

Abstract
More than 200 mutations in the β-myosin gene (MYH7) that cause clinically distinct cardiac and/or skeletal myopathies have been reported, but to date no comprehensive statistical analysis of these mutations has been performed. As a part of this review, we developed a new interactive database and research tool called MyoMAPR (Myopathic Mutation Analysis Profiler and Repository). We report that the distribution of mutations along the β-myosin gene is not homogeneous, and that myosin is a highly constrained molecule with an uncommon sensitivity to amino acid substitutions. Increasing knowledge of the characteristics of MH7 mutations may provide a valuable resource for scientists and clinicians studying diagnosis, risk stratification and treatment of disease associated with these mutations.
Introduction
The ~ 23-kb long human MYH7 gene, located on chromosome 14 (GenBank accession No. AJ238393), contains 40 exons that direct the synthesis of the 1935 amino acid β-myosin heavy chain, the major myosin human isoform expressed in both cardiac and slow skeletal muscle fibers. Mutations in the MYH7 gene cause two types of cardiac pathology: familial hypertrophic cardiomyopathy (FHC) (Tardiff 2005) and familial dilated cardiomyopathy (DCM) (Chang and Potter 2005). Recently, MYH7 mutations have been also associated with myosin storage myopathy or hyaline body myopathy and Laing-type distal myopathy (MPD1) (Laing and Nowak 2005, Oldfors 2007).To date, more than 200 mutations causing cardiac and distal skeletal myopathies have been found in the MYH7 gene, but their biochemical characteristics and their potential impact on myosin structure have not yet been systematically reviewed. To facilitate this analysis, we developed a new software tool called MyoMAPR (Myopathic Mutation Analysis Profiler and Repository) and carried out a comprehensive bioinformatics review of disease-causing mutations in MYH7. At present, MyoMAPR containing 203 missense mutations with relevant references, is regularly updated, and is publicly available at http://bmf.colorado.edu/myomapr along with complete explanations of its key features.
Overall Distribution of Mutations
MyoMAPR was first used to map mutations along the two major domains of myosin: the motor and rod regions. The motor domain/rod junction was set at proline 838, which corresponds to the beginning of the predicted coiled-coil structure (McLachlan and Karn 1982), and missense mutations were scored as similar or dissimilar based on the physical/chemical characteristics before and after the amino acid substitutions. Scoring was carried out according to the following similarity groups: nonpolar amino acids (A, V, L, I, P, F, W, M, G); uncharged polar amino acids (S, T, C, Y, N, Q); negatively charged polar amino acids (D, E); positively charged amino acids (K, R, H). There are 9 deletions and 3 nonsense mutations reported in the literature (see MyoMAPR, Indels/Stop Codon tab). Interestingly, 6 of the 7 single amino acid deletions affect charged amino acids (3 E, 3 K), suggesting that they may affect charge periodicity. However, we have excluded these mutations from our study, since the small sample sizes prevent more detailed conclusions.
MyoMAPR analysis revealed several features of myosin mutations. First, more mutations are found in the motor domain than in the rod domain (Figure 1A). Second, mutations are not homogeneously scattered through either myosin domain, but appear to cluster in discrete regions (Figure 1B). Throughout the whole molecule, mutations that conserve amino acid properties are more frequent than statistically expected (assuming that mutations occur at random throughout the molecule, see the MyoMAPR reference tab menu for an explanation of the statistical method used); conversely, dissimilar mutations are under-represented (Figure 1C). However, the ratios of similar to dissimilar mutations differ between the motor domain and the rod domain (0.8 vs. 0.233 respectively) (Figure 1D and E). Thus, these results suggest that the MYH7 gene sequence/structure is not a homogeneous substrate for mutagenesis, because some regions appear to be targeted differently and more frequently than others.
Figure 1.

Mapping of myosin motor domain and rod domain mutations. Black columns correspond to the total number of observed mutations (similar plus dissimilar), dotted and striped columns correspond to the number of similar and dissimilar observed mutations, respectively. White columns correspond to the theoretical number of expected mutations. (A) Distribution of total mutations present in the myosin motor domain and rod. * indicates that the observed number of mutations is significantly different than expected (p < 0.01). (B) This analysis was carried out using a scanning smoothing window of 56 amino acids. The x-axis corresponds to the amino acid position; the y-axis corresponds to the number of mutations. The boundary between the motor domain and rod is shown at the top of the graph. (C) Number of similar and dissimilar mutations present in the whole myosin molecule. * indicates that the observed number of mutations is significantly different than expected (p < 0.05). (D) Number of similar and dissimilar mutations present in the motor domain. * indicates that the observed number of mutations is significantly different than expected (p < 0.01) (E) Number of similar and dissimilar mutations present in the rod. * indicates that the observed number of mutations is significantly different than expected (p < 0.05).
