

Abstract
Hepatocyte apoptosis in addition to oxidative stress could be a key component in the pathogenesis of nonalcoholic steatohepatitis (NASH). However, the underlying mechanisms of hepatocellular apoptotic response associated with oxidative stress have not been investigated in high-fat diet (HFD)–induced NASH models. In this study, Sprague-Dawley rats were fed either a Lieber-DeCarli control diet (CD; 35% energy from fat) or a HFD (71% energy from fat) for 6 wk. Pathologic lesions, lipid peroxidation products, and apoptotic hepatocytes in the liver were examined. The expressions of hepatic tumor necrosis factor-α (TNFα) and protein concentrations of cleaved caspase-3, cytochrome p4502E1 (CYP2E1), phosphorylated c-Jun NH2-terminal kinase (JNK), Bax, Bcl-2, and Bcl-xl were measured. Results showed that the key histological features of NASH, including steatosis, inflammatory cell infiltration, and ballooning degeneration of hepatocytes, were induced by HFD feeding, with increased hepatic TNFα mRNA expression. HFD-fed rats had elevated lipid peroxidation products and CYP2E1 protein in the liver. The apoptotic hepatocytes were significantly greater in livers of rats fed HFD than in those fed CD, and these were associated with a higher level of cleaved caspase-3. In addition, HFD feeding increased both hepatic phosphorylated JNK and pro-apoptotic Bax but did not affect anti-apoptotic Bcl-2 and Bcl-xl compared with CD feeding. These data indicate that the increased oxidative stress and its associated JNK activation as well as an imbalance of pro- and anti-apoptotic proteins in the Bcl-2 family all contribute to high hepatocyte apoptosis that may play an important role in the pathogenesis of NASH in this model.
Introduction
Nonalcoholic fatty liver disease is the most common form of chronic liver disease in the United States (1) and encompasses a wide spectrum of liver damage ranging from steatosis to nonalcoholic steatohepatitis (NASH)6 and then to cirrhosis (2). Liver injury in NASH resembles alcoholic steatohepatitis and is usually characterized by fat accumulation, infiltration of inflammatory cells, and a varying extent of ballooning degeneration of hepatocytes in the absence of significant alcohol consumption (3). Studies have shown that NASH patients are at high risk for progression to cirrhosis (4,5), which, in turn, is the most common risk factor for hepatocellular carcinoma.
Understanding the pathogenic mechanisms for NASH is important to apply effective preventive and/or treatment strategies against this disease. A “2 hit” model for NASH pathogenesis was proposed in 1998 (6), in which fat accumulation was considered as the first hit and increased oxidative stress was proposed to be the major second hit. Emerging data recently have shown a high rate of hepatocyte apoptosis in NASH patients, with the magnitude of apoptosis correlating with hepatic inflammation instead of simple steatosis. This implies that apoptosis could be involved in NASH causation (7,8). In addition, the association between increased oxidative stress and a high rate of cellular apoptosis has been reported in hepatocytes (9). Furthermore, increased oxidative stress itself can mediate a variety of cellular responses leading to diverse outcomes such as cell growth and apoptosis. Among these responses, activation of the mitogen-activated protein kinase plays an important role in initiating oxidative damage-induced cellular events (10). The c-Jun NH2-terminal kinase (JNK) pathway represents 1 subgroup of mitogen-activated protein kinase; the role of sustained JNK activation for cellular apoptosis has been well established (11). Recent in vitro evidence showed that Bcl-2 family members, including both antiapoptotic proteins (e.g. Bcl-2 or Bcl-xl) and proapoptotic proteins (e.g. Bax) could be major candidates for JNK regulation of cell apoptosis (12–14). For example, JNK activation is able to either upregulate pro-apoptotic proteins or inhibit antiapoptotic proteins, leading to an imbalance of the Bcl-2 family (15). The ratio between pro- and antiapoptotic proteins can, to some extent, determine or influence the cell death or survival.
