

Abstract
In its role as a tumor suppressor, WT1 transactivates several genes which are regulators of cell growth and differentiation pathways. For instance, WT1 induces the expression of the cell cycle regulator p21, the growth regulating glycoprotein amphiregulin, the proapoptotic gene Bak and the Ras/MAPKinase inhibitor, sprouty1. Here we show that WT1 transactivates another important negative regulator of the Ras/MAPKinase pathway, MKP3. In a WT1-inducible cell line that exhibits decreased cell growth and increased apoptosis upon expression of WT1, microarray analysis showed that MKP3 is the most highly induced gene. This was confirmed by real time PCR where MKP3 and other members of the FGF8 syn expression group which includes sprouty1 and the ets family of transcription factors were induced rapidly following WT1 expression. WT1 induction was associated with a block in the phosphorylation of ERK in response to EGF stimulation, an effect mediated by MKP3. In the presence of a dominant negative MKP3, WT1 could no longer block phosphorylation of ERK. Lastly when MKP3 expression is down regulated by shRNA, WT1 is less able to block Ras mediated transformation of 3T3 cells.
Keywords: WT1, Wilms tumor, MAP Kinase Phosphatase 3 (MKP3), oncogene
Introduction
WT1 encodes a transcription factor required for normal kidney development that is mutated in 10-15% of sporadic Wilms tumors. Constitutional mutation of WT1 has been described in three developmental syndromes, WAGR, Denys-Drash (DDS) and Frasier (1), which are associated with genitourinary malformations and Wilms tumor. Bona fide WT1 target genes, activated or repressed by the protein, are gradually being identified. Much of this work has been carried out by gain of function studies in heterologous cell systems such as NIH 3T3 fibroblasts (2) or Saos-2 osteosarcoma cells (3). Nevertheless these studies have yielded WT1 targets validated by their expression in the developing kidney and their roles in growth control. WT1 has both activation and repression domains located in the N-terminus, however, our previous work suggested that the growth suppression activity of WT1 was most closely linked to its ability to induce gene expression (4).
WT1 target genes may be thematically grouped. WT1 can induce the expression of genes implicated in renal development and the mesenchymal-epithelial transition such as E-cadherin (2), podocalyxin (5), nephrin (6) and Wnt4 (7). Hence, the growth regulatory role of WT1 may be tightly linked to its role in guiding development. Depending on the cellular context, WT1, through its actions on apoptotic regulators such as Bcl2 (8, 9), Bfi1 (10), Bak and several BH3-only proteins (11), can increase or decrease apoptosis. And, like the archetypal tumor suppressor p53, WT1 can inhibit cell growth through activation of p21WAF1/CIP1 (12), a common feature of differentiating cells. WT1 also regulates signaling by growth factor receptors. WT1 induces the expression of amphiregulin, which is secreted by the metanephric mesenchyme to stimulate the growth of the ureteric tree (3). Similarly WT1 induces the expression of TrkB, a growth factor receptor critical for coronary vasculature development (13). However, WT1 can also down-regulate growth factor pathways. For example, WT1 represses expression of the IGF1 receptor (14), the EGF receptor (15, 16), connective tissue growth factor (17), and vascular endothelial growth factor (18).
We previously reported that Sprouty1 (Spry1), an inhibitor of FGFR signaling through the Ras/MAPK pathway, is a direct target gene of WT1 and is required for normal glomeruli formation (19). Here we show that the MAP kinase phosphatase 3 (MKP3), another important inhibitor of the Ras pathway, is also regulated by WT1. MKP3 is a member of the dual specificity phosphatase (DSP) family that dephosphorylates both serine/threonine and tyrosine residues, leading to the inactivation of MAP kinases (20). MKP3 is a highly specific inhibitor of ERK1/2 but has little effect on JNK/SAPK and p38 MAPKs (21, 22). Like Spry1, MKP3 is detected in the ureter tips of the developing kidney, and in the developing limb buds and lung, mirroring places of high FGF8 expression (23, 24).
Within the cell MKP3 is localized to the cytoplasm (21) where it binds specifically and tightly to the C-terminus of ERK1/2 (25) and is engaged in a complicated negative-positive feedback loop with ERK1/2. Upon growth factor stimulation ERK1/2 is phosphorylated, translocates to the nucleus and phosphorylates nuclear targets such as Elk1. MKP3 has a low basal activity, but after ERK2 pathway activation, MKP3 is transcriptionally up-regulated (21, 26) and its enzymatic activity is stimulated (27, 28). Activated MKP3 binds to, dephosphoryates and sequesters ERK in the cytoplasm in an inactive form. In turn, MKP3 is regulated by ERK2 (27-29). Upon binding to MKP3, ERK2 phosphorylates MKP3 leading to its proteasomal degradation. In this way ERK1/2 exerts a positive feedback on its own activity by promoting the degradation of one of its major inactivators (30). MKP3 gene expression in the mouse, Drosophila, Xenopus and Zebrafish is localized to areas of high EGFR and FGFR signaling, suggesting that MKP3 constitutes an evolutionarily conserved negative feedback loop on the activity of the Ras/MAPK signaling pathway (21, 31, 32). The fact that WT1 transactivates two negative regulators of the Ras/MAPK pathway suggests that this is a major target of WT1 function.