Mutations in the Motor Domain
The distribution of mutations in the functional sub-domains of the MYH7 motor domain (S1) was next examined (see MyoMAPR for a detailed 3D representation). The sub-domains play fundamental roles in the regulation of myosin mechano-chemical activity during the contractile cycle and include the ATP-binding pocket and the actin-myosin interface that represent the core regions of the motor domain. The switch II, relay and SH1 helix are defined as functional joints, and articulate the structural rearrangements of the motor domain. The converter amplifies and transmits the motor domain structural rearrangements to the neck domain (or lever arm) containing the light chain binding sites. The two 25/50-kDa and 50/20-kDa loops appear to regulate myosin kinetic properties such as ATPase activity and actin-myosin sliding. Analysis of these functional elements for disease-causing mutations did not reveal any significant difference in the distribution of total, similar and dissimilar mutations, although the converter appears to accumulate more mutations than expected (Figure 2A). Nevertheless, when the functional domains were considered as a whole and compared with the “structural-domains” consisting of the remainder of the motor domain, we found that total and similar mutations were more frequent than expected in the functional domains, while the reverse was observed in the structural domains (Figure 2B). Interestingly, the motor domain mutations causing dilated cardiomyopathy [I201T (Villard et al. 2005), A223T (Daehmlow et al. 2002), T412N (Villard et al. 2005), S532P (Kamisago et al. 2000), A550V (Villard et al. 2005), S642L (Daehmlow et al. 2002), F764L (Kamisago et al. 2000)], accumulate in the sub-domain associated with the actin-myosin interface (Figure 2C). They represent 25% of the total mutations found in this particular sub-domain. Thus, the functional sub-domain sequences appear to be hotspots for mutations, and could contribute to the uneven distribution of mutations observed along the whole molecule.
Figure 2.

Analysis of mutations located in the motor domain. Black columns correspond to the total number of observed mutations (similar plus dissimilar), dotted and striped columns correspond to the number of similar and dissimilar observed mutations, respectively. White columns correspond to the theoretical number of expected mutations. (A) Distribution of mutations in the motor domain sub-domains. For this analysis, the 50/20 loop was included in the actin-myosin interface sub-domain. (B) Comparison between mutations present in the motor domains functional sub-domains (F) and the outer structural region (S). * indicates that the observed number of mutations is significantly different than expected (p < 0.01). (C) Motor domain distribution of mutations causing dilated cardiomyopathy. Motor domain minus A-M/C corresponds to the motor domain minus the actin-myosin interface and converter regions. * indicates that the observed number of mutations is significantly different than expected (p < 0.01).
Mutations in the Rod Domain
The highly repetitive amino acid sequence of the rod α-helical coiled-coil structure has been previously studied and modeled in fine detail. The structural unit of the rod corresponds to a 28-amino acid repeat. It has a dipolar charge distribution and is composed of four cyclical sequence patterns of seven residues called “heptad repeats”. Residues in the heptad repeat are designated d, e, f, g, a, b, c. Nonpolar, hydrophobic residues that are responsible for coiled-coil formation and stability are generally found in a and d core positions. Conversely, charged residues occupy mostly e and g positions. The entire rod divides into 38, 28-amino acid complete repeats. It has been suggested that interactions between charge clusters on adjacent coiled-coils could be the main mechanism promoting thick filament formation. Determinants promoting filament formation are located in the light meromyosin region (LMM) of the α-helical coiled-coil C-terminal region. In addition, two hinge-like regions assisting the interactions of the motor domain with actin are in sub-fragment 2 (S2), located at the junction between the motor domain and rod.
Effects of Mutations on Myosin predicted Coiled-Coil Structure
Mutations could interfere with either the formation of the coiled-coil structure, and/or the higher order assembly of myofibrils. To discriminate between these two phenotypes, mutations were first analyzed by Coils, Paircoil and Paircoil2, three different algorithms that can be used to score the impact of mutations on the myosin rod coiled-coil profile (see MyoMAPR for references). Just a small subset of observed mutations are predicted to disrupt the coiled-coil structure by Coils and Paircoil but not Paircoil2 (Table 1). In particular, amino acid substitutions that introduce proline or tryptophan in essentially any position of the heptad repeat are predicted to be the most damaging. Remarkably, none of the mutations that result in the inclusion of proline in the rod coiled-coil [(R1500P (Meredith et al. 2004), A1663P (Meredith et al. 2004), L1706P (Meredith et al. 2004) and L1793P (Dye et al. 2006)] elicit a cardiac phenotype, but instead cause distal skeletal myopathies, despite the fact that the gene is expressed in both cardiac and skeletal muscle.
Table 1. Mutations that could disrupt the α-helical coiled-coil structure of the rod.
Mutations were analyzed using both Paircoil and Coils algorithms. Paircoil predictions were generated with the Probability Cutoff equal to 0.5. Coils predictions were generated with two different coiled-coil database derived scoring matrixes (MTK, MTDK) and three scanning amino acid windows (14, 21, 28 amino acid). +/− corresponds to mutations having a mild disruptive effect on the predicted coiled-coil structure of myosin rod.