The development of animal models for NASH provides a useful tool to investigate potential mechanisms involved in its pathogenesis. Although typical histological features are reproduced in a commonly used NASH model induced by a methionine- and choline-deficient diet (16), rodents fed this diet lose large amounts of body weight in a short period of time, which is rare in patients with NASH. Recently, the use of high-fat diets (HFD) has developed several nutritional animal models for NASH (17–19). For example, mice fed by infusing a HFD for 9 wk develop the histological and pathogenic features of NASH (17). However, some of the major biochemical changes in the liver in this model did not mimic those observed in NASH patients [e.g. the decreased rather than increased cytochrome p4502E1 (CYP2E1) protein]. Baumgardner et al. (18) developed another nutritional NASH model by feeding Sprague-Dawley rats a high-polyunsaturated fat (70% corn oil) diet through total enteral nutrition. In this model, compared with the controls, the hepatic pathological score of hepatocellular ballooning and lobular inflammation reached significance after 65 d. Although these 2 models seem to resemble human NASH, they involved forced overfeeding (85 and 17% in excess of standard intake, respectively) rats by surgical i.g. feeding techniques. Furthermore, both models require technical expertise and specialized equipment that may hamper their widespread use and introduce additional stress to the animals. In contrast, another nutritional model of NASH developed by Lieber et al. (19) for rats reproduces most histopathological (e.g. steatosis, inflammation) and major biochemical changes [e.g. high hepatic tumor necrosis factor-α (TNFα) and CYP2E1] in patients with NASH by ad libitum consumption of the Lieber-DeCarli HFD. In addition, the diet used in this model is commercially available and contains all essential nutrients in adequate amounts.
Because the potential mechanisms of the cellular apoptotic response associated with increased oxidative stress have not been investigated in HFD-induced NASH models, we fed rats in the present study the Lieber-DeCarli HFD for 6 wk to examine whether it can induce apoptosis by increasing oxidative stress and JNK activation as well as causing an imbalance of pro- and antiapoptotic proteins within the Bcl-2 family.
Materials and Methods
Rats and diets
Eight-week-old male Sprague-Dawley rats (Charles River) were randomly assigned to 2 groups (n = 6/group) and they consumed ad libitum either a Lieber-DeCarli liquid control diet (CD; 35% energy from fat) or a Lieber-DeCarli liquid HFD (71% energy from fat) (Dyets). The diet compositions have been described previously (19). Because the liquid diet provides an adequate amount of water, additional water was not given. Each rat was housed individually in temperature- and humidity-controlled rooms and was kept on a 12-h-light:12-h-dark cycle. The Institutional Animal Care and Use Committee at the USDA Human Nutrition Research Center on Aging at Tufts University approved all animal protocols. The total experimental period included 1 wk of diet adaptation followed by a 6-wk period of feeding the different experimental diets. We measured the body weight of each rat weekly. All animals were killed under deep anesthesia and the liver was promptly excised and removed. Two small portions of liver right lobe were fixed in 10% neutral-buffered formalin for histological examination; the remaining liver was snap-frozen in liquid nitrogen and stored at −80°C for subsequent analysis.
Histological examinations
Formalin-fixed and paraffin-embedded liver tissues were processed routinely for hematoxylin and eosin (H&E) staining. Liver histology was examined under a light microscope and then graded according to the magnitude of steatosis, inflammation, and ballooning degeneration of hepatocytes as described before (2,20). Briefly, the degree of steatosis was graded 0–4 based on the average percent of fat-accumulated hepatocytes per field at ×200 magnification under H&E staining (grading: 0 = <5%, 1 = 5-25%, 2 = 26-50%, 3 = 51-75%, 4 = >75%). Inflammation was evaluated by the number of inflammatory cells counted in 10 random fields at ×200 magnification. The mean of these numbers was calculated and regarded as inflammatory cells/mm2. Hepatocellular ballooning degeneration was evaluated as either negative (absent) or positive (present).
