

Abstract
Since the introduction of highly active anti-retroviral therapy, survival from HIV has markedly improved, but less of an effect has been found for HIV-associated neurocognitive disorders. Based on our previous findings, we hypothesized that increased production of osteopontin might contribute to the persistence of CNS dysfunctions. We found increased osteopontin in brains of humans and monkeys during HIV/SIV encephalitis. In the cerebrospinal fluid, osteopontin was elevated in HIV infected individuals regardless of neuropsychological status. However in the plasma, osteopontin levels were significantly increased in individuals with HIV-associated dementia. Additionally, a longitudinal study of monkeys revealed that plasma osteopontin increased prior to the development of SIV-induced neurological and clinical abnormalities. Thus, plasma osteopontin levels significantly correlate with HIV-induced CNS dysfunction in the current era of efficacious antiviral treatment, and suggest that developing interventions to modulate osteopontin production or signaling might be beneficial in the prevention or treatment of HIV-induced CNS disorders.
Keywords: HIV, SIV, dementia, encephalitis, osteopontin, macrophage, cerebrospinal, brain, central nervous system
Introduction
HIV positive patients frequently exhibit a wide range of neurological problems due to the neuropathological events that result from HIV-1 infection itself, as opposed to opportunistic infections. Neurological symptoms associated with HIV can include cognitive impairments, motor disturbances, and behavioral changes [1]. HIV-associated neurocognitive abnormalities have been classified in two categories: minor cognitive/motor disorder, resulting in complaints and problems that can compromise everyday functioning; and dementia, characterized by more severe impairments [2]; an update to this nosology has recently been proposed [3]. HIV-associated dementia commonly occurs at late stages of viral infection when the immune system is compromised, whereas HIV-associated minor cognitive/motor disorder occurs at early stages of infection as well.
Neurological complications of HIV infection persist in the current era of highly active anti-retroviral therapy (HAART) [4]. Fortunately, following HAART introduction, the incidence of HIV-associated dementia has decreased from an estimated 20% to less than 5% of advanced adult AIDS cases. However the minor cognitive/motor disorder develops in 20% of symptomatic HIV seropositive adults, even those treated with HAART. The condition is associated with shortened survival and significant effects on quality of life, such as job-related skills and employment, and its presence is predictive of future development of HIV encephalitis [5, 6]. Thus, despite HAART, the occurrence of clinical neurological dysfunction still occurs.
The etiology of the neuropathogenesis observed in HIV infection is complex. Viral infection of the brain is thought to be an initiating event, but other mechanisms secondary to virus infection, are involved in the brain dysfunction. Macrophages, the major target for HIV in the brain, infiltrate the brain and can produce numerous products potentially harmful to the CNS, are prime candidates in mediating indirect damage to the CNS initiated by HIV infection [7].
Ongoing research efforts have focused on the identification of dependable correlates for HIV-associated neurocognitive abnormalities. One clue was found in the blood where an increase in the monocyte subset that express CD16 in addition to CD69 had been found in HIV-associated dementia, and supernatants from such cells showed neurotoxic properties [8]. However, with the onset of HAART, this correlation no longer held [9].
Other research has focused on the cerebrospinal fluid (CSF), and findings have been reported for diverse metabolites and proteins [10-15]. However, these correlations were demonstrated mainly in the era before the availability of HAART. In the current era of treatment the relationship between CSF markers and neurologic status is altered [16-18], and this lack of biomarkers has hindered work on HIV-induced CNS disorders, which continue to afflict infected individuals.
Infection of macaques with simian immunodeficiency virus remains the best model of HIV disease in humans. Progressive lymphoid changes, opportunistic infections, a wasting syndrome and central nervous system disease characterize the syndrome in both humans and monkeys [19]. Using the SIV model, our previous gene array expression analysis of the brains of rhesus monkeys with SIV encephalitis [20] revealed a significant increase in osteopontin, an extracellular protein involved in differentiation and immune cell activation as well as cell attachment and migration [21]. We subsequently showed that osteopontin increased monocyte retention and protected monocytes from apoptosis, and proposed these mechanisms as an underlying cause of macrophage accumulation during HIV-1 infection [22].
We then hypothesized that increased expression of osteopontin may correlate with the development of the neurological disease associated with HIV-1 infection. To test this, we examined the expression of osteopontin in both plasma and cerebral spinal fluid in humans as well as SIV-infected monkeys. Our results clearly demonstrate a correlation of plasma osteopontin levels with severity of neurologic disease.
Methods
Rhesus macaques
Rhesus monkeys were infected with derivatives of SIVmac251 [23, 24]. Animals were categorized as: SIV encephalitis (n=7 for the RNA analysis, n=6 for ELISA analysis) and SIV without encephalitis, which is subdivided into: simian AIDS without encephalitis (n=4 for ELISA analysis) and SIV positive without encephalitis and without simian AIDS (n=12 for the RNA analysis, n=4 for ELISA analysis). Additionally, for quantitative real-time PCR analysis, we utilized RNA from uninfected controls animals (n=9).
RNA isolation
RNA was purified from frontal lobe samples using TRIzol reagent (Invitrogen, Carlsbad, CA), and then further purified utilizing the RNeasy mini kit (Qiagen, Valencia, CA). RNA was quantified by 260 nm UV absorption.