| Algorithms
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Paircoil | Coils
|
|||||||
| MTK
|
MTIDK
|
|||||||
| Heptad position | Mutation | 14 | 21 | 28 | 14 | 21 | 28 | |
| d | A901G | − | +/− | − | − | +/− | − | − |
| L1793P | + | + | − | − | + | +/− | − | |
|
| ||||||||
| e | A1379T | − | − | +/− | − | − | +/− | − |
|
| ||||||||
| f | R1500P | + | + | + | + | + | + | − |
| R1500W | − | + | + | + | − | − | − | |
| R1845W | − | + | + | + | + | + | − | |
|
| ||||||||
| g | A1663P | + | + | + | + | + | + | +/− |
| R1712W | − | + | + | + | + | + | − | |
|
| ||||||||
| a | L1706P | + | + | + | + | + | + | + |
| K1354D | − | +/− | +/− | +/− | +/− | +/− | +/− | |
|
| ||||||||
| b | A1834M | − | − | +/− | +/− | − | +/− | +/− |
|
| ||||||||
| c | D1096Y | − | + | + | − | + | + | − |
| R1420W | − | + | + | + | − | − | − | |
Potential Effects of Mutations on Myosin Assembly
No particular mutation features were observed when the entire rod was considered. (Figure 3 panels A–F). However, significant differences became apparent when individual repeats of 28 amino acids and rod domains were considered. The rod was divided into three functional domains: 1) the rod-hinge domain spanning amino acids 838 to 1126 (~ repeat 1 to 10) and containing the first hinge located at the motor domain/rod junction, 2) the hinge domain spanning amino acids 1127 to 1279 (~ repeat 10 to 16) and containing the second hinge (Lu and Wong 1985), and 3) the LMM spanning amino acids 1279 to 1935 (~ repeat 16 to 38) and containing the major determinants directing myosin assembly. We initially found that mutations are not equally distributed in each of the 38 repeats: their density is higher in the first 3 repeats corresponding to the first hinge of S2 (Figure 4A). Biochemical scoring showed that mutation enrichment is mainly due to dissimilar amino acid substitutions (Figure 4B). The total number of mutations per repeat drops after the first hinge and remains quite constant along the remaining rod, including the second hinge (Figure 4A). By comparing the charge profile of individual repeats, we also found that the first three repeats show the highest degree of diversity (Figure 4C), in agreement with their specialized function (Blankenfeldt et al. 2006, Li et al. 2003, Root et al. 2006). Interestingly, MyoMAPR analysis of Shannon entropy that measures the diversity of a system (Shannon 1948) revealed that the first three repeats exhibit low sequence conservation, indicating that a more permissive amino acid selection pressure is acting on the beginning of S2.
Figure 3.
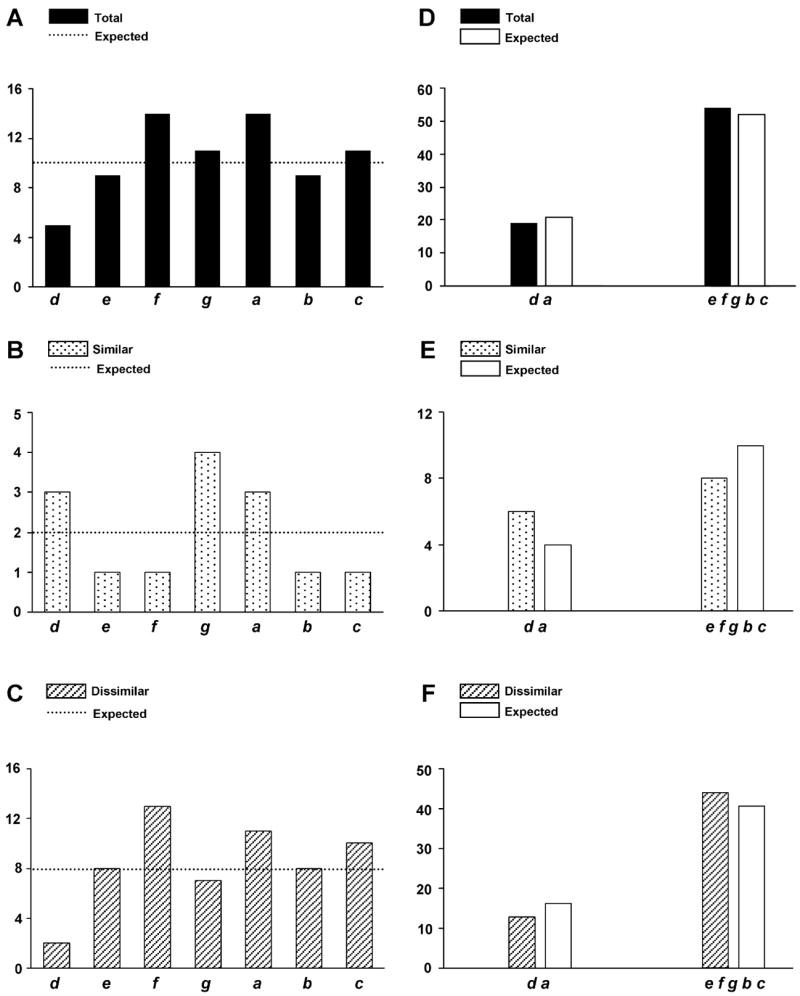
Analysis of mutations located in the heptad repeats of the rod. Black columns correspond to the total number of observed mutations (similar plus dissimilar), dotted and striped columns correspond to the number of similar and dissimilar observed mutations respectively. White columns and dotted lines correspond to the theoretical number of expected mutations. d, e, f, g, a, b, c indicate the heptad repeat positions. Distribution of total (A), similar (B) and dissimilar (C) mutations in each position of the heptad repeat. Comparison of total (D), similar (E) and dissimilar (F) mutations in the internal d and a positions and the exposed e, f, g, b, c positions.