Quantification of apoptotic hepatocytes
Apoptotic hepatocytes were identified in rat liver by the terminal dUTP nick end labeling (TUNEL) assay. An In Situ Cell Death Detection kit, Tetramethyl rhodamine red (Roche Diagnostics) was used for detection of DNA strand breaks in apoptotic cells by a fluorescence microscope. The assay uses an optimized terminal transferase to label free 3′ OH ends in DNA strands with TMR-labeled dUTP and can detect early stages of DNA fragmentation in apoptotic cells. Briefly, deparaffinized liver sections were boiled in 100 mmol/L citrate buffer (pH 6.0) for 20 min and subsequently in 3% H2O2 for 15 min at room temperature to quench endogenous peroxidase activity. After these pretreatments, TMR-dUTP and optimized terminal transferase were incorporated into the slides and incubated for 1 h at 37°C in the dark. Tissue sections were viewed under a fluorescence microscope. The number of cells with TUNEL-positive nuclei was determined by manual counting from 20 randomly selected fields at ×400 magnification per liver sample. Results were expressed as the mean number of TUNEL-positive apoptotic hepatocytes per microscopic field.
Hepatic lipid peroxidation
Lipid peroxides are unstable and decompose to form a complex series of compounds. Malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) are major aldehydic metabolites of lipid peroxidation and have been used to reflect lipid peroxidation (21–23). The lipid peroxidation colorimetric microplate assay (Oxford Biochemical Research) was used to measure MDA plus 4-HNE in the liver.
Gene expression by real-time PCR
Total RNA was isolated from the liver by TriPure Isolation Reagent (Roche Diagnostics) according to the instruction. cDNA was then prepared from the RNA samples using Moloney murine leukemia virus RT (Invitrogen) and an automated thermal cycler (Bio-Rad Laboratories). After quantification and qualification, the PCR for mRNA detection was carried out in each well using 20 μL reaction mixture containing 10 μL 2× SYBR Green Supermix, 0.4 μL of 10 μmol/L primer mix (including forward and reverse primers), and 2.5 μL cDNA diluted in Rnase-free water. Cycling conditions were 50°C for 2 min and 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. Gene-specific primer sequences were designed using the Primer Express version 2.0 software (Applied Biosystems). PCR results were then normalized to the levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and calculated by reference to the mean values for the control group using the comparative cycle threshold method. For each sample and each gene, PCR were carried out in duplicate and repeated twice. Gene expression was analyzed using the following pairs of primers: TNFα (forward, CCAGACCCTCACACTCAGATCA; reverse, TCCGCTTGGTGGTTTGCTA), Bax (forward, AACAACATGGAGCTGCAGAGG; reverse, GAAGTTGCCGTCTGCAAACAT), and GAPDH (forward, AGTGCCAGCCTCGTCTCATAG′; reverse, CCTTGACTGTGCCGTTGAACT).
Protein concentrations by western blotting
Whole liver homogenate was prepared from fresh liver sample as previously described (24,25). Liver protein extracts (40 μg) were resolved on SDS-PAGE gel electrophoresis. After blocking the membrane, immunoblotting was performed based on the manufacturer's instruction for each primary antibody against CYP2E1 (Chemicon International), total and phosphorylated JNK, cleaved caspase-3 (Cell Signaling Technology), Bcl-xl, Bcl-2, and Bax (Santa Cruz Biotechnology). Membranes were then incubated with the secondary antibodies against rabbit or mouse (Bio-Rad Laboratory). The blots were developed by using the ECL Western blotting system (Amersham) and analyzed by computerized densitometry (Bio-Rad GS-710, Bio-Rad Laboratories). Anti-GAPDH antibody was used as internal control for equal loading of proteins.
Statistical analysis
SPSS (version 14.0) and Graph Pad PRISM (version 3.0) software were used for statistical analyses. Results were presented as means ± SEM. Nonparametric Wilcoxon's tests were used to compare grading of steatosis and inflammation between CD and HFD groups and other variables were compared using unpaired Student's t tests. Differences were considered significant at P < 0.05.
Results
General characteristics
The 2 groups did not differ in body weight or absolute (g) or relative (g/100g body) liver weight at the end of the experiment (data not shown).