Reverse Transcription
RNA was incubated with random primers and SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA); followed by heat-inactivation and treatment with RNaseH (New England BioLabs, Ipswich, MA).
Quantitative Real-Time PCR (qRT-PCR)
Specific RNA transcripts were quantified through the use of real-time PCR using dual-labeled (FAM-TAMRA) hydrolysis probes; reactions were optimized and validated by dilutional analysis. For osteopontin, the primers were agaagttccgcagacctgac and gctttccacatgtgaggtga, the probe cagtaccctgatgctacagacgagg. For TATA-box binding protein, the primers were aaagaccattgcacttcgtg and ggttcgtggctctcttatcc, the probe tcccaagcggtttgctgcag.
Primers and probes were obtained from Eurogentec (San Diego, CA). Real time PCR reactions were performed in duplicate using Platinum qPCR UDG Supermix (Invitrogen) in a Stratagene MX3000 machine (Stratagene, La Jolla, CA). To compute the relative amounts of osteopontin mRNA in the samples, the average cycle threshold (Ct) of the primary signal of TATA-box binding protein (as a housekeeping gene) was subtracted from that for the osteopontin to give the change in Ct (dCt), and relative units were calculated as 2-dCt.
Immunohistochemistry
Formalin-fixed, paraffin-embedded rhesus brain sections were deparaffinized with xylene and hydrated in graded alcohols. Immunohistochemical staining followed a basic indirect protocol using a citrate antigen retrieval method. The primary antibody utilized was directed towards osteopontin (Panomics, Redwood City, CA; 1:2), detected with PicturePlus universal secondary antibody-horseradish peroxidase polymer reagent (Zymed, San Francisco, CA), developed with NovaRed chromogen (Vector Laboratories, Burlingame, CA), and finally followed by hematoxylin counterstain (Sigma-Aldrich, St. Louis, MO). Control slides included omission of primary antibodies and irrelevant antibodies. Image capture was performed with a Spot RT Color CCD camera and Spot RT software, version 3.4.2 for MacOS (Spot Diagnostic Instruments, Sterling Heights, MI) using a Leica Diaplan microscope (Leica Inc., Deerfield, IL).
Immunofluorescence
Prior to immunofluorescence experiments, formalin-fixed, paraffin-embedded rhesus brain slides were exposed to a dual black light/ natural sunshine light for 18 h to minimize autofluorescence [25]. To examine whether osteopontin was expressed in infiltrating or perivascular macrophages, or activated microglia, we utilized an antibody directed toward CD163 (Novocastra, Newcastle upon Tyne, UK; 1:100) [26] in conjunction with osteopontin. These sections were pretreated as for the immunohistochemistry.. Sections were blocked with 10% normal goat serum (NGS) in CAS block (0.5% casein, 0.05% thimerosol in PBS) for 30 min and incubated overnight with primary antibodies. For the osteopontin, the secondary antibody was biotinylated anti-rabbit IgG (H+L) (Vector; 1:100, 30 mins) followed by streptavidin AlexaFluor 488 conjugate (Molecular Probes, Eugene, OR; 1:500, 30 min in the dark). For the CD163, the secondary antibody was anti-mouse AlexaFluor 594 (Molecular Probes, 1:500). For the detection of DNA/nuclei using DAPI, sections were overlaid using 25 μl of Vectashield mounting media containing 1.5 μg/μl of DAPI (Vector, Burlingame, CA) and observed by fluorescence microscopy (Axiovert 200 inverted microscope; Zeiss; Oberkochen, Germany for osteopontin (green), DAPI (blue) and CD163 (red). Green, red and blue images were merged using AxioVision software (Zeiss).
Human plasma and CSF samples
EDTA-anticoagulated plasma, and CSF centrifuged free of cells were obtained from patients that were HIV negative, and three groups of HIV infected individuals based on neurocognitive diagnosis: neuropsychologically normal (similar to Memorial Sloan-Kettering classification (MSKCC) =0), minor cognitive/motor disorder (similar to MSKCC=1), and HIV-associated dementia (similar to MSKCC= 2, 3, or 4). All plasma and CSF were received from the four different sites of the National NeuroAIDS Tissue Consortium (NNTC) (HIV negative n=17, neuropsychologically normal n=33, minor cognitive/motor disorder n=37, and HIV-associated dementia n=25). The NNTC utilizes a series of 14 neuropyschological tests to determine neurocognitive diagnosis [27, 28]. HIV negative plasma and CSF were received from investigators at one of the NNTC sites, whereas all sites supplied HIV positive samples. Viral loads were converted from copies/ml to log10 copies RNA/ml. For plasma viral loads any values less than 400 copies/ml or undetectable were reported as <2.6 log10 copies RNA/ml. For CSF viral loads any values reported as less than 50 copies/ml or undetectable were reported as <1.7 log10 copies RNA/ml.
Rhesus monkey plasma collection
Rhesus macaques had blood samples serially collected under ketamine anesthesia. For plasma isolation, blood was placed in EDTA-treated tubes and centrifuged for separation from cells and erythrocytes.