Materials and Methods
Plasmids
To generate the MKP3-luc reporter, a 2 Kb section of the human MKP3 promoter (Ensemble Gene ID ENSG00000139318) from -2140 to +20, relative to the start of transcription, was PCR-amplified from human genomic DNA with gene-specific primers (5′-ATAGGTACCCGAACACGCTCCTCCAGG-3′ and 5′ TTTAAGCTTAATCCCTCCCTCCAAGGC), introducing KpnI and HindIII sites and cloned into pGL2-Basic (Promega). Flag-tagged wildtype and mutant hMKP3 were subcloned from GST-hMKP3 and GST-hMKP3 (C294S) (gift of Ming-Ming Zhou, Mount Sinai) and ligated to pCMV-Flag (Invitrogen). pBIG2i-WT1A, a doxycycline-inducible vector, was previously described (33). AU5-RasR12 was provided by Andrew Chen (Mount Sinai) and WT1A and WT1A-112 expression vectors were previously described (4).
Cell culture
WT1A doxycycline-inducible cell lines were established in HEK293 and CCG99-11 cells by transfection with pBIG2i-WT1A using Lipofectamine Plus (33). Transformants were selected in 100ug/ml or 400ug/ml of hygromycin B, expanded and WT1 expression was induced in 2ug/ml or 1ug/ml doxycycline, respectively. Saos-WT1 inducible clones (11) were maintained in DMEM containing 10% fetal bovine serum (FBS), 0.5mg/ml G418 and 1ug/ml tetracycline and induced by tetracycline withdrawal. NIH3T3 cells were maintained in DMEM containing 10% newborn calf serum, and HEK293 and 293T cells were maintained in DMEM containing 10% FBS.
Quantitative reverse transcription PCR Analysis
RNA was extracted using RNeasy (Qiagen, Valencia, CA) and RT-PCR was performed using iScript II cDNA Synthesis kit (Biorad, Hercules, CA) and Quantitect SYBR Green PCR kit (Qiagen, Valencia, CA). Synthesis of PCR products was monitored by the DNA Engine Opticon System (MJ Research, Waltham, MA) and normalized to GAPDH amplification. The primers were: hGAPDH (5′-CCAAAATCAAGTGGGGCGATG-3′ and 5′-AAAGGTGGAGGAGTGGGTGTCG-3′), hDUSP6 (5′-CAACAGGGTTCCAGCACAGCAG-3′ and 5′-GCCAGACACATTCCAGCAAGGAG-3′) mDUSP6 (5′-TCGGGCTGCTGCTCAAGAAAC-3′ and 5′-CGGTCAAGGTCAGACTCAATGTCC-3′), hETV1 (5′-TCCCTCCATCGCAGTCCATACC-3′ and 5′-TCCTTCCCTTGGCATCGTCG-3′), hETV4 (5′-TCAAACAGGAACAGACGGACTTCG-3 and 5′-TCAGGGACAACGCAGACATCATC-3′) and hETV5 (5′-CCTGATGATGAACAGTTTGTCCCAG-3′ and 5′-CCATAGTTAGCACCAAGAGCCTGC -3′).
Immunoblotting
Cell lysates were prepared in 1% NP-40 lysis buffer [150mM NaCl, 50mM Tris (pH 6.9), 1% NP-40, 1mM PMSF, 10mM NaF, 15mM EDTA, 1mM Na3VO4, 20mM Na4P2O7, protease inhibitor cocktail (Roche Diagnostics)]. Immunoblotting was performed with the following antibodies: WT1(C-19) (Santa Cruz), GAPDH (Chemicon International), pERK1/2(E-4) (Santa Cruz), ERK1/2(#06-182) (Upstate), AU5 (Covance) and Flag(F3165) (Sigma). For induction of p-ERK, CCG-5.1 and 7.1 were treated with EGF (40ng/ml) for 10 min at 37°C prior to lysis.
Luciferase Assay
MKP3-luc (100ng), a control Renilla reporter (5ng) and increasing concentrations of WT1A expression vectors (1ug) were transfected into 293T cells using Superfect (Qiagen). Cells were harvested after 48 hours and assayed using the Dual-Luciferase Reporter Assay System (Promega). Results were normalized to total protein concentration determined by Bradford assay.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed (33) with anti-WT1 (C19), a matched IgG isotype or no primary antibody at a concentration of 3μg per 1.5×107 cells. The PCR reaction contained 2 ul of either immunoprecipitated sample or 2% of input DNA, 0.4μM of each primer, and 12.5μl of 2X SYBR Green Mix (Qiagen) in a 25μl reaction. After 40 cycles of amplification, the PCR product was resolved on a 1% agarose gel. The primer sequences for MKP3 promoter regions are as follows: MKP3-1F: 5′CATTTCCCAAACCAGGGAAGAG-3′, MKP3-1R: 5′-AAGAGGCCGCGCTTTGTCCCA-3′; MKP3-2F: 5-′GCCAACTGTAACCAATCGTCGA-3′ MKP3-2R: 5′-GTTCGGGGGAATCTATATCTCTC-3′; and for β-actin are: F: 5′-CCTCTTCCTCAATCTCGCTCT-3′; R: 5′-CTCGAGCCATAAAAGGCAACT-3′.