Histopathological lesions
Feeding the HFD for 6 wk caused remarkable fat accumulation and infiltration of a mixed population of inflammatory cells in the liver, as well as ballooning degeneration of hepatocytes characterized by cell swelling with empty intracellular content, indicating cell necrosis (Fig. 1B–D). In contrast, only mild steatosis and scattered inflammatory cells were found in the liver of rats fed CD in which no ballooning degeneration of hepatocyte was observed (Fig. 1A). These hepatic lesions were further verified semiquantitatively by the grading of steatosis, inflammation, and hepatocyte ballooning degeneration. The magnitudes of both steatosis and inflammation in HFD were significantly higher than those in the CD group (Table 1). The higher inflammatory response in the liver induced by the HFD was accompanied by a hepatic TNFα mRNA level that was twice that of the CD-fed rats.
FIGURE 1.
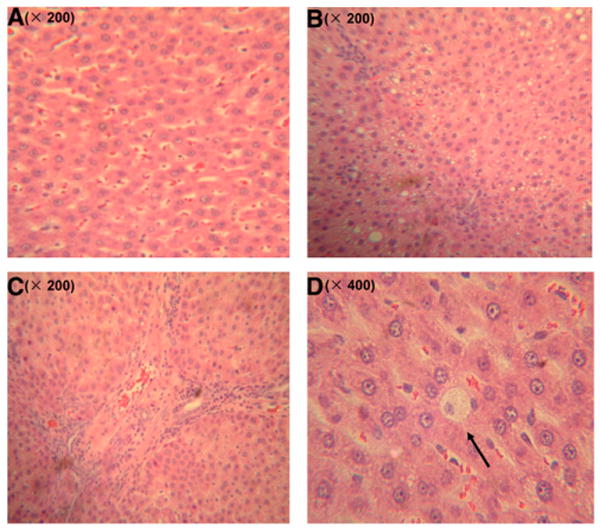
Histopathology of rats fed HFD or CD for 6 wk. Sections were stained with H&E. (A) Liver section of rats fed the CD had mild steatosis and scattered inflammatory cells. (B) Rats fed the HFD had abundant accumulation of fat droplets in hepatocytes. (C) Rats fed the HFD displayed remarkable infiltration of mixed inflammatory cells. (D) The presence of ballooning degeneration of hepatocytes in HFD-fed group.
TABLE 1.
Histological evaluation of rats fed CD or HFD for 6 wk1
| Group | Steatosis (grade) | Inflammation | Ballooning degeneration |
|---|---|---|---|
| cells/mm2 | |||
| CD | 0.3 ± 0.1 | 3.4 ± 0.5 | − |
| HFD | 1.4 ± 0.1* | 14.9 ± 1.3* | + |
Values are means ± SEM, n = 6.
Different from CD, P < 0.05.
Cellular apoptosis
Only a few TUNEL-positive hepatocytes were identified in the livers of the CD-fed rats. In contrast, TUNEL-positive hepatocytes were readily observed in livers from the HFD-fed rats (Fig. 2A). The number of TUNEL-positive apoptotic hepatocytes was greater in rats fed the HFD (3.3 ± 0.4 apoptotic cells/× 400 field) than in those fed the CD (1.3 ± 0.1 apoptotic cells/× 400 field; P < 0.05). The results of the TUNEL assay were further supported by a greater protein concentration of cleaved caspase-3, the active form of a key executor of cellular apoptosis (Fig. 2B), in livers from HFD-fed rats compared with the CD group.
FIGURE 2.