Cytokine measurement
The levels of osteopontin in plasma and CSF from humans and monkeys, and CCL2 and IL-6 in monkey plasma, were measured using a human osteopontin ELISA (Immuno-Biological Laboratories, IBL-America; Minneapolis, MN), human CCL2 ELISA (R & D Systems) or human IL-6 ELISA (R & D Systems) respectively. Samples were analyzed in duplicate according to the manufacturer’s instructions.
Statistical analysis
For the osteopontin ELISA data in human plasma and CSF, outlier analysis and test of fit to normality was performed using Statgraphics Centurion build 15.02 (StatPoint Inc., Herndon, VA). Outliers were defined as data points which lie more than 3.0 times the interquartile range and as having median absolute deviation (MAD) Z score greater than 3.5. Outliers were identified using standard scores, Grubb’s test, and the result verified by Dixon’s test where applicable. Fit to normality was tested using the Shapiro-Wilk test and standardized skew. Using the raw values, 4 outliers were identified, and 4/8 samples did not fit the assumption of normality. The log10 transformation was tested and a better fit to normality was obtained where only one sample did not fit (one sample from the neuropsychologically normal group in CSF) and only 2 log10 values (one large in plasma HIV-associated dementia group and one small in CSF HIV-associated dementia group) met our outlier criterion.
For other statistical analyses we used Prism version 4.0b (GraphPad Software, Inc., San Diego, CA) and Delta Graph version 5.7.3 (Red Rock Software, Salt Lake City, UT) software.
Regulatory Approval
All animal studies were performed under approval from the Scripps Institutional Animal Care and Use Committee, and followed Scripps and NIH guidelines. Human studies were performed under informed consent by the patients. Samples were taken under the guidelines the approval of regulatory committees at the institution where the samples were procured, and the studies reported here performed under the guidelines of the Scripps Human Subjects Committee.
Results
Identification of osteopontin upregulation during SIV encephalitis
In order to validate prior DNA array results indicating upregulation of osteopontin in SIV encephalitis [20], we performed quantitative real-time PCR analysis on RNA isolated from the frontal lobes of control uninfected animals, SIV infected animals without encephalitis, and SIV infected animals that spontaneously developed SIV encephalitis. We indeed found that osteopontin mRNA is significantly upregulated in the brain of SIV encephalitic animals (Figure 1).
Figure 1. Osteopontin mRNA is upregulated in the frontal lobe of animals with encephalitis compared to uninfected animals.

Quantitative real-time PCR analysis was utilized to examine osteopontin mRNA expression in the frontal lobe of control uninfected monkeys (n=9), SIV-infected animals without encephalitis (SIVE-, n=12) and those with SIV encephalitis (SIVE+, n=7). Osteopontin mRNA is significantly upregulated in the brains of monkeys with SIVE compared to uninfected controls and SIV without encephalitis animals.
Next, to examine the cellular source of osteopontin in the brain, we performed immunohistochemistry, and found that osteopontin was indeed detectable in SIV encephalitic brains, especially at perivascular regions (Figure 2A), but not in uninfected animals or SIV-infection monkeys without encephalitis (data not shown). Osteopontin was also expressed in the brains of human patients who suffered from HIV-associated dementia (Figure 2B). Double-label immunofluorescence was performed to determine the cell types expression osteopontin in brains of monkeys with SIV encephalitis (Figure 2C-E). Co-localization of CD163 (a scavenger receptor expressed on macrophages) and osteopontin was seen (Figure 2E, arrows), indicating that these cells are expressing osteopontin during encephalitis. Osteopontin was thus expressed in the brains of monkeys and humans with encephalitis, and is produced by macrophages, key cells in HIV neuropathogenesis.
Figure 2. Osteopontin is expressed in both brains of monkeys with SIV encephalitis and humans with HIV encephalitis.
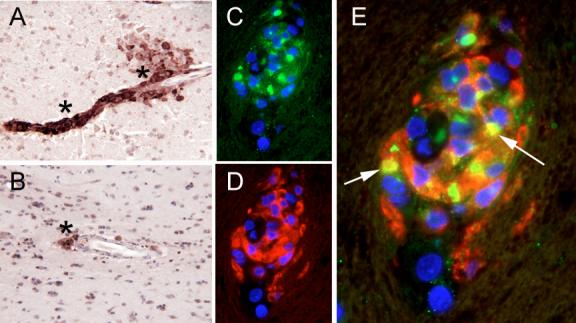
(A,B) Immunohistochemical analysis revealed the presence of osteopontin (*) in the brains of monkeys with SIV encephalitis (A) and the brains of human with encephalitis (B). CD163+ perivascular cells express osteopontin in the brains of monkeys with SIV encephalitis. (C-E) Immunofluorescence was performed on brain of monkeys with SIV encephalitis. (C) Osteopontin is shown in green and DAPI in blue. (D) CD163 is shown in red with DAPI in blue. (E) Merged image, the arrows point to co-expression of CD163 and osteopontin (yellow). All images were taken through a 63× magnification lens.
