

Abstract
Mutations in the bestrophin-1 (Best1) gene are linked to several kinds of macular degeneration in both humans and dogs. Although bestrophins have been shown clearly to be Cl− ion channels, it is controversial whether Cl− channel dysfunction can explain the diseases. It has been suggested that bestrophins are multifunctional proteins: they may regulate voltage-gated Ca2+ channels in addition to functioning as Cl− channels. Here, we show that human Best1 gene (hBest1) differentially modulates CaV1.3 (L-type) voltage-gated Ca2+ channels through association with the CaVβ subunit. In transfected human embryonic kidney 293 cells, hBest1 inhibited CaV1.3. Inhibition of CaV1.3 was not observed in the absence of the β subunit. Also, the hBest1 C terminus binds to CaVβ subunits, suggesting that the effect of hBest1 was mediated by the CaVβ subunit. The region of hBest1 responsible for the effect was localized to a region (amino acids 330–370) in the cytoplasmic C terminus that contains a predicted src-homology-binding domain that is not present in other bestrophin subtypes. Mutation of Pro330 and Pro334 abolished the effects of hBest1 on CaV1.3. The effect was specific to hBest1; it was not observed with mouse Best1 (mBest1), mBest2, or mBest3. Wild-type hBest1 and the disease-causing mutants R92S, G299R, and D312N inhibited CaV currents the same amount, whereas the A146K and G222E mutants were less effective. We propose that hBest1 regulates CaV channels by interacting with the CaVβ subunit and altering channel availability. Our findings reveal a novel function of bestrophin in regulation of CaV channels and suggest a possible mechanism for the role of hBest1 in macular degeneration.
Keywords: macular degeneration, retinal pigment epithelium, ion channel, calcium, src-homology, chloride
Introduction
The human Best1 gene (hBest1) encodes the founding member of a protein family called “bestrophins” (Marquardt et al., 1998; Petrukhin et al., 1998; Hartzell et al., 2008). hBest1 protein is highly expressed in the basolateral plasma membrane of retinal pigment epithelium (RPE) (Marmorstein et al., 2000). Mutations in hBest1 are linked to several kinds of macular degeneration, including Best vitelliform macular dystrophy (BVMD) (Marquardt et al., 1998; Petrukhin et al., 1998), adult-onset vitelliform dystrophy (Kramer et al., 2000; Seddon et al., 2001), autosomal dominant vitreoretinochoroidopathy (Yardley et al., 2004), and canine multifocal retinopathy (Guziewicz et al., 2007).
Considerable evidence supports the idea that bestrophins are Cl− channels (Hartzell et al., 2008). RNA interference knockdown of bestrophins in Drosophila S2 cells (Chien et al., 2006; Chien and Hartzell, 2007) and in epithelial cells (Kunzelmann et al., 2007; Barro Soria et al., 2008) reduces endogenous Cl