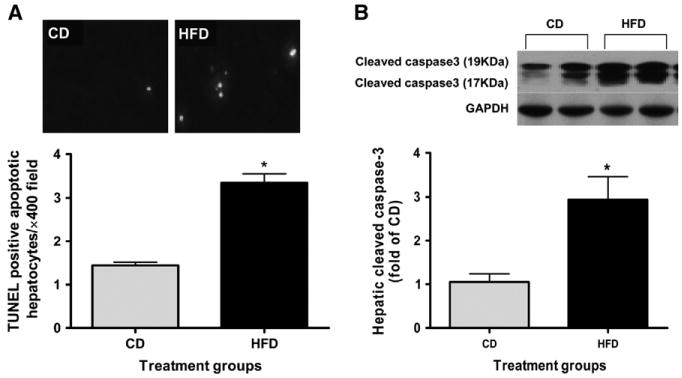
Cell apoptosis in liver of rats fed HFD or CD for 6 wk. (A) TUNEL-positive apoptotic hepatocytes showing red under fluorescence microscope were quantified from 20 randomly selected fields at ×400 magnification. (B) Representative western blot of cleaved caspase-3 and GAPDH proteins expression (inset). Values are means ± SEM, n = 6. *Different from CD, P < 0.05.
Oxidative stress
HFD feeding in the rats resulted in increased hepatic lipid peroxidation. The concentration of lipid peroxidation products in the HFD group was 1.8-fold of that in the CD group (Fig. 3A). In addition, the hepatic protein concentration of CYP2E1 in response to HFD feeding was 1.6-fold of that in CD-fed rats (P < 0.05) (Fig. 3B). CYP2E1 has been shown to be one of the most potent microsome cytochrome to generate free radicals (25).
FIGURE 3.
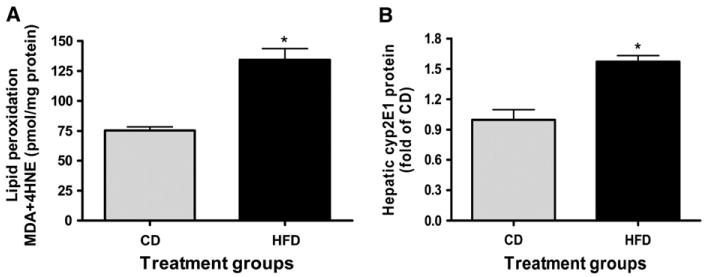
Lipid peroxidation products (A) and CYP2E1 protein (B) in liver of rats fed HFD or CD for 6 wk. Values are means ± SEM, n = 6. *Different from CD, P < 0.05.
JNK activation and Bcl-2 family
The hepatic total JNK protein concentration did not differ between the 2 groups. However, phosphorylated JNK protein in the HFD-fed rats was 2.2-fold that of rats fed the CD (Fig. 4A) (P < 0.05). Of the Bcl-2 family members, the proapoptotic Bax protein concentration was significantly elevated after feeding the HFD for 6 wk (Fig. 4B) with no change in its transcript level (data not shown). The antiapoptotic Bcl-2 and Bcl-xl protein concentrations also did not differ between the 2 groups (data not shown).
FIGURE 4.

JNK signaling (A) and Bcl-2 family (B) proteins in liver of rats fed HFD or CD for 6 wk. (A) Representative western blot of total JNK and phosphorylated JNK. (B) Representative western blot of Bax and GAPDH proteins. Values are means ± SEM, n = 6. *Different from CD, P < 0.05.
Discussion
The present study clearly demonstrated a high rate of hepatocyte apoptosis in a NASH rat model induced by a HFD. Our observation is in agreement with findings from both human studies of NASH patients (7,8) and an animal NASH model induced by i.g. feeding (18), which suggests that increased apoptosis could contribute to NASH pathogenesis. Moreover, the biological importance of HFD-induced hepatocyte apoptosis in this model is more appropriate, because the rats consumed the diets ad libitum, similar to human eating patterns, rather than being force-fed. The higher cleaved caspase-3 and apoptotic hepatocytes in HFD-induced NASH were associated with high oxidative stress, as reflected by significantly increased MDA and 4-HNE, indicating a potential major contribution to hepatocyte apoptosis from increased oxidative stress. In addition, this increased oxidative stress associated with high apoptosis could, at least partially, result from the induction of CYP2E1 by HFD feeding. CYP2E1 has been shown to be invariably elevated and responsible for generation of free radicals in the liver of NASH patients (26,27). The induction of CYP2E1 could be due to the production of acetone, a well-known inducer for CYP2E1 activation (28), as demonstrated in rats fed a liquid diet with a high fat:carbohydrate ratio (29).