Plasma, but not CSF, osteopontin level correlates with neurological status in HIV infected individuals
Although plasma is most easily sampled clinically, the CSF provides a window into the CNS, which is accessible to clinical testing. In order to examine whether plasma or CSF osteopontin levels may help to indicate the presence of HIV-induced CNS disease, we investigated the levels of osteopontin in these biofluids of individuals with and without HIV infection. Examination of osteopontin in the plasma and CSF was performed in four specific groups of individuals: HIV negative individuals, HIV positive individuals with normal neuropsychological tests, HIV positive individuals with the minor cognitive-motor disorder and HIV positive patients with dementia. All samples were from patients who were alive and all HIV+ individuals were neuropsychologically tested and classified into these categories based on validated, reproducible criteria [27, 28]. Other than the neuropsychological classification, the groups were fairly well matched for demographic and clinical parameters (Table 1). HIV positive subjects were mostly middle-aged men with moderate immunosuppression on combination antiretroviral therapy, although about half had detectable plasma viral loads. Neither the plasma nor CSF viral loads were significantly different between the HIV infected but neuropsychologically normal, minor cognitive/motor disorder and HIV-associated dementia groups.
Table 1.
Characteristics of the study population. The number of subjects, median age and interquartile range (IQR) (in years), median and IQR of the blood absolute CD4 cell count (per μl), use of HAART when sample was taken, and viral load in plasma and CSF (log10/ml), with percent undetectable and IQR. nd - not determined, NA - not applicable, Neg: HIV negative population, NPN: HIV+ but neuropsychologically normal, MCMD: HIV+ with minor cognitive/motor disorder, and HAD: HIV+ with HIV-associated dementia
| Neg | NPN | MCMD | HAD | |
|---|---|---|---|---|
| # of subjects | 17 | 33 | 37 | 25 |
| Age (median) | 39 | 42 | 44 | 41 |
| Age (IQR) | 29.5-47.5 | 37-50.5 | 40-53.5 | 38-50.5 |
| Male sex | 65% | 82% | 89% | 92% |
| CD4 count (median) | nd | 192 | 202.5 | 191 |
| CD4 count (IQR) | nd | 85-337.5 | 91-390 | 109.5-300.5 |
| On HAART | NA | 82% | 81% | 76% |
| Viral Load | ||||
| Plasma (<2.6) | NA | 45% | 54% | 38% |
| Plasma (IQR) | NA | <2.6-4.5 | <2.6-4.75 | <2.6-4.8 |
| CSF (<1.7) | NA | 67% | 51% | 53% |
| CSF (IQR) | NA | <1.7-2.45 | <1.7-3.0 | <1.7-2.4 |
Analysis of plasma osteopontin levels revealed a significant difference between the groups (Figure 3A; ANOVA, p<0.0001). More specifically, both groups with deficits were increased. The plasma osteopontin levels in the minor cognitive/motor disorder group were significantly different than those in the HIV negative group (Tukey’s Multiple Comparison test; p<0.01), whereas osteopontin levels in the HIV-associated dementia group were significantly different than those found in the HIV negative as well as the HIV+ neuropsychologically normal group (Tukey’s Multiple Comparison test; p<0.001 and p<0.05 respectively). These results demonstrate that osteopontin levels correlate with neurological status. The osteopontin levels in the plasma were lowest in the HIV negative individuals and the highest in the patients with HIV-associated dementia. In the CSF, osteopontin levels were again significantly different between the groups (Figure 3B; ANOVA, p<0.0001). However, the differences among the groups were limited to all the HIV+ groups (neuropsychologically normal, minor cognitive/motor disorder and HIV-associated dementia) having significantly higher levels than did the HIV negative group (p<0.01, p<0.001 and p<0.01 respectively). Thus the presence of HIV infection itself, regardless of neuropsychological status, correlated with increased CSF osteopontin. Thus, CSF osteopontin levels, while elevated by HIV infection, are not a good marker of neurologic disease status, whereas plasma osteopontin levels revealed a significant correlation.
Figure 3. Osteopontin plasma levels correlate to HIV neurological diagnosis.

Osteopontin (OPN) was measured in the plasma and CSF of individuals that were HIV negative (Neg), neuropsychologically normal (NPN), minor cognitive/motor disorder (MCMD) or HIV-associated dementia (HAD), and the values within the groups shown as box and whisker plots, indicating the median, the 25th and 75th percentiles, and the largest and smallest values. The groups are significantly different by ANOVA, bars and asterisks indicate significant difference by post-hoc testing, as described in the results. (A) Plasma. (B) CSF.
A longitudinal study of SIV-infected rhesus revealed that osteopontin in the plasma is predictive of the onset of symptoms
Following this demonstration that plasma osteopontin levels in patients infected with HIV was further increased with severe clinical neurological diagnosis, HIV-associated dementia, we assayed plasma osteopontin levels over the course of disease in an experimental model system, the SIV-infected rhesus monkey, in order to assess whether the increase in plasma osteopontin maybe useful in predicting the development of CNS disease. We thus analyzed serial plasma osteopontin levels of SIV-infected monkeys with SIV encephalitis (SIVE+, Figure 4A) and without SIV encephalitis (SIVE-, Figure 4B), the latter both with (Figure 4B, closed symbols) and without (Figure 4B, open symbols) the development of simian AIDS. The increase in osteopontin in animals that developed encephalitis (34.8 ± 19.1 ng/ml per week, mean ± SD) was significantly greater than that found in those with simian AIDS without encephalitis (6.2 ± 4.4) or those infected with SIV but without simian AIDS (1.3 ± 3.2) (ANOVA, p=0.004, with post-hoc Tukey’s tests, p<0.05 and p<0.01, respectively). Plasma osteopontin levels thus correlated with the development of AIDS with encephalitis, as opposed to the development of AIDS without encephalitis or SIV infection itself. Furthermore, since the animals did not receive therapy, they were sacrificed soon after showing symptoms of disease. In most of the animals, the increase in osteopontin occurred 1-3 weeks prior to clinical disease.