Figure 4.

Analysis of observed mutations located in single repeats of the rod. (A) Distribution of total mutations in the each of the 38 repeats of the rod. The x-axis corresponds to repeat number; the y-axis corresponds to the number of mutations. The boundaries between the rod-hinge, the hinge and the LMM domains are shown at the top of the graph. p < 0.01 indicates that the distribution is statistical different from the random expectation. (B) Distribution of dissimilar observed mutations represented as striped columns in each of the 38 repeats of the rod. The dotted line corresponds to the theoretical number of expected mutations, the x-axis to repeat number, and the y-axis to the number of mutations. p < 0.01 indicates that the observed number of mutations in each repeat is significantly different than expected. (C) Charge profile comparison between repeats of the rod. The degrees of homology are imaged in the heat map according to the following color relationship: red (highest) →yellow →green→ blue (lowest).
When the statistical analysis of mutations in the three domains of the rod was compared, we found that the rod-hinge domain has more total and dissimilar mutations than expected, while both the hinge and the LMM showed the reverse behavior (Figure 5). Within individual rod domains, similar dissimilar and total mutations were equally represented in each position of the heptad repeat (Figure 6 panel A, B, C, left), with no preference in their distribution in the internal versus exposed positions (Figure 6, panels A, B, C right). Nonetheless, comparison between domains uncovered significant differences in the number and nature of mutations in the d, a, c positions, and in internal versus exposed positions (Figure 7A, B). The analysis of mutations causing a primary dilated cardiomyopathy [T1019N (Villard et al. 2005), R1193S (Villard et al. 2005), E1426K (Villard et al. 2005), R1500W (Karkkainen et al. 2004), R1634S (Villard et al. 2005)] revealed no emphasis on particular domains of the rod or the heptad repeat, but a high frequency (4/5) of dissimilar amino acid changes. Intriguingly, mutations causing skeletal muscle myopathies R1500P [(Meredith et al. 2004), A1663P (Meredith et al. 2004), L1706P (Meredith et al. 2004), L1793P (Dye et al. 2006), R1845W (Tajsharghi et al. 2003), E1883K (Tajsharghi et al. 2007), H1901L (Bohlega et al. 2004)] were present only in the rod domain, where they were randomly distributed (Figure 8).
Figure 5.

Analysis of mutations located in the rod-hinge, hinge and LMM domains. Black columns correspond to the total number of observed mutations (similar plus dissimilar), dotted and striped columns correspond to the number of similar and dissimilar observed mutations respectively. White columns correspond to the theoretical number of expected mutations. * indicates that the number of observed mutations is significantly different than expected (p < 0.01).
Figure 6.
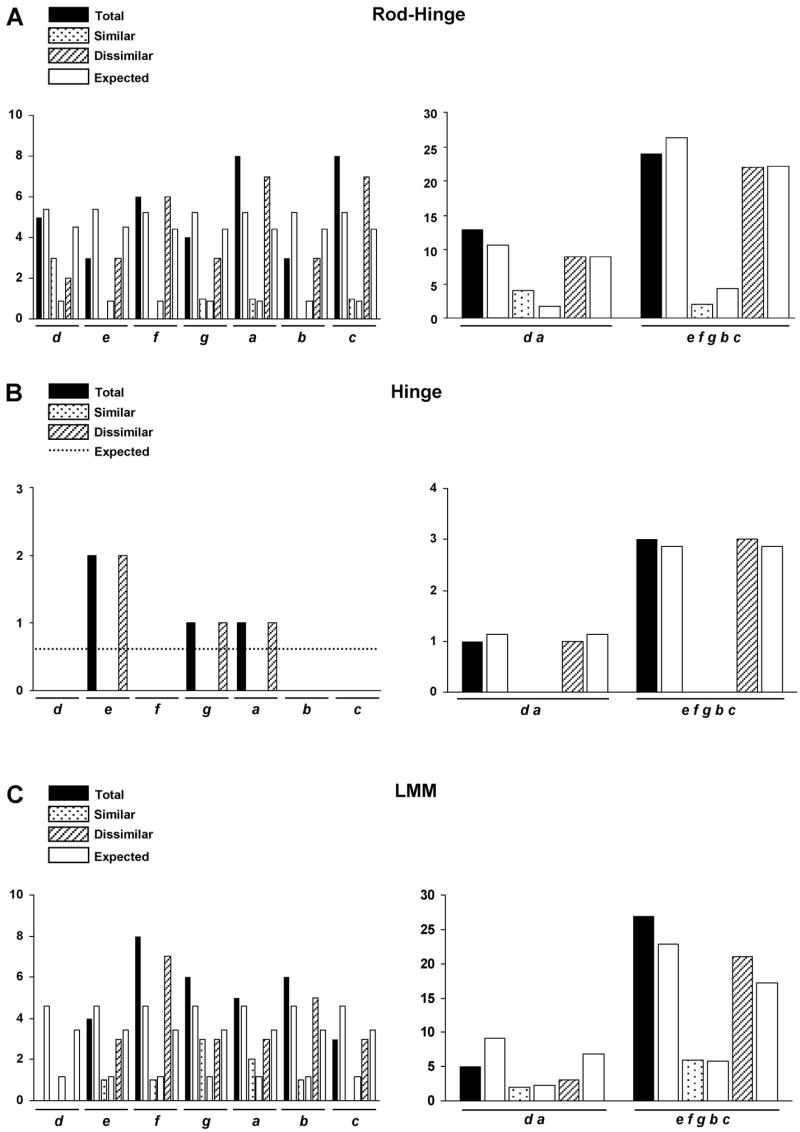
Analysis of mutations located in the heptad repeats of the rod-hinge, hinge, and LMM domains. Black columns correspond to the total number of observed mutations (similar plus dissimilar), dotted and striped columns correspond to the number of similar and dissimilar observed mutations respectively. White columns and the dotted line correspond to the theoretical number of expected mutations. d, e, f, g, a, b, c indicate the heptad repeat positions. Distribution of total, similar and dissimilar mutations in each position of the heptad repeat in the rod-hinge (A left), hinge (B left), and LMM (C left). Distribution of total, similar, and dissimilar mutations in the internal d and a positions and the exposed e, f, g, b, c positions in the rod-hinge (A right), hinge (B right), and LMM (C right).