Many previous studies have demonstrated that JNK activation contributes to stress-induced apoptosis in several cell types, including hepatocytes (30). Our results of higher phosphorylated JNK, but not total JNK, in this NASH model indicated that JNK activation could be involved in NASH pathogenesis. Our results were also in agreement with a recent study investigating the function of JNK activation in a methionine- and choline-deficient diet–induced NASH model, in which ablation of JNK1 led to significant improvement of hepatic inflammation (31). All these observations suggest an essential role of JNK activation in the development of NASH.
Although the exact molecular mechanisms that account for the regulation of JNK in cellular apoptosis remain unclear, one potential target of JNK regulation is the Bcl-2 family. In the present study, we observed a significant increase of Bax protein concentration in the livers of rats fed the HFD. This increase of Bax proapoptotic protein without changes of Bcl-2/Bcl-xl antiapoptotic proteins may lead to an imbalance within the Bcl-2 family, which then mediates mitochondrial dysfunction and cell apoptosis. A critical role of Bax was recently demonstrated by the observation that activated JNK did not cause the release of mitochondrial cytochrome c and cell apoptosis in Bax −/− fibroblasts (32). JNK regulation of Bax protein can occur at multiple levels from transcriptional to posttranslational levels. For example, Bax mRNA expression was upregulated in cell apoptosis via stimulation of the c-Jun, a well-known downstream functional target of JNK activation (33). However, in our study, the Bax transcript level did not differ between the 2 groups. This is similar to a previous study in which JNK-dependent regulation of Bax was essential to mediate cell apoptosis in response to cadmium treatment, but only Bax protein level was significantly increased without change of its mRNA expression in fibroblast (34). Therefore, it is possible that JNK activation may stabilize Bax protein depending on the specific stimulus and cellular context. This clearly needs further investigation.
In our study, HFD feeding-induced apoptosis was accompanied with a significant increase of hepatic expression of TNFα, which suggested that increased apoptosis could be an important mechanism leading to inflammation in the liver. It has been shown that engulfment of apoptotic bodies of hepatocytes by isolated rat Kupffer cells, resident macrophages of the liver implicated in a wide spectrum of liver inflammation and damage (35–37), stimulated a death ligand and inflammatory cytokine expression such as Fas ligand and TNFα (38). Furthermore, the inhibition of hepatocyte apoptosis with a caspase inhibitor significantly reduced death ligand expression by Kupffer cells (38). A more mechanistic investigation in this model using isolated hepatocyte vs. Kupffer cells is currently being conducted in this laboratory.
In summary, we demonstrated that hepatocyte apoptosis was significantly increased in NASH induced by a liquid HFD. Our findings indicate that the increased oxidative stress and its associated JNK activation as well as an imbalance of pro- and antiapoptotic proteins in the Bcl-2 family all contributed to high hepatocyte apoptosis that may play a significant role in the pathogenesis of NASH in this model. The present study also provides useful information regarding potential molecular targets for NASH prevention and treatment.
Acknowledgments
The authors thank Dr. Donald E. Smith for his assistance with animal care and handling.
Footnotes
NIH grant R01CA104932 and USDA under agreement of NO 1950-51000-064S. Any opinions, findings, conclusion, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the views of the USDA and NIH.
Author disclosures: Y. Wang, L. M. Ausman, R. M. Russell, A. S. Greenberg, and X.-D. Wang, no conflicts of interest.
Abbreviations used: CD, control diet; CYP2E1, cytochrome P450 2E1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; H&E, hematoxylin and eosin; HFD, high-fat diet; 4-HNE, 4-hydroxynonenal; JNK, c-Jun NH2-terminal kinase; MDA, malondialdehyde; NASH, nonalcoholic steatohepatitis; TNFα, tumor necrosis factor-α; TUNEL, terminal dUTP nick end labeling.