Figure 4. Plasma osteopontin correlates with development of SIV encephalitis.
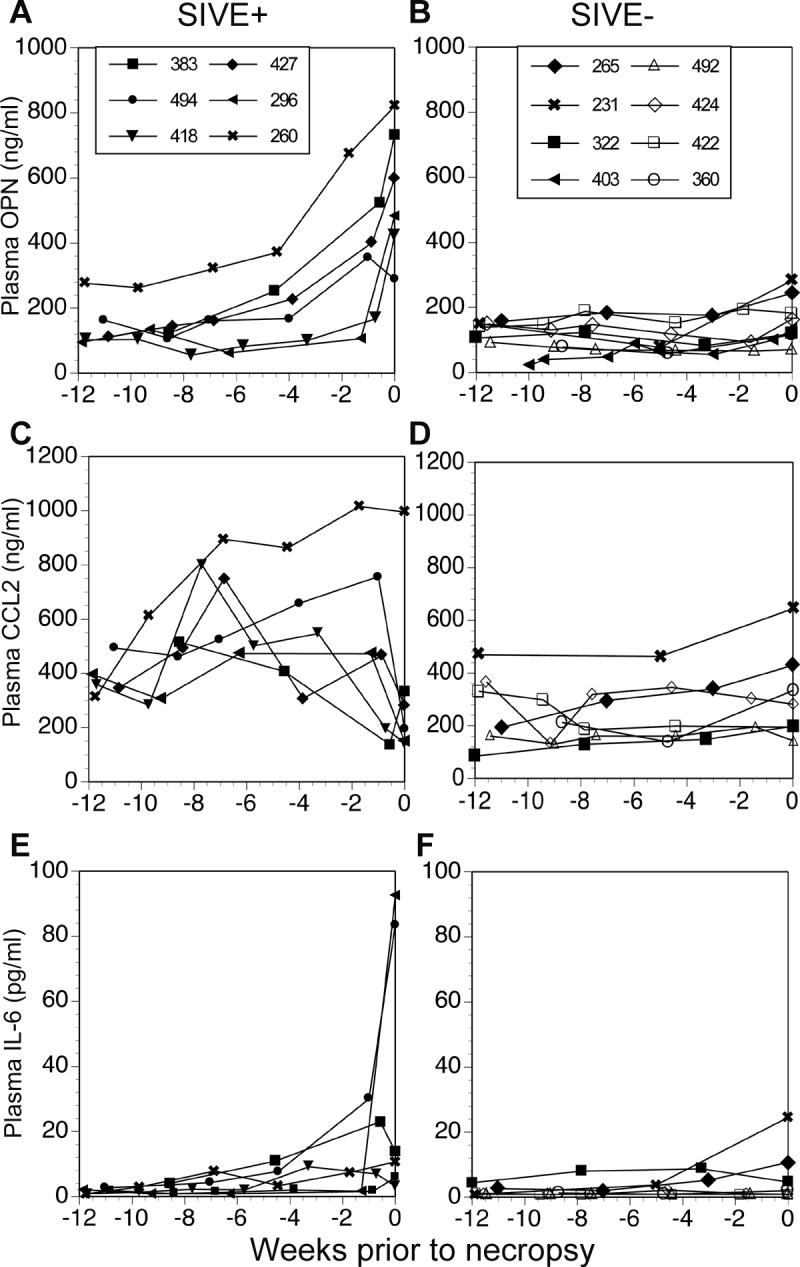
Cytokine plasma levels were examined in a longitudinal study after SIV infection. Graphs are plotted as plasma levels versus weeks prior to necropsy. (A) Plasma osteopontin (OPN) levels in animals with a pathological diagnosis of SIV encephalitis (SIVE+). (B) Plasma osteopontin levels in non-encephalitic animals (SIVE-), which included animals that were infected with SIV, developed AIDS but without encephalitis (closed symbols) and animals that were infected with SIV, but were sacrificed healthy (open symbols). (C) Plasma CCL2 levels in the animals with SIV encephalitis. (D) Plasma CCL2 levels in non-encephalitic animals. (E) Plasma IL-6 levels in the animals with SIV encephalitis. (F) Plasma IL-6 levels in non-encephalitic animals.
To assess if other potentially inflammatory cytokines were also increased in the plasma, we assayed the levels of CCL2 and IL-6 in the same animals (Figure 4C-F). CCL2 levels varied inconsistently through the course of disease in the SIV encephalitic animals (Figure 4C) and IL-6 was only highly increased in 2 out of 6 SIV encephalitic animals at necropsy (Figure 4E). CCL2 and IL-6 levels remained stable in all non-SIV encephalitic cases (Figure 4D and 4F). Thus the increase in plasma osteopontin did not reflect a general increase in pro-inflammatory markers, and osteopontin was the best biomarker in the plasma to predict the onset of CNS disease.