Figure 7.


Comparison of rod-hinge, hinge, and LMM mutations located in each position of the heptad repeats (A), and in the internal d and a positions and the exposed e, f, g, b, c positions (B). Black columns correspond to the total number of observed mutations (similar plus dissimilar), dotted and striped columns correspond to the number of similar and dissimilar observed mutations respectively. White columns correspond to the theoretical number of expected mutations. Circled d, e, f, g, a, b, c indicate the heptad repeat positions.* indicates that the observed number of mutations is significantly different than expected (p < 0.01, except for the a position with p < 0.05).
Figure 8.

Distribution of mutations causing distal skeletal myopathies. The black columns correspond to the total number of observed mutations (similar plus dissimilar). White columns correspond to the theoretical number of expected mutations. * indicates that the number of observed mutations is significantly different than expected (p < 0.05).
Mutations Changing Residue Charge
The effect of mutations on maintaining or altering the amino acid charges was then examined. When the whole myosin molecule was analyzed, we found that there were generally fewer neutral-to-neutral changes and more positive-to-neutral changes than expected (Figure 9A). While the same charge pattern was observed for the motor domain at positions E497D (Arad et al. 2005), E743D (Davis et al. 2001) and D778E (Andersen et al. 2001, Havndrup et al. 2003; Richard et al. 2003) (Figure 9B) and for the rod at position K875R (Girolami et al. 2006), the most important differences were observed for charge transitions occurring at the negative residues of the rod (Figure 9C). Although equally represented in every position of the heptad repeat, positive-to-neutral, negative-to-neutral and particularly negative-to-positive changes were significantly enriched in the rod. Accordingly, the E to K change is the most common amino acid substitution observed in the 203 mutations (Table 2). Further analysis of the three rod domains revealed that the rod-hinge domain shows a pattern of charge transitions similar to the one observed for whole rod, whereas the LMM shows a reduction in negative to neutral transitions (Figure 10A, B, C). The influence of charge reversals on the myosin charge profile, and the periodicity as determined by Fourier transform, revealed that these mutations can cause a local but significant perturbation in the rod charge character that could interfere with myosin assembly/thick filament stability (data not shown).
Figure 9.
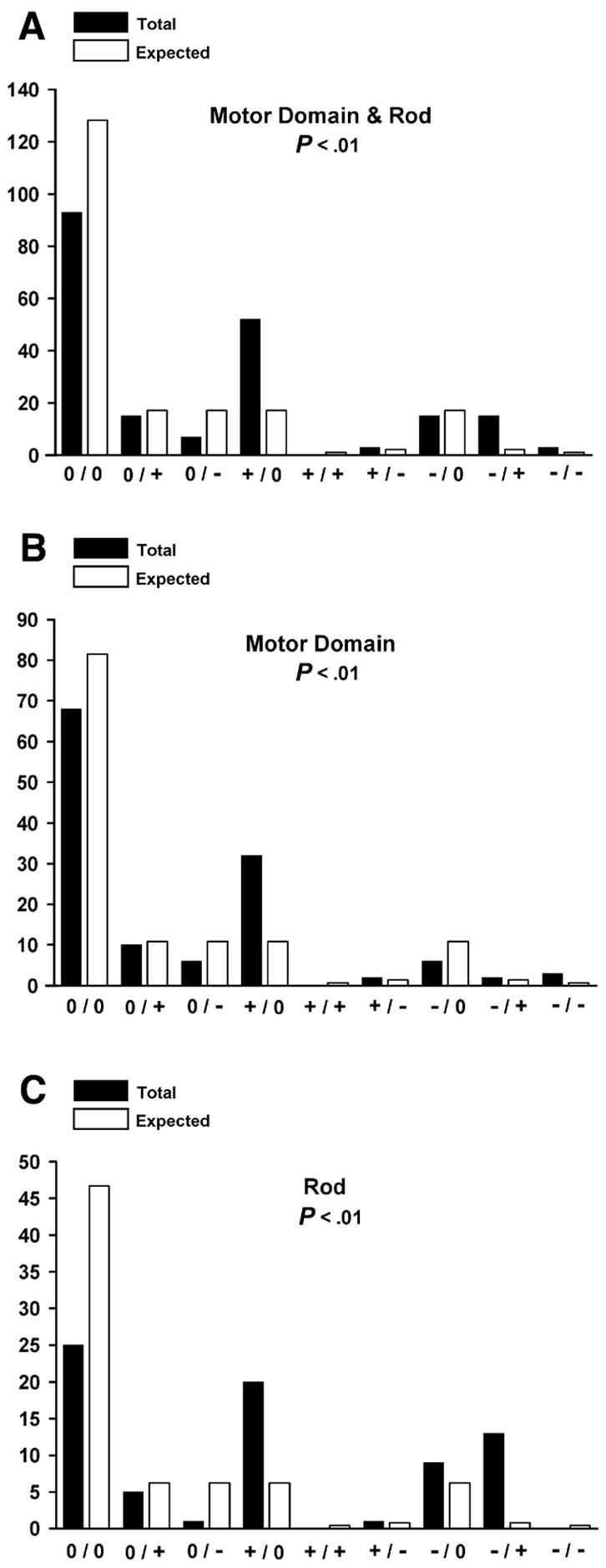
Amino acid charge transition analysis. Black columns correspond to the number of observed charge transitions; white columns correspond to the theoretical number of expected charge transitions. Entire myosin molecule (A), motor domain (B) and rod (C). p < 0.01 indicates that all numbers of transitions observed are significantly different than expected.