RNA interference
shRNAs targeting MKP3 (mouse: 5′-AGCTCAACCTGTCCATGAA-3; human: 5′-GCTCAATCTGTCGATGAAC-3′) were ligated to the pSiren-RetroQ (puromycin selection) vector (Clonetech, Palo Alto Ca). A negative control shRNA was provided by Clontech.
Foci formation Assay
NIH3T3 cells were transfected with the indicated combinations of AU5-Ras R12, WT1, Flag-MKP3, control shRNA and mMKP3 shRNA using Lipofectamine plus (Invitrogen). At 24 hrs post transfection, plates were split 1:3 and cultured for 10 days till foci formed. Cells were fixed (10% methanol, 10% acetic acid), stained with 0.4% crystal violet in 10% ethanol and foci were counted blindly. For Western blotting, cells were harvested at 48 hrs post transfection and processed as above. In shRNA experiments, cells were selected in puromycin (1.0 ug/ml) for 4 days following transfection. Surviving cells were grown for an additional 10 days before staining.
Results
Wild-type WT1A activates transcription of MKP3
Microarray analysis of Saos-2 cells induced to express WT1A (33) indicated that MKP3/Dusp6 was a highly induced gene. Real-time PCR analysis confirmed that expression of WT1A for 18, 24 and 48 hrs led to an increase in expression of MKP3, ranging from 12 to 90 fold, while WT1A-112, a point mutant of WT1 defective for transcriptional activation, minimally induced MKP3 expression (3-fold). MKP3 is a member of a syn-expression group regulated by FGF8 during limb bud development (34). Members of this group include Sef, a negative regulator of FGF signaling, and the Ets transcription factors, ETV1, ETV4 and ETV5. In our system, these Ets genes were significantly induced by WT1, as assessed by real time PCR (Figure 1A) and corroborated to varying degrees by microarray analysis (e.g. ETV1: 11.5 fold; ETV5: 13 fold). The Sef gene was not represented on the microarray chip; however, Spry1, a relative to Sef, was also significantly induced by WT1 (11).
Figure 1.

Induction of MKP3 and FGF8 syn expression group genes by WT1. Real time PCR analysis of indicated genes in Saos-WT1 inducible cell line following withdrawal of tetracycline. Data were calculated as fold induction relative to pre-WT1-induction condition after normalizing with GAPDH. Western blots corresponding to each induction experiment shows expression of induced WT1 over time.
MKP3 mRNA was induced three-fold within 6 hours of WT1 induction (Figure 1B), suggesting that it might be a direct transcriptional target of WT1. To test this hypothesis, an MKP3 promoter reporter plasmid, encompassing two putative WT1 binding regions, was co-expressed in 293T cells with expression vectors for wild-type or mutant WT1A. Wild-type WT1A activated the promoter in a dose-dependent manner whereas the mutant WT1A-112 did not (Figure 2B). Chromatin immunoprecipitation from the Wilms tumor cell line, CCG99-11, expressing endogenous WT1, confirmed that WT1 binds to regions 1 and 2 within the MKP3 promoter (Figure 2C). Enrichment was stronger for region 1, which contains 3 canonical WT1 binding sites including the more potent WTE site, than for region 2, which covers only one EGR1-type site.
Figure 2.
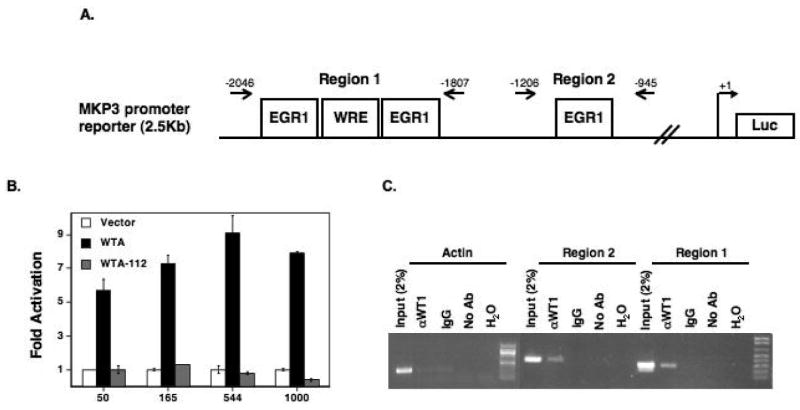
Regulation of MKP3 promoter activity by WT1 in kidney cell lines. (A) Schematic diagram of the MKP3 promoter fragment highlighting the location of putative WT1 binding sites and PCR primers. (B) Luciferase assays of 293T cells transfected with the MKP3 promoter and increasing concentrations of a vector expressing wild-type or mutant WT1. Data is representative of 3 experiments; each performed in triplicate and normalized to protein concentrations. (C) Chromatin immunoprecipitation from the Wilms tumor cell line, CCG99-11, using a WT1 antibody, IgG or no antibody. PCR primers detect MKP3 promoter regions. Actin primers were used as control.