Discussion
Our study is a unique combination and cross validation of work in humans and the rhesus macaque animal model. We have demonstrated that osteopontin mRNA as well as protein was upregulated in the brains of rhesus monkeys with SIV encephalitis. In human, although in HIV infection increased CSF osteopontin levels were found relative to those in uninfected individuals, it was the plasma (but not CSF) osteopontin levels that correlated with neurocognitive diagnosis. In HIV infected individuals, plasma osteopontin levels were significantly increased in those with HIV-associated dementia compared to those without neuropsychological abnormalities. Furthermore, the osteopontin plasma levels in those with minor cognitive/motor disorder and HIV-associated dementia were increased relative to those in HIV negative individuals. Finally, a longitudinal study of the infected rhesus revealed that osteopontin in the plasma is a dependable correlate of SIV encephalitis. It is also predictive, as osteopontin levels were increased prior to termination in rhesus monkeys with encephalitis.
From the clinical point of view, one goal is to be able to use osteopontin in conjunction with other biomarkers in the plasma to diagnose and/or predict clinical symptomatology associated with HIV-associated neurocognitive disorders. In order to advance this goal further, a longitudinal study of the plasma of HIV infected individuals needs to be performed in conjunction with assessment of other molecules and neuropsychological evaluation. Interestingly, it was recently demonstrated that plasma osteopontin levels were elevated prior to increased disease activity in relapsing-remitting multiple sclerosis patients [29]. In a similar manner, plasma osteopontin levels in individuals without dementia could be followed, in conjunction with other markers, to investigate osteopontin levels indicative of a progression towards HIV-associated dementia.
Somewhat surprisingly, although HIV infection itself led to increased CSF osteopontin levels, it was the plasma levels that correlated with neurocognitive status. The increased plasma osteopontin levels could potentially provide a mechanism for CNS disease in the setting of HIV infection. The combination of HIV-induced increased expression of osteopontin in the CNS, along with the development of increased levels in the plasma of a subset of infected individuals, may drive the development of neurological disease. Mechanistically, our previous work indicated that the potential role of osteopontin in the plasma would be to protect monocytes from apoptosis, whereas in the brain osteopontin could decrease the number of macrophages returning to the circulation [22], yielding increased numbers of macrophages in the brain, which is the best pathological correlate of HIV-associated dementia [30]. A relevant analogy has recently been reported in a mouse model of obesity. The disease, induced by a high fat diet, is characterized by elevated levels of plasma osteopontin, and increased osteopontin-containing macrophages in adipose tissue. The use of osteopontin knockout mice revealed a central role for osteopontin in this model [31]. Similarly, we find that in CNS disease induced by HIV and SIV there is increased plasma osteopontin and increased brain macrophages, which express osteopontin. However we note that in addition to differences in absolute levels, osteopontin is functionally regulated by glycosylation, phosphorylation and proteolytic cleavage [32-35] and changes in these processes may also alter the quantification of osteopontin by ELISA.
Herein, we have shown correlational findings between plasma osteopontin and HIV neurological status and we intend to conduct future studies that examine whether there are any causal relationships between increased osteopontin and CNS disease. The potential contribution of osteopontin to HIV neuropathogenesis, versus it serving as a marker of macrophage activation, awaits further investigation. Interestingly, both peroxisomes proliferator-activator receptor (PPAR)α and PPARγagonists have been shown to reduce osteopontin production by macrophages [36, 37]. We note that although we have identified macrophages as a source of osteopontin in the brain, there may be multiple cellular sources of the osteopontin circulating in the plasma. Still, both fibrates, PPARα agonists, and thiazolidinediones, PPARγ agonists, have been utilized in HIV infected individuals to treat HAART-induced lipodystrophy. Although no data have been reported for thiazolidinedione treatment, benfibrate treatment of patients with type 2 diabetes led to reduced levels of plasma osteopontin [36]. It would be of interest to assess whether similar reductions can occur in HIV infected individuals, and the influence of such treatment on CNS disease.
In conclusion, we have shown that osteopontin is increased in the CSF of HIV infected individuals, and osteopontin exhibits a stair-step increase across diagnostic categories of HIV-associated neurocognitive disorders in the plasma. These findings are consistent with a pathophysiological model in which osteopontin amplifies brain macrophage accumulation, the pathological substrate of HIV-associated dementia. Furthermore, longitudinal analysis of osteopontin levels in SIV-infected monkeys suggested the value of osteopontin as a plasma biomarker in correlation with neurological disease associated with AIDS. Thus, plasma osteopontin levels may be used, along with other criteria, to help predict neurological disease course in AIDS. Means of lowering osteopontin production or its signaling can be attempted therapeutically to prevent or ameliorate this disorder.
Acknowledgements
We thank Claudia Flynn, Jason Lee, Ryan Ojakian, Debbie Watry, and Michelle Zandonatti for technical assistance, Dr. Caroline Lanigan for the outlier analysis and Dr. J. Lindsay Whitton for use of the fluorescence microscopy system.