Table 2.
Motor domain and rod most frequent amino acids substitutions.
| Motor Domain & Rod | Motor Domain | Rod |
|---|---|---|
| E → K = 15 | R → H = 6 | E → K = 13 |
| R → C = 9 | A → V= 5 | R → W = 5 |
| R → H = 8 | G → R = 5 | R → C = 4 |
| R → W = 8 | I → T= 5 | A → T = 3 |
| V → M = 8 | R → C = 5 | E → Q = 3 |
| A → T = 7 | K → N = 4 | |
| A → V = 6 | N → S = 4 | |
| K → N = 6 | A → T = 4 | |
| R → G = 5 | R → Q = 4 | |
| I → T = 5 | ||
| M → T = 4 | ||
| L → V = 4 |
Figure 10.

Amino acid charge transition analysis. Black columns correspond to the number of observe transitions; white columns correspond to the theoretical number of expected charge transitions. rod-hinge (A), hinge (B) and LMM (C). p < 0.01 indicates that all numbers of transitions observed are significantly different than expected.
“Wild-Type” Mutations
MyoMAPR includes a myosin-weighted consensus sequence, generated from the alignment of 115 myosin sequences, which reflects the frequency of each amino acid at each alignment position over evolutionary history. Sequences that differ the most from other aligned sequences were given more weight, because they have been evolving independently for a longer time (Gerstein et al. 1994). Less than 10% of mutations were present in the weighted consensus sequence, with a frequency ranging between 1 to 32% (data not shown). Thus, some amino acid substitutions that cause a disease in the β-myosin background are present in the wild type gene of other species, or in other myosin isoforms. However, detailed analysis of amino acid covariation in other proteins has revealed that harmful effects of mutations on protein structure and function can in some cases be eliminated by compensatory change(s) in other positions of the molecule (Kondrashov et al. 2002). Thus, it remains to be determined whether the subset of mutations that are found in the consensus sequence are permitted because they have induced covariation in other non-conserved residues.
Lack of Common Mutation Trait
The lack of a predominant pattern among the remaining 90% of mutations indicates that amino acid replacements not included in the consensus sequence are not well tolerated, and may result in a cardiac phenotype whose severity is determined by the type of amino acid change (genotype-phenotype correlation), as well as by genetic and environmental factors (Bos et al. 2007, Daw et al. 2007, Marian and Roberts 2001, Tsoutsman et al. 2006). This hypothesis is also supported by the ratio between synonymous and nonsynonymous myosin single nucleotide polymorphisms found in healthy individuals: 34 synonimous substitutions [(Blair et al. 2002, Erdmann et al. 2003, Hougs et al. 2005, Van Driest et al. 2004, Villard et al. 2005)], over only 4 nonsynonymous substitutions [(A393T (Woo et al. 2003), I909V (Woo et al. 2003), S1491C (Blair et al. 2002, Hougs et al. 2005, Van Driest et al. 2004, Villard et al. 2005), K1919N (Van Driest et al. 2004)]. However, it is tempting to speculate that mutations that display low penetrance, late onset of cardiomyopathy, and almost normal life expectancy may have represented common polymorphisms when the life expectancy was much shorter than today. Mildly deleterious substitutions linked to delayed phenotypes may have survived because they were selectively neutral or nearly neutral alleles, which have a small but nonzero probability of fixation in the population (Kimura 1979). Remarkably, we found that many substitutions between similar amino acids are pathological, and almost all types of substitution have been observed. The only amino acids that were never affected by mutations were cysteines and tryptophans in the motor domain (each representing 0.96% of the sequence), and phenyalanines (0.82%), isoleucines (3.1%), tryptophans (0.18%), and tyrosines (0.64%) in the rod. Amino acid changes introducing a tyrosine (motor domain), or an alanine or isoleucine (rod) were the only absent replacements.