Expression of WT1 attenuates the activation of ERK1/2
Because WT1 trans-activated MKP3 and spry1, both inhibitors of the Ras/MAPK pathway (19), we interrogated the effect of WT1 on the phosphorylation state of ERK1/2 in the Saos-2 cells by Western blot and detected a decrease in p-ERK1/2, but not total ERK1/2, levels upon induction of WT1 (data not shown). This phenomenon, coupled with the fact that growth factor-induced MKP3 expression antagonizes MAPK signaling in the developing vertebrate limb (34), prompted us to study the effect of MKP3 induction by WT1 on the phosphorylation state of ERK1/2 in kidney-related cell lines. CCG99-11 Wilms tumor-derived cells and HEK293 kidney cells were engineered to over-express WT1A in an inducible manner. Induction of WT1 in the CCG99-11 cells significantly attenuated the ability of EGF to induce phosphorylation of ERK1/2 (Figure 3A). Of note, a previous study showed that WT1 repressed the EGFR expression at the transcriptional level in U2OS and Saos-2 cells resulting in apoptosis (16). However, our expression array profiles from CCG5.1 cells showed that the expression levels of all 15 EGFR probe sets on HG U133Plus2 were not changed after WT1 induction (data not shown), suggesting that the decrease or lack of EGF response was not due to a decrease in EGFR. The HEK293 inducible-cell line clones exhibited a high level of basal p-ERK1/2 without EGF stimulation (Figure 3B lanes 1 and 5). Nonetheless, this basal p-ERK1/2 was depressed upon induction of WT1 (Figure 3B, lanes 2 and 6 and 3C, lane 2).
Figure 3.
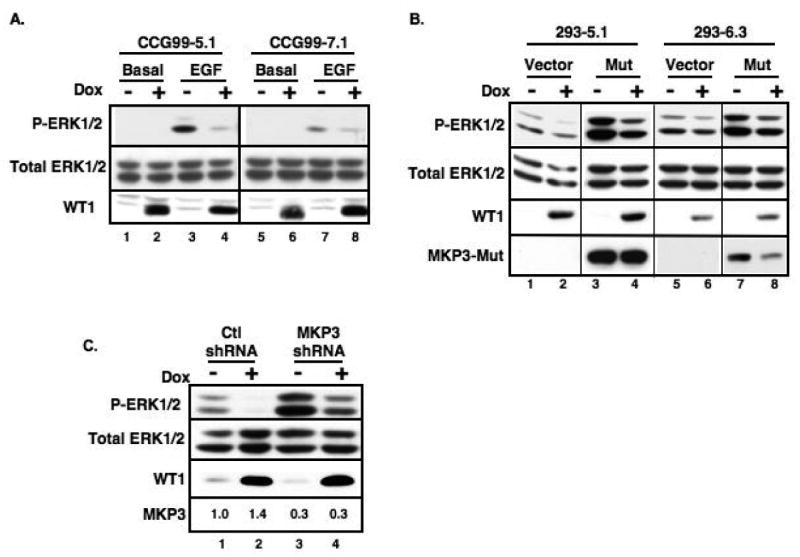
WT1 regulates the level of phosphorylated ERK1/2 through activation of MKP3. (A) WT1-inducible CCG99-11 clones were incubated with or without doxycycline (Dox) in the presence or absence of EGF. HEK293 clones were transfected with either (B) Flag-tagged MKP3 C294S mutant or (C) MKP3 shRNA vectors prior to induction with Dox. Western blots were performed to detect total and phosphorylated ERK1/2, WT1 and MKP3. Relative expression of endogenous MKP3 mRNA, as determined by qPCR, is indicated.
To explore the role of MKP3 in the pathway between WT1 and ERK1/2, a catalytically inactive dominant negative form of MKP3 (C294S) which binds to p-ERK1/2 but lacks phosphatase activity (34, 35), was transfected into the HEK293 inducible clones. Under this condition, the level of basal p-ERK1/2 was dramatically increased (Figure 3B, lanes 3 and 7) and WT1 was less effective at blocking ERK1/2 phosphorylation (compare lanes 1-2 (2.5:1) with lanes 3-4 (1.3:1)), consistent with the notion that MKP3 mediates, at least in part, the effect of WT1 on p-ERK1/2. ShRNA mediated knockdown of MKP3 (Figures 4 and 3C, lanes 3 and 4) resulted in a large increase in basal p-ERK1/2 levels (Figure 3C, lane 3). Furthermore, when MKP3 was depleted, the inhibitory effect of WT1 on p-ERK1/2 levels was attenuated (Figure 3C, compare lanes 1-2 with lanes 3-4).
Figure 4.
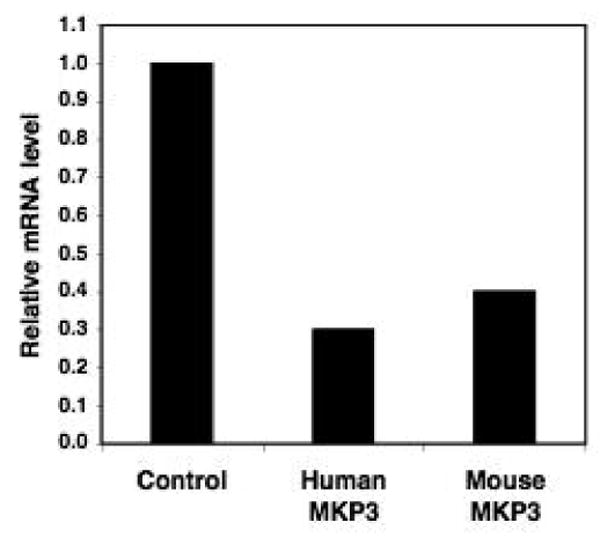
Reduction of MKP3 expression by shRNA. HEK293 and 3T3 cell lines were transfected with human and mouse MKP3-specific short hairpin interfering RNA (shRNA) vectors, respectively, or with control vector and selected for 4 days. Expression level of MKP3 was determined by qPCR analysis.
