et al. Hepcidin, a new iron regulatory peptide. Blood Cells Mol Dis. 2002;29:327–335. doi: 10.1006/bcmd.2002.0573. [DOI] [PubMed] [Google Scholar]
- 63.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 64.Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA. 2001;98:8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nicolas G, Bennoun M, Porteu A, et al. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci USA. 2002;99:4596–4601. doi: 10.1073/pnas.072632499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110:1037–1044. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 68.Nemeth E, Valore EV, Territo M, et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101:2461–2463. doi: 10.1182/blood-2002-10-3235. [DOI] [PubMed] [Google Scholar]
- 69.Kemna EH, Pickkers P, Nemeth E, van der Hoeven H, Swinkels D. Time-course analysis of hepcidin, serum iron, and plasma cytokine levels in humans injected with LPS. Blood. 2005;106:1864–1866. doi: 10.1182/blood-2005-03-1159. [DOI] [PubMed] [Google Scholar]
- 70.Detivaud L, Nemeth E, Boudjema K, et al. Hepcidin levels in humans are correlated with hepatic iron stores, hemoglobin levels, and hepatic function. Blood. 2005;106:746–748. doi: 10.1182/blood-2004-12-4855. [DOI] [PubMed] [Google Scholar]
- 71.Papanikolaou G, Tzilianos M, Christakis JI, et al. Hepcidin in iron overload disorders. Blood. 2005;105:4103–4105. doi: 10.1182/blood-2004-12-4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frazer DM, Wilkins SJ, Becker EM, et al. Hepcidin expression inversely correlates with the expression of duodenal iron transporters and iron absorption in rats. Gastroenterology. 2002;123:835–844. doi: 10.1053/gast.2002.35353. [DOI] [PubMed] [Google Scholar]
- 73.Frazer DM, Inglis HR, Wilkins SJ, et al. Delayed hepcidin response explains the lag period in iron absorption following a stimulus to increase erythropoiesis. Gut. 2004;53:1509–1515. doi: 10.1136/gut.2003.037416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 75.Fleming RE, Sly WS. Mechanisms of iron accumulation in hereditary hemochromatosis. Annu Rev Physiol. 2002;64:663–680. doi: 10.1146/annurev.physiol.64.081501.155838. [DOI] [PubMed] [Google Scholar]
- 76.Fleming RE, Ahmann JR, Migas MC, et al. Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc Natl Acad Sci USA. 2002;99:10653–10658. doi: 10.1073/pnas.162360699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nemeth E, Roetto A, Garozzo G, et al. Hepcidin is decreased in TFR2 hemochromatosis. Blood. 2005;105:1803–1806. doi: 10.1182/blood-2004-08-3042. [DOI] [PubMed] [Google Scholar]
- 78.Kawabata H, Fleming RE, Gui D, et al. Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood. 2005;105:376–381. doi: 10.1182/blood-2004-04-1416. [DOI] [PubMed] [Google Scholar]
- 79.Bridle KR, Frazer DM, Wilkins SJ, et al. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet. 2003;361:669–673. doi: 10.1016/S0140-6736(03)12602-5. [DOI] [PubMed] [Google Scholar]
- 80.Ahmad KA, Ahmann JR, Migas MC, et al. Decreased liver hepcidin expression in the Hfe knockout mouse. Blood Cells Mol Dis. 2002;29:361–366. doi: 10.1006/bcmd.2002.0575. [DOI] [PubMed] [Google Scholar]
- 81.Muckenthaler M, Roy CN, Custodio AO, et al. Regulatory defects in liver and intestine implicate abnormal hepcidin and Cybrd1 expression in mouse hemochromatosis. Nat Genet. 2003;34:102–107. doi: 10.1038/ng1152. [DOI] [PubMed] [Google Scholar]
- 82.Lee P, Peng H, Gelbart T, et al. The IL-6- and lipopolysaccharide-induced transcription of hepcidin in HFE-, transferrin receptor 2-, and beta 2-microglobulin-deficient hepatocytes. Proc Natl Acad Sci USA. 2004;101:9263–9265. doi: 10.1073/pnas.0403108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Makui H, Soares RJ, Jiang W, Constante M, Santo MM. Contribution of Hfe expression in macrophages to the regulation of hepatic hepcidin levels and iron loading. Blood. 2005 doi: 10.1182/blood-2005-02-0629. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Parkkila S, Niemela O, Britton RS, et al. Molecular aspects of iron absorption and HFE expression. Gastroenterology. 2001;121:1489–1496. doi: 10.1053/gast.2001.29617. [DOI] [PubMed] [Google Scholar]
- 85.Trinder D, Olynyk JK, Sly WS, Morgan EH. Iron uptake from plasma transferrin by the duodenum is impaired in the Hfe knockout mouse. Proc Natl Acad Sci USA. 2002;99:5622–5626. doi: 10.1073/pnas.082112299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Frazer DM, Anderson GJ. The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Mol Dis. 2003;30:288–297. doi: 10.1016/s1079-9796(03)00039-1. [DOI] [PubMed] [Google Scholar]
- 87.Nicolas G, Viatte L, Lou DQ, et al. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet. 2003;34:97–101. doi: 10.1038/ng1150. [DOI] [PubMed] [Google Scholar]
- 88.Pietrangelo A. Non-HFE hemochromatosis. Hepatology. 2004;39:21–29. doi: 10.1002/hep.20007. [DOI] [PubMed] [Google Scholar]
- 89.Camaschella C, Roetto A, Cali A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet. 2000;25:14–15. doi: 10.1038/75534. [DOI] [PubMed] [Google Scholar]
- 90.De Domenico I, Ward DM, Nemeth E, et al. The molecular basis of ferroportin-linked hemochromatosis. Proc Natl Acad Sci USA. 2005;102:8955–8960. doi: 10.1073/pnas.0503804102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Drakesmith H, Schimanski LM, Ormerod E, et al. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood. 2005;106:1092–1097. doi: 10.1182/blood-2005-02-0561. [DOI] [PubMed] [Google Scholar]
- 92.Gordeuk VR. African iron overload. Semin Hematol. 2002;39:263–269. doi: 10.1053/shem.2002.35636. [DOI] [PubMed] [Google Scholar]
- 93.Gordeuk VR, Caleffi A, Corradini E, et al. Iron overload in Africans and African-Americans and a common mutation in the SCL40A1 (ferroportin 1) gene. Blood Cells Mol Dis. 2003;31:299–304. doi: 10.1016/s1079-9796(03)00164-5. [DOI] [PubMed] [Google Scholar]
- 94.Barton JC, Acton RT, Rivers CA, et al. Genotypic and phenotypic heterogeneity of African Americans with primary iron overload. Blood Cells Mol Dis. 2003;31:310–319. doi: 10.1016/s1079-9796(03)00166-9. [DOI] [PubMed] [Google Scholar]
- 95.Beutler E, Barton JC, Felitti VJ, et al. Ferroportin 1 (SCL40A1) variant associated with iron overload in African-Americans. Blood Cells Mol Dis. 2003;31:305–309. doi: 10.1016/s1079-9796(03)00165-7. [DOI] [PubMed] [Google Scholar]