Conclusions
Collectively, our data suggest that β-myosin possesses a uniform and high sensitivity to mutations and suggest that, in vivo, the molecule could behave as a single large functional domain shaped by the multiple interactions that take place in the sarcomere. The functional interdependence between the myosin motor domain and rod is also supported by phylogenetic analysis showing coevolution of the two regions (Korn 2000), and by experimental data employing Dictyostelium/Acanthamoeba chimeric constructs (Liu et al. 2000). Moreover, FHC patients carrying β-myosin mutations in the motor domain and rod show comparable clinical phenotypes (Van Driest et al. 2004). The homogeneous and extreme sensitivity of myosin to amino acid replacements is most likely due to the unique structural relationships occurring among the ~300-myosin molecules that network to form a single thick filament. In this context, the effects of mutations can be easily amplified and become additively deleterious to sarcomere organization, performance, and half-life.
Acknowledgments
We would like to thanks Ada Buvoli and Bob Thompson for discussion and critical reading of the manuscript. M. H. was supported by NIH/CU Molecular Biophysics Training Program T32GM065103.This work was partially supported by NIH grant 5R01 HL085573-01 and 5R01HL85573-2 to L. A. L. Analyses were run using the Keck RNA Bioinformatics Facility at CU Boulder.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen PS, Havndrup O, Bundgaard H, et al. Myosin light chain mutations in familial hypertrophic cardiomyopathy: phenotypic presentation and frequency in Danish and South African populations. J Med Genet. 2001;38:E43. doi: 10.1136/jmg.38.12.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arad M, Penas-Lado M, Monserrat L, et al. Gene mutations in apical hypertrophic cardiomyopathy. Circulation. 2005;112:2805–2811. doi: 10.1161/CIRCULATIONAHA.105.547448. [DOI] [PubMed] [Google Scholar]
- Blair E, Redwood C, de Jesus Oliveira M, et al. Mutations of the light meromyosin domain of the beta-myosin heavy chain rod in hypertrophic cardiomyopathy. Circ Res. 2002;90:263–269. doi: 10.1161/hh0302.104532. [DOI] [PubMed] [Google Scholar]
- Blankenfeldt W, Thoma NH, Wray JS, Gautel M, Schlichting I. Crystal structures of human cardiac beta-myosin II S2-Delta provide insight into the functional role of the S2 subfragment. Proc Natl Acad Sci U S A. 2006;103:17713–17717. doi: 10.1073/pnas.0606741103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlega S, Abu-Amero SN, Wakil SM, et al. Mutation of the slow myosin heavy chain rod domain underlies hyaline body myopathy. Neurology. 2004;62:1518–1521. doi: 10.1212/01.wnl.0000123255.92062.37. [DOI] [PubMed] [Google Scholar]
- Bos JM, Ommen SR, Ackerman MJ. Genetics of hypertrophic cardiomyopathy: one, two, or more diseases? Curr Opin Cardiol. 2007;22:193–199. doi: 10.1097/HCO.0b013e3280e1cc7f. [DOI] [PubMed] [Google Scholar]
- Chang AN, Potter JD. Sarcomeric protein mutations in dilated cardiomyopathy. Heart Fail Rev. 2005;10:225–235. doi: 10.1007/s10741-005-5252-6. [DOI] [PubMed] [Google Scholar]
- Daehmlow S, Erdmann J, Knueppel T, et al. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;298:116–120. doi: 10.1016/s0006-291x(02)02374-4. [DOI] [PubMed] [Google Scholar]
- Davis JS, Hassanzadeh S, Winitsky S, et al. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell. 2001;107:631–641. doi: 10.1016/s0092-8674(01)00586-4. [DOI] [PubMed] [Google Scholar]
- Daw EW, Chen SN, Czernuszewicz G, et al. Genome-wide mapping of modifier chromosomal loci for human hypertrophic cardiomyopathy. Hum Mol Genet. 2007;16:3463–3471. doi: 10.1093/hmg/ddm202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye DE, Azzarelli B, Goebel HH, Laing NG. Novel slow-skeletal myosin (MYH7) mutation in the original myosin storage myopathy kindred. Neuromuscul Disord. 2006;16:357–360. doi: 10.1016/j.nmd.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Erdmann J, Daehmlow S, Wischke S, et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet. 2003;64:339–349. doi: 10.1034/j.1399-0004.2003.00151.x. [DOI] [PubMed] [Google Scholar]
- Gerstein M, Sonnhammer EL, Chothia C. Volume changes in protein evolution. J Mol Biol. 1994;236:1067–1078. doi: 10.1016/0022-2836(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Girolami F, Olivotto I, Passerini I, et al. A molecular screening strategy based on beta-myosin heavy chain, cardiac myosin binding protein C and troponin T genes in Italian patients with hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown) 2006;7:601–607. doi: 10.2459/01.JCM.0000237908.26377.d6. [DOI] [PubMed] [Google Scholar]
- Havndrup O, Bundgaard H, Andersen PS, et al. Outcome of clinical versus genetic family screening in hypertrophic cardiomyopathy with focus on cardiac beta-myosin gene mutations. Cardiovasc Res. 2003;57:347–357. doi: 10.1016/s0008-6363(02)00711-3. [DOI] [PubMed] [Google Scholar]