Tumor suppressor activity of WT1 utilizes MKP3
We previously showed that WT1 inhibited the oncogenicity of activated Ras in an NIH3T3 focus forming assay (36). We re-confirmed that WT1 blocked the tumor promoting activity of Ras and had no tumor promoting activity itself (Figure 5A). Similarly, flag-tagged MKP3, when co-expressed with Ras, blocked Ras-mediated foci formation (Figure 5A). The MKP3 C294S mutant could not be used in this assay because it still interferes with the translocation of active ERK1/2 to the nucleus, even though it lacks phosphatase activity (34, 35). Therefore an shRNA targeting murine MKP3 was developed that depleted endogenous MKP3 mRNA by 60% (Figure 4). This moderate depletion did not promote foci formation and did not affect Ras-induced foci formation (Figure 5B). However, in the presence of the MKP3 shRNA, WT1 was less effective in inhibiting focus formation by Ras.
Figure 5.
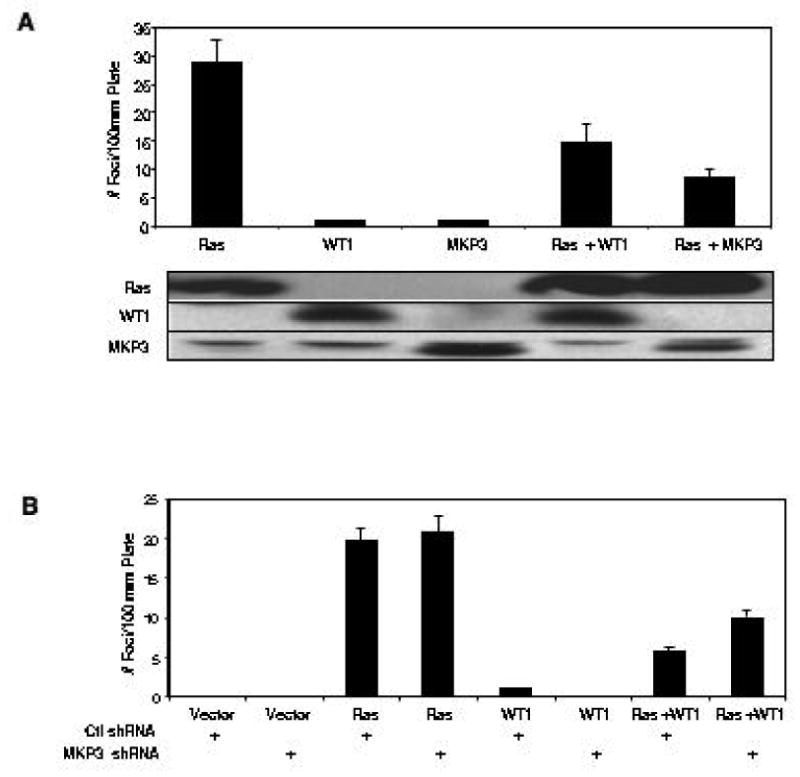
Suppression of Ras-induced foci formation by WT1 is mediated by MKP3. (A) NIH3T3 cells were transfected with AU5-Ras in the presence or absence of WT1 or Flag-tagged MKP3 and allowed to grow for 10 days. Foci were counted and plotted as mean number of foci/10cm plate. The relative expression of each protein, as determined by Western blot, is shown. (B) Knock down of endogenous MKP3 expression by shRNA partially blocked the WT1-mediated suppression of foci formation. Experiments were carried out independently three times in triplicate.
Discussion
In this study we identified MKP3 as a novel target gene of WT1 that mediates WT1 inhibition of ERK1/2 activation. We found that induction of WT1 blocked activation of ERK1/2 in Saos-2 and other cell lines. This was associated with a striking elevation of MKP3 levels. We showed that overexpression of WT1 induced expression of MKP3 in vivo and bound to and transactivated the promoter of MKP3. Furthermore using shRNA methodology, we demonstrated that MKP3 is an intermediary in the pathway between WT1 and ERK phosphorylation. We have previously shown that WT1 augments levels of Spry1 that can inhibit ERK1/2 activation by growth factors as well (19). Both MKP3 and Spry1 appear to be direct targets of WT1. Whether other counter-regulatory molecules are directly bound and activated by WT1 or are indirectly activated through induction of growth factors remains to be determined.
WT1 has been described as both a tumor suppressor and tumor promoter, depending upon the cell type or the developmental stage in which WT1 is expressed (reviewed in (37)). For example, at murine embryonic day 9, WT1 expression is required for maintenance of the metanephric mesenchyme (reviewed in (38)). In the absence of WT1 expression, the metanphric mesenchyme undergoes apoptosis and the ureteric buds fail to grow. However by day 11, if WT1 expression is knocked-down by siRNA, nephrogenesis is severely impaired and abnormal cell proliferation occurs (39). Overproliferation of cells that do not undergo apoptosis and persist in the form of nephrogenic rests can become the basis of a tumor. During hematopoiesis in adult tissues, overexpression of WT1 leads to growth arrest and reduced colony formation, suggesting a role as tumor suppressor (40). Furthermore, 10-15% of AML patients have mutations in WT1 and overexpression of WT1 in a WT1-negative myeloblastic leukemia cells results in decreased cell growth (37). Conversely, other studies reported WT1 up-regulation in the majority of cases of acute leukemia. In model AML systems, a knockdown of WT1 was associated with decreased growth and increased apoptosis. In addition, overexpression of WT1 in K562 erythroleukemia cells actually protected the cells against induction of apoptosis by chemotherapy agents whereas WT1 specific siRNA retarded proliferation of the cell line (41), suggesting a role as an oncogene.