Literature Cited
- 1.Ong JP, Younossi ZM. Epidemiology and natural history of NAFLD and NASH. Clin Liver Dis. 2007;11:1–16. doi: 10.1016/j.cld.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–9. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 3.James OF, Day CP. Non-alcoholic steatohepatitis (NASH): a disease of emerging identity and importance. J Hepatol. 1998;29:495–501. doi: 10.1016/s0168-8278(98)80073-1. [DOI] [PubMed] [Google Scholar]
- 4.Ludwig J, McGill DB, Lindor KD. Review: nonalcoholic steatohepatitis. J Gastroenterol Hepatol. 1997;12:398–403. doi: 10.1111/j.1440-1746.1997.tb00450.x. [DOI] [PubMed] [Google Scholar]
- 5.Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology. 1990;11:74–80. doi: 10.1002/hep.1840110114. [DOI] [PubMed] [Google Scholar]
- 6.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–5. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 7.Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, Gores GJ. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–43. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 8.Ribeiro PS, Cortez-Pinto H, Sola S, Castro RE, Ramalho RM, Baptista A, Moura MC, Camilo ME, Rodrigues CM. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol. 2004;99:1708–17. doi: 10.1111/j.1572-0241.2004.40009.x. [DOI] [PubMed] [Google Scholar]
- 9.Singh R, Czaja MJ. Regulation of hepatocyte apoptosis by oxidative stress. J Gastroenterol Hepatol. 2007;22 1:S45–8. doi: 10.1111/j.1440-1746.2006.04646.x. [DOI] [PubMed] [Google Scholar]
- 10.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 11.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 12.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 13.Maundrell K, Antonsson B, Magnenat E, Camps M, Muda M, Chabert C, Gillieron C, Boschert U, Vial-Knecht E, et al. Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1. J Biol Chem. 1997;272:25238–42. doi: 10.1074/jbc.272.40.25238. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19:8469–78. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19:142–9. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 16.Koteish A, Diehl AM. Animal models of steatosis. Semin Liver Dis. 2001;21:89–104. doi: 10.1055/s-2001-12932. [DOI] [PubMed] [Google Scholar]
- 17.Deng QG, She H, Cheng JH, French SW, Koop DR, Xiong S, Tsukamoto H. Steatohepatitis induced by intragastric overfeeding in mice. Hepatology. 2005;42:905–14. doi: 10.1002/hep.20877. [DOI] [PubMed] [Google Scholar]
- 18.Baumgardner JN, Shankar K, Hennings L, Badger TM, Ronis MJ. A new model for nonalcoholic steatohepatitis in the rat utilizing total enteral nutrition to overfeed a high-polyunsaturated fat diet. Am J Physiol Gastrointest Liver Physiol. 2008;294:G27–38. doi: 10.1152/ajpgi.00296.2007. [DOI] [PubMed] [Google Scholar]
- 19.Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C, Ponomarenko A, DeCarli LM. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79:502–9. doi: 10.1093/ajcn/79.3.502. [DOI] [PubMed] [Google Scholar]
- 20.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–74. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 21.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 22.Chitchumroonchokchai C, Bomser JA, Glamm JE, Failla ML. Xanthophylls and alpha-tocopherol decrease UVB-induced lipid peroxidation and stress signaling in human lens epithelial cells. J Nutr. 2004;134:3225–32. doi: 10.1093/jn/134.12.3225. [DOI] [PubMed] [Google Scholar]
- 23.Andreadou I, Iliodromitis EK, Mikros E, Bofilis E, Zoga A, Constantinou M, Tsantili-Kakoulidou A, Kremastinos DT. Melatonin does not prevent the protection of ischemic preconditioning in vivo despite its antioxidant effect against oxidative stress. Free Radic Biol Med. 2004;37:500–10. doi: 10.1016/j.freeradbiomed.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 24.Chung J, Chavez PR, Russell RM, Wang XD. Retinoic acid inhibits hepatic Jun N-terminal kinase-dependent signaling pathway in ethanol-fed rats. Oncogene. 2002;21:1539–47. doi: 10.1038/sj.onc.1205023. [DOI] [PubMed] [Google Scholar]