These studies were supported by NIH grants NS045534 and MH073490, and the NIMH Core Support Program for Mental Health/AIDS Research MH62261 (the TSRI SNAPS) and MH62512 (the UCSD HNRC). The NNTC is supported by the NIMH and NINDS contract NIH-N01MH32002. THB was supported by NRSA post-doctoral fellowship F32 NS048830. This is manuscript #18560 from The Scripps Research Institute.
Footnotes
The authors declare no conflicts of interest.
References
- 1.Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- 2.Janssen RS, Cornblath DR, Epstein LG, Foa RP, McArthur J, Price RW. Nomenclature and research case definitions for neurologic manifestations of human immunodeficiency virus-type 1 (HIV-1) infection. Report of a Working Group of the American Academy of Neurology AIDS Task Force. Neurology. 1991;41:778–785. doi: 10.1212/wnl.41.6.778. [DOI] [PubMed] [Google Scholar]
- 3.Antinori A, Arendt G, Becker JT, et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69:1789–99. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005;4:543–55. doi: 10.1016/S1474-4422(05)70165-4. [DOI] [PubMed] [Google Scholar]
- 5.Heaton RK, Velin RA, McCutchan JA, et al. Neuropsychological impairment in human immunodeficiency virus- infection: implications for employment. Psychosom Med. 1994;56:8–17. doi: 10.1097/00006842-199401000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Cherner M, Masliah E, Ellis RJ, et al. Neurocognitive dysfunction predicts postmortem findings of HIV encephalitis. Neurology. 2002;59:1563–7. doi: 10.1212/01.wnl.0000034175.11956.79. [DOI] [PubMed] [Google Scholar]
- 7.Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–94. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- 8.Pulliam L, Gascon R, Stubblebine M, McGuire D, McGrath MS. Unique monocyte subset in patients with AIDS dementia. Lancet. 1997;349:692–5. doi: 10.1016/S0140-6736(96)10178-1. [DOI] [PubMed] [Google Scholar]
- 9.Kusdra L, McGuire D, Pulliam L. Changes in monocyte/macrophage neurotoxicity in the era of HAART: implications for HIV-associated dementia. Aids. 2002;16:31–8. doi: 10.1097/00002030-200201040-00005. [DOI] [PubMed] [Google Scholar]
- 10.Gelbard HA, Nottet HS, Swindells S, et al. Platelet-activating factor: a candidate human immunodeficiency virus type 1-induced neurotoxin. J Virol. 1994;68:4628–35. doi: 10.1128/jvi.68.7.4628-4635.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heyes MP, Rubinow D, Lane C, Markey SP. Cerebrospinal fluid quinolinic acid concentrations are increased in acquired immune deficiency syndrome. Ann Neurol. 1989;26:275–7. doi: 10.1002/ana.410260215. [DOI] [PubMed] [Google Scholar]
- 12.Cinque P, Bestetti A, Marenzi R, et al. Cerebrospinal fluid interferon-gamma-inducible protein 10 (IP-10, CXCL10) in HIV-1 infection. J Neuroimmunol. 2005 doi: 10.1016/j.jneuroim.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Brew BJ, Dunbar N, Pemberton L, Kaldor J. Predictive markers of AIDS dementia complex: CD4 cell count and cerebrospinal fluid concentrations of beta 2-microglobulin and neopterin. J Infect Dis. 1996;174:294–8. doi: 10.1093/infdis/174.2.294. [DOI] [PubMed] [Google Scholar]
- 14.Gisslen M, Hagberg L, Brew BJ, Cinque P, Price RW, Rosengren L. Elevated cerebrospinal fluid neurofilament light protein concentrations predict the development of AIDS dementia complex. J Infect Dis. 2007;195:1774–8. doi: 10.1086/518043. [DOI] [PubMed] [Google Scholar]
- 15.Kelder W, McArthur JC, Nance-Sproson T, McClernon D, Griffin DE. Beta-chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus- associated dementia. Ann Neurol. 1998;44:831–5. doi: 10.1002/ana.410440521. [DOI] [PubMed] [Google Scholar]
- 16.Cysique LA, Brew BJ, Halman M, et al. Undetectable Cerebrospinal Fluid HIV RNA and beta-2 Microglobulin Do Not Indicate Inactive AIDS Dementia Complex in Highly Active Antiretroviral Therapy-Treated Patients. J Acquir Immune Defic Syndr. 2005;39:426–429. doi: 10.1097/01.qai.0000165799.59322.f5. [DOI] [PubMed] [Google Scholar]
- 17.Enting RH, Foudraine NA, Lange JM, et al. Cerebrospinal fluid beta2-microglobulin, monocyte chemotactic protein-1, and soluble tumour necrosis factor alpha receptors before and after treatment with lamivudine plus zidovudine or stavudine. J Neuroimmunol. 2000;102:216–21. doi: 10.1016/s0165-5728(99)00219-2. [DOI] [PubMed] [Google Scholar]
- 18.Sevigny JJ, Albert SM, McDermott MP, et al. Evaluation of HIV RNA and markers of immune activation as predictors of HIV-associated dementia. Neurology. 2004;63:2084–90. doi: 10.1212/01.wnl.0000145763.68284.15. [DOI] [PubMed] [Google Scholar]