These varying assignments of WT as a tumor suppressor or oncogene may be due to the variety of its interacting proteins. For example, co-expression of p53 with WT1 can block the pro-apoptotic activity of p53 and convert WT1 from a trans-activator to a trans-repressor (42, 43). Furthermore, multiple WT1 isoforms and post-translational modifications such as phosphorylation and sumoylation can regulate WT1 function. Phosphorylation of WT1 causes redistribution of the protein to the cytoplasm and blocks the ability of WT1 to bind DNA (44). Mutant WT1 molecules can dimerize with the wild-type form through an N-terminal association domain and act as a dominant negative inhibitor of WT1 function (45).
Given its role as a negative regulator of ERK1/2, MKP3 can be considered a putative tumor suppressor gene. Marchetti and colleagues (46) found that MKP3 blocked oncogenic Ras activity in vivo. This group transformed a doxycycline-inducible MKP3-cell line with HA-Ras and injected it into nude mice. Mice treated with doxycline experienced a delay in tumor emergence and reduced growth. MKP3 maps to 12q22, a locus which is either lost or hypermethylated in human pancreatic cancer (47, 48). Furthermore, in situ carcinomas express elevated levels of MKP3 while MKP3 is down-regulated in invasive carcinomas. MKP3 is a target of palytoxin, a skin tumor promoting agent (49), which up-regulates the activity of ERK by inducing the loss of MKP3 protein, in the presence of oncogenic Ras, through an unknown mechanism. Collectively, these data suggest that induction of MKP3 may represent a therapeutic goal in cancer.
Acknowledgments
Supported by NIH grant R01CA102270 to JDL and Postdoctoral Fellowship (#PF-05-252-01-MGO) from the American Cancer Society to MKHK
References
- 1.Rivera MN, Haber DA. Wilms' tumour: connecting tumorigenesis and organ development in the kidney. Nat Rev Cancer. 2005;5:699–712. doi: 10.1038/nrc1696. [DOI] [PubMed] [Google Scholar]
- 2.Hosono S, Gross I, English MA, et al. E-cadherin is a WT1 target gene. J Biol Chem. 2000;275:10943–53. doi: 10.1074/jbc.275.15.10943. [DOI] [PubMed] [Google Scholar]
- 3.Lee SB, Huang K, Palmer R, et al. The Wilms tumor suppressor WT1 encodes a transcriptional activator of amphiregulin. Cell. 1999;98:663–73. doi: 10.1016/s0092-8674(00)80053-7. [DOI] [PubMed] [Google Scholar]
- 4.English MA, Licht JD. Tumor-associated WT1 missense mutants indicate that transcriptional activation by WT1 is critical for growth control. J Biol Chem. 1999;274:13258–63. doi: 10.1074/jbc.274.19.13258. [DOI] [PubMed] [Google Scholar]
- 5.Palmer RE, Kotsianti A, Cadman B, et al. WT1 regulates the expression of the major glomerular podocyte membrane protein Podocalyxin. Curr Biol. 2001;11:1805–9. doi: 10.1016/s0960-9822(01)00560-7. [DOI] [PubMed] [Google Scholar]
- 6.Guo G, Morrison DJ, Licht JD, et al. WT1 activates a glomerular-specific enhancer identified from the human nephrin gene. J Am Soc Nephrol. 2004;15:2851–6. doi: 10.1097/01.ASN.0000143474.91362.C4. [DOI] [PubMed] [Google Scholar]
- 7.Sim EU, Smith A, Szilagi E, et al. Wnt-4 regulation by the Wilms' tumour suppressor gene, WT1. Oncogene. 2002;21:2948–60. doi: 10.1038/sj.onc.1205373. [DOI] [PubMed] [Google Scholar]
- 8.Heckman C, Mochon E, Arcinas M, et al. The WT1 protein is a negative regulator of the normal bcl-2 allele in t(14;18) lymphomas. J Biol Chem. 1997;272:19609–14. doi: 10.1074/jbc.272.31.19609. [DOI] [PubMed] [Google Scholar]
- 9.Mayo MW, Wang CY, Drouin SS, et al. WT1 modulates apoptosis by transcriptionally upregulating the bcl-2 proto-oncogene. Embo J. 1999;18:3990–4003. doi: 10.1093/emboj/18.14.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simpson LA, Burwell EA, Thompson KA, et al. The antiapoptotic gene A1/BFL1 is a WT1 target gene that mediates granulocytic differentiation and resistance to chemotherapy. Blood. 2006;107:4695–702. doi: 10.1182/blood-2005-10-4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrison DJ, English MA, Licht JD. WT1 induces apoptosis through transcriptional regulation of the proapoptotic Bcl-2 family member Bak. Cancer Res. 2005;65:8174–82. doi: 10.1158/0008-5472.CAN-04-3657. [DOI] [PubMed] [Google Scholar]
- 12.Englert C, Maheswaran S, Garvin AJ, et al. Induction of p21 by the Wilms' tumor suppressor gene WT1. Cancer Res. 1997;57:1429–34. [PubMed] [Google Scholar]