- 25.Chung J, Liu C, Smith DE, Seitz HK, Russell RM, Wang XD. Restoration of retinoic acid concentration suppresses ethanol-enhanced c-Jun expression and hepatocyte proliferation in rat liver. Carcinogenesis. 2001;22:1213–9. doi: 10.1093/carcin/22.8.1213. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez FJ. Role of cytochromes P450 in chemical toxicity and oxidative stress: studies with CYP2E1. Mutat Res. 2005;569:101–10. doi: 10.1016/j.mrfmmm.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 27.Weltman MD, Farrell GC, Hall P, Ingelman-Sundberg M, Liddle C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology. 1998;27:128–33. doi: 10.1002/hep.510270121. [DOI] [PubMed] [Google Scholar]
- 28.Miller KW, Yang CS. Studies on the mechanisms of induction of N-nitrosodimethylamine demethylase by fasting, acetone, and ethanol. Arch Biochem Biophys. 1984;229:483–91. doi: 10.1016/0003-9861(84)90179-6. [DOI] [PubMed] [Google Scholar]
- 29.Yoo JS, Ning SM, Pantuck CB, Pantuck EJ, Yang CS. Regulation of hepatic microsomal cytochrome P450IIE1 level by dietary lipids and carbohydrates in rats. J Nutr. 1991;121:959–65. doi: 10.1093/jn/121.7.959. [DOI] [PubMed] [Google Scholar]
- 30.Marderstein EL, Bucher B, Guo Z, Feng X, Reid K, Geller DA. Protection of rat hepatocytes from apoptosis by inhibition of c-Jun N-terminal kinase. Surgery. 2003;134:280–4. doi: 10.1067/msy.2003.237. [DOI] [PubMed] [Google Scholar]
- 31.Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, Czaja MJ. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–72. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- 32.Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar-Sagi D, Davis RJ. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol Cell Biol. 2002;22:4929–42. doi: 10.1128/MCB.22.13.4929-4942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandal M, Olson DJ, Sharma T, Vadlamudi RK, Kumar R. Butyric acid induces apoptosis by up-regulating Bax expression via stimulation of the c-Jun N-terminal kinase/activation protein-1 pathway in human colon cancer cells. Gastroenterology. 2001;120:71–8. doi: 10.1053/gast.2001.20897. [DOI] [PubMed] [Google Scholar]
- 34.Papadakis ES, Finegan KG, Wang X, Robinson AC, Guo C, Kayahara M, Tournier C. The regulation of Bax by c-Jun N-terminal protein kinase (JNK) is a prerequisite to the mitochondrial-induced apoptotic pathway. FEBS Lett. 2006;580:1320–6. doi: 10.1016/j.febslet.2006.01.053. [DOI] [PubMed] [Google Scholar]
- 35.Kojima Y, Suzuki S, Tsuchiya Y, Konno H, Baba S, Nakamura S. Regulation of pro-inflammatory and anti-inflammatory cytokine responses by Kupffer cells in endotoxin-enhanced reperfusion injury after total hepatic ischemia. Transpl Int. 2003;16:231–40. doi: 10.1007/s00147-002-0536-4. [DOI] [PubMed] [Google Scholar]
- 36.Souto EO, Miyoshi H, Dubois RN, Gores GJ. Kupffer cell-derived cyclooxygenase-2 regulates hepatocyte Bcl-2 expression in choledocho-venous fistula rats. Am J Physiol Gastrointest Liver Physiol. 2001;280:G805–11. doi: 10.1152/ajpgi.2001.280.5.G805. [DOI] [PubMed] [Google Scholar]
- 37.Wheeler MD, Kono H, Yin M, Nakagami M, Uesugi T, Arteel GE, Gabele E, Rusyn I, Yamashina S, et al. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic Biol Med. 2001;31:1544–9. doi: 10.1016/s0891-5849(01)00748-1. [DOI] [PubMed] [Google Scholar]
- 38.Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, Gores GJ. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. 2003;38:1188–98. doi: 10.1053/jhep.2003.50472. [DOI] [PubMed] [Google Scholar]