- 19.Burudi EM, Fox HS. Simian immunodeficiency virus model of HIV-induced central nervous system dysfunction. Adv Virus Res. 2001;56:435–68. doi: 10.1016/s0065-3527(01)56035-2. [DOI] [PubMed] [Google Scholar]
- 20.Roberts ES, Zandonatti MA, Watry DD, et al. Induction of Pathogenic Sets of Genes in Macrophages and Neurons in NeuroAIDS. Am J Pathol. 2003;162:2041–57. doi: 10.1016/S0002-9440(10)64336-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin YH, Yang-Yen HF. The osteopontin-CD44 survival signal involves activation of the phosphatidylinositol 3-kinase/Akt signaling pathway. J Biol Chem. 2001;276:46024–30. doi: 10.1074/jbc.M105132200. [DOI] [PubMed] [Google Scholar]
- 22.Burdo TH, Wood MR, Fox HS. Osteopontin prevents monocyte recirculation and apoptosis. J Leukoc Biol. 2007;81:1504–11. doi: 10.1189/jlb.1106711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burdo TH, Marcondes MC, Lanigan CM, Penedo MC, Fox HS. Susceptibility of Chinese rhesus monkeys to SIV infection. Aids. 2005;19:1704–6. doi: 10.1097/01.aids.0000186823.76230.33. [DOI] [PubMed] [Google Scholar]
- 24.Lane TE, Buchmeier MJ, Watry DD, Jakubowski DB, Fox HS. Serial passage of microglial SIV results in selection of homogeneous env quasispecies in the brain. Virology. 1995;212:458–65. doi: 10.1006/viro.1995.1503. [DOI] [PubMed] [Google Scholar]
- 25.Baschong W, Suetterlin R, Laeng RH. Control of autofluorescence of archival formaldehyde-fixed, paraffin- embedded tissue in confocal laser scanning microscopy (CLSM) J Histochem Cytochem. 2001;49:1565–72. doi: 10.1177/002215540104901210. [DOI] [PubMed] [Google Scholar]
- 26.Roberts ES, Masliah E, Fox HS. CD163 identifies a unique population of ramified microglia in HIV encephalitis (HIVE) J Neuropathol Exp Neurol. 2004;63:1255–64. doi: 10.1093/jnen/63.12.1255. [DOI] [PubMed] [Google Scholar]
- 27.Woods SP, Rippeth JD, Frol AB, et al. Interrater reliability of clinical ratings and neurocognitive diagnoses in HIV. J Clin Exp Neuropsychol. 2004;26:759–78. doi: 10.1080/13803390490509565. [DOI] [PubMed] [Google Scholar]
- 28.Carey CL, Woods SP, Gonzalez R, et al. Predictive validity of global deficit scores in detecting neuropsychological impairment in HIV infection. J Clin Exp Neuropsychol. 2004;26:307–19. doi: 10.1080/13803390490510031. [DOI] [PubMed] [Google Scholar]
- 29.Vogt MH, Floris S, Killestein J, et al. Osteopontin levels and increased disease activity in relapsing-remitting multiple sclerosis patients. J Neuroimmunol. 2004;155:155–60. doi: 10.1016/j.jneuroim.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Glass JD, Fedor H, Wesselingh SL, McArthur JC. Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol. 1995;38:755–62. doi: 10.1002/ana.410380510. [DOI] [PubMed] [Google Scholar]
- 31.Nomiyama T, Perez-Tilve D, Ogawa D, et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest. 2007 doi: 10.1172/JCI31986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ashkar S, Weber GF, Panoutsakopoulou V, et al. Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science. 2000;287:860–4. doi: 10.1126/science.287.5454.860. [DOI] [PubMed] [Google Scholar]
- 33.Weber GF, Zawaideh S, Hikita S, Kumar VA, Cantor H, Ashkar S. Phosphorylation-dependent interaction of osteopontin with its receptors regulates macrophage migration and activation. J Leukoc Biol. 2002;72:752–61. [PubMed] [Google Scholar]
- 34.Christensen B, Kazanecki CC, Petersen TE, Rittling SR, Denhardt DT, Sorensen ES. Cell type-specific post-translational modifications of mouse osteopontin are associated with different adhesive properties. J Biol Chem. 2007;282:19463–72. doi: 10.1074/jbc.M703055200. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto N, Sakai F, Kon S, et al. Essential role of the cryptic epitope SLAYGLR within osteopontin in a murine model of rheumatoid arthritis. J Clin Invest. 2003;112:181–8. doi: 10.1172/JCI17778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamachi T, Nomiyama T, Gizard F, et al. PPARalpha agonists suppress osteopontin expression in macrophages and decrease plasma levels in patients with type 2 diabetes. Diabetes. 2007;56:1662–70. doi: 10.2337/db06-1177. [DOI] [PubMed] [Google Scholar]
- 37.Oyama Y, Akuzawa N, Nagai R, Kurabayashi M. PPARgamma ligand inhibits osteopontin gene expression through interference with binding of nuclear factors to A/T-rich sequence in THP-1 cells. Circ Res. 2002;90:348–55. doi: 10.1161/hh0302.105098. [DOI] [PubMed] [Google Scholar]