- 13.Wagner N, Wagner KD, Theres H, et al. Coronary vessel development requires activation of the TrkB neurotrophin receptor by the Wilms' tumor transcription factor Wt1. Genes Dev. 2005;19:2631–42. doi: 10.1101/gad.346405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Idelman G, Glaser T, Roberts CT, Jr, et al. WT1-p53 interactions in insulin-like growth factor-I receptor gene regulation. J Biol Chem. 2003;278:3474–82. doi: 10.1074/jbc.M211606200. [DOI] [PubMed] [Google Scholar]
- 15.Liu XW, Gong LJ, Guo LY, et al. The Wilms' tumor gene product WT1 mediates the down-regulation of the rat epidermal growth factor receptor by nerve growth factor in PC12 cells. J Biol Chem. 2001;276:5068–73. doi: 10.1074/jbc.M008776200. [DOI] [PubMed] [Google Scholar]
- 16.Englert C, Hou X, Maheswaran S, et al. WT1 suppresses synthesis of the epidermal growth factor receptor and induces apoptosis. Embo J. 1995;14:4662–75. doi: 10.1002/j.1460-2075.1995.tb00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stanhope-Baker P, Williams BR. Identification of connective tissue growth factor as a target of WT1 transcriptional regulation. J Biol Chem. 2000;275:38139–50. doi: 10.1074/jbc.M004901200. [DOI] [PubMed] [Google Scholar]
- 18.Graham K, Li W, Williams BR, et al. Vascular endothelial growth factor (VEGF) is suppressed in WT1-transfected LNCaP cells. Gene Expr. 2006;13:1–14. doi: 10.3727/000000006783991953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gross I, Morrison DJ, Hyink DP, et al. The receptor tyrosine kinase regulator Sprouty1 is a target of the tumor suppressor WT1 and important for kidney development. J Biol Chem. 2003;278:41420–30. doi: 10.1074/jbc.M306425200. [DOI] [PubMed] [Google Scholar]
- 20.Karlsson M, Mathers J, Dickinson RJ, et al. Both nuclear-cytoplasmic shuttling of the dual specificity phosphatase MKP-3 and its ability to anchor MAP kinase in the cytoplasm are mediated by a conserved nuclear export signal. J Biol Chem. 2004;279:41882–91. doi: 10.1074/jbc.M406720200. [DOI] [PubMed] [Google Scholar]
- 21.Muda M, Theodosiou A, Rodrigues N, et al. The dual specificity phosphatases M3/6 and MKP-3 are highly selective for inactivation of distinct mitogen-activated protein kinases. J Biol Chem. 1996;271:27205–8. doi: 10.1074/jbc.271.44.27205. [DOI] [PubMed] [Google Scholar]
- 22.Groom LA, Sneddon AA, Alessi DR, et al. Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, a novel cytosolic dual-specificity phosphatase. Embo J. 1996;15:3621–32. [PMC free article] [PubMed] [Google Scholar]
- 23.Klock A, Herrmann BG. Cloning and expression of the mouse dual-specificity mitogen-activated protein (MAP) kinase phosphatase Mkp3 during mouse embryogenesis. Mech Dev. 2002;116:243–7. doi: 10.1016/s0925-4773(02)00153-3. [DOI] [PubMed] [Google Scholar]
- 24.Dickinson RJ, Eblaghie MC, Keyse SM, et al. Expression of the ERK-specific MAP kinase phosphatase PYST1/MKP3 in mouse embryos during morphogenesis and early organogenesis. Mech Dev. 2002;113:193–6. doi: 10.1016/s0925-4773(02)00024-2. [DOI] [PubMed] [Google Scholar]
- 25.Nichols A, Camps M, Gillieron C, et al. Substrate recognition domains within extracellular signal-regulated kinase mediate binding and catalytic activation of mitogen-activated protein kinase phosphatase-3. J Biol Chem. 2000;275:24613–21. doi: 10.1074/jbc.M001515200. [DOI] [PubMed] [Google Scholar]
- 26.Camps M, Chabert C, Muda M, et al. Induction of the mitogen-activated protein kinase phosphatase MKP3 by nerve growth factor in differentiating PC12. FEBS Lett. 1998;425:271–6. doi: 10.1016/s0014-5793(98)00250-6. [DOI] [PubMed] [Google Scholar]
- 27.Camps M, Nichols A, Gillieron C, et al. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science. 1998;280:1262–5. doi: 10.1126/science.280.5367.1262. [DOI] [PubMed] [Google Scholar]
- 28.Farooq A, Chaturvedi G, Mujtaba S, et al. Solution structure of ERK2 binding domain of MAPK phosphatase MKP-3: structural insights into MKP-3 activation by ERK2. Mol Cell. 2001;7:387–99. doi: 10.1016/s1097-2765(01)00186-1. [DOI] [PubMed] [Google Scholar]
- 29.Zhou B, Zhang J, Liu S, et al. Mapping ERK2-MKP3 binding interfaces by hydrogen/deuterium exchange mass spectrometry. J Biol Chem. 2006;281:38834–44. doi: 10.1074/jbc.M608916200. [DOI] [PubMed] [Google Scholar]