- Hougs L, Havndrup O, Bundgaard H, et al. One third of Danish hypertrophic cardiomyopathy patients with MYH7 mutations have mutations [corrected] in MYH7 rod region. Eur J Hum Genet. 2005;13:161–165. doi: 10.1038/sj.ejhg.5201310. [DOI] [PubMed] [Google Scholar]
- Kamisago M, Sharma SD, DePalma SR, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- Karkkainen S, Helio T, Jaaskelainen P, Miettinen R, et al. Two novel mutations in the beta-myosin heavy chain gene associated with dilated cardiomyopathy. Eur J Heart Fail. 2004;6:861–868. doi: 10.1016/j.ejheart.2004.04.017. [DOI] [PubMed] [Google Scholar]
- Kimura M. Model of effectively neutral mutations in which selective constraint is incorporated. Proc Natl Acad Sci U S A. 1979;76:3440–3444. doi: 10.1073/pnas.76.7.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov AS, Sunyaev S, Kondrashov FA. Dobzhansky-Muller incompatibilities in protein evolution. Proc Natl Acad Sci U S A. 2002;99:14878–14883. doi: 10.1073/pnas.232565499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn ED. Coevolution of head, neck, and tail domains of myosin heavy chains. Proc Natl Acad Sci U S A. 2000;97:12559–12564. doi: 10.1073/pnas.230441597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing NG, Nowak KJ. When contractile proteins go bad: the sarcomere and skeletal muscle disease. Bioessays. 2005;27:809–822. doi: 10.1002/bies.20269. [DOI] [PubMed] [Google Scholar]
- Li Y, Brown JH, Reshetnikova L, Blazsek A, Farkas L, Nyitray L, Cohen C. Visualization of an unstable coiled coil from the scallop myosin rod. Nature. 2003;424:341–345. doi: 10.1038/nature01801. [DOI] [PubMed] [Google Scholar]
- Liu X, Shu S, Yamashita RA, Xu Y, Korn ED. Chimeras of Dictyostelium myosin II head and neck domains with Acanthamoeba or chicken smooth muscle myosin II tail domain have greatly increased and unregulated actin-dependent MgATPase activity. Proc Natl Acad Sci U S A. 2000;97:12553–12558. doi: 10.1073/pnas.230441497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu RC, Wong A. The amino acid sequence and stability predictions of the hinge region in myosin subfragment 2. J Biol Chem. 1985;260:3456–3461. [PubMed] [Google Scholar]
- Marian AJ, Roberts R. The molecular genetic basis for hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2001;33:655–670. doi: 10.1006/jmcc.2001.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan AD, Karn J. Periodic charge distributions in the myosin rod amino acid sequence match cross-bridge spacings in muscle. Nature. 1982;299:226–231. doi: 10.1038/299226a0. [DOI] [PubMed] [Google Scholar]
- Meredith C, Herrmann R, Parry C, et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1) Am J Hum Genet. 2004;75:703–708. doi: 10.1086/424760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfors A. Hereditary myosin myopathies. Neuromuscul Disord. 2007;17:355–367. doi: 10.1016/j.nmd.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- Root DD, Yadavalli VK, Forbes JG, Wang K. Coiled-coil nanomechanics and uncoiling and unfolding of the superhelix and alpha-helices of myosin. Biophys J. 2006;90:2852–2866. doi: 10.1529/biophysj.105.071597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon M. A mathematical theory of communication. Bell System Tech J. 1948;27:379–423. 623–656. [Google Scholar]
- Tajsharghi H, Oldfors A, Macleod DP, Swash M. Homozygous mutation in MYH7 in myosin storage myopathy and cardiomyopathy. Neurology. 2007;68:962. doi: 10.1212/01.wnl.0000257131.13438.2c. [DOI] [PubMed] [Google Scholar]
- Tajsharghi H, Thornell LE, Lindberg C, Lindvall B, Henriksson KG, Oldfors A. Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol. 2003;54:494–500. doi: 10.1002/ana.10693. [DOI] [PubMed] [Google Scholar]
- Tardiff JC. Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev. 2005;10:237–248. doi: 10.1007/s10741-005-5253-5. [DOI] [PubMed] [Google Scholar]
- Tsoutsman T, Lam L, Semsarian C. Genes, calcium and modifying factors in hypertrophic cardiomyopathy. Clin Exp Pharmacol Physiol. 2006;33:139–145. doi: 10.1111/j.1440-1681.2006.04340.x. [DOI] [PubMed] [Google Scholar]
- Van Driest SL, Jaeger MA, Ommen SR, et al. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:602–610. doi: 10.1016/j.jacc.2004.04.039. [DOI] [PubMed] [Google Scholar]
- Villard E, Duboscq-Bidot L, Charron P, et al. Mutation screening in dilated cardiomyopathy: prominent role of the beta myosin heavy chain gene. Eur Heart J. 2005;26:794–803. doi: 10.1093/eurheartj/ehi193. [DOI] [PubMed] [Google Scholar]
- Woo A, Rakowski H, Liew JC, et al. Mutations of the beta myosin heavy chain gene in hypertrophic cardiomyopathy: critical functional sites determine prognosis. Heart. 2003;89:1179–1185. doi: 10.1136/heart.89.10.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]