- 30.Marchetti S, Gimond C, Chambard JC, et al. Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol Cell Biol. 2005;25:854–64. doi: 10.1128/MCB.25.2.854-864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim M, Cha GH, Kim S, et al. MKP-3 has essential roles as a negative regulator of the Ras/mitogen-activated protein kinase pathway during Drosophila development. Mol Cell Biol. 2004;24:573–83. doi: 10.1128/MCB.24.2.573-583.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gomez AR, Lopez-Varea A, Molnar C, et al. Conserved cross-interactions in Drosophila and Xenopus between Ras/MAPK signaling and the dual-specificity phosphatase MKP3. Dev Dyn. 2005;232:695–708. doi: 10.1002/dvdy.20227. [DOI] [PubMed] [Google Scholar]
- 33.Kim MK, Mason JM, Li CM, et al. A pathologic link between Wilms tumor suppressor gene, WT1, and IFI16. Neoplasia. 2008;10:69–78. doi: 10.1593/neo.07869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawakami Y, Rodriguez-Leon J, Koth CM, et al. MKP3 mediates the cellular response to FGF8 signalling in the vertebrate limb. Nat Cell Biol. 2003;5:513–9. doi: 10.1038/ncb989. [DOI] [PubMed] [Google Scholar]
- 35.Brunet A, Roux D, Lenormand P, et al. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. Embo J. 1999;18:664–74. doi: 10.1093/emboj/18.3.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo XN, Reddy JC, Yeyati PL, et al. The tumor suppressor gene WT1 inhibits ras-mediated transformation. Oncogene. 1995;11:743–50. [PubMed] [Google Scholar]
- 37.Yang L, Han Y, Saurez Saiz F, et al. A tumor suppressor and oncogene: the WT1 story. Leukemia. 2007;21:868–76. doi: 10.1038/sj.leu.2404624. [DOI] [PubMed] [Google Scholar]
- 38.Roberts SG. Transcriptional regulation by WT1 in development. Curr Opin Genet Dev. 2005;15:542–7. doi: 10.1016/j.gde.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 39.Davies JA, Ladomery M, Hohenstein P, et al. Development of an siRNA-based method for repressing specific genes in renal organ culture and its use to show that the Wt1 tumour suppressor is required for nephron differentiation. Hum Mol Genet. 2004;13:235–46. doi: 10.1093/hmg/ddh015. [DOI] [PubMed] [Google Scholar]
- 40.Ellisen LW, Carlesso N, Cheng T, et al. The Wilms tumor suppressor WT1 directs stage-specific quiescence and differentiation of human hematopoietic progenitor cells. Embo J. 2001;20:1897–909. doi: 10.1093/emboj/20.8.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hossain A, Nixon M, Kuo MT, et al. N-terminally truncated WT1 protein with oncogenic properties overexpressed in leukemia. J Biol Chem. 2006;281:28122–30. doi: 10.1074/jbc.M512391200. [DOI] [PubMed] [Google Scholar]
- 42.Maheswaran S, Park S, Bernard A, et al. Physical and functional interaction between WT1 and p53 proteins. Proc Natl Acad Sci U S A. 1993;90:5100–4. doi: 10.1073/pnas.90.11.5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maheswaran S, Englert C, Bennett P, et al. The WT1 gene product stabilizes p53 and inhibits p53-mediated apoptosis. Genes Dev. 1995;9:2143–56. doi: 10.1101/gad.9.17.2143. [DOI] [PubMed] [Google Scholar]
- 44.Ye Y, Raychaudhuri B, Gurney A, et al. Regulation of WT1 by phosphorylation: inhibition of DNA binding, alteration of transcriptional activity and cellular translocation. Embo J. 1996;15:5606–15. [PMC free article] [PubMed] [Google Scholar]
- 45.Reddy JC, Hosono S, Licht JD. The transcriptional effect of WT1 is modulated by choice of expression vector. J Biol Chem. 1995;270:29976–82. doi: 10.1074/jbc.270.50.29976. [DOI] [PubMed] [Google Scholar]
- 46.Marchetti S, Gimond C, Roux D, et al. Inducible expression of a MAP kinase phosphatase-3-GFP chimera specifically blunts fibroblast growth and ras-dependent tumor formation in nude mice. J Cell Physiol. 2004;199:441–50. doi: 10.1002/jcp.10465. [DOI] [PubMed] [Google Scholar]
- 47.Furukawa T, Sunamura M, Motoi F, et al. Potential tumor suppressive pathway involving DUSP6/MKP-3 in pancreatic cancer. Am J Pathol. 2003;162:1807–15. doi: 10.1016/S0002-9440(10)64315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu S, Furukawa T, Kanai N, et al. Abrogation of DUSP6 by hypermethylation in human pancreatic cancer. J Hum Genet. 2005;50:159–67. doi: 10.1007/s10038-005-0235-y. [DOI] [PubMed] [Google Scholar]
- 49.Warmka JK, Mauro LJ, Wattenberg EV. Mitogen-activated protein kinase phosphatase-3 is a tumor promoter target in initiated cells that express oncogenic Ras. J Biol Chem. 2004;279:33085–92. doi: 10.1074/jbc.M403120200. [DOI] [PubMed] [Google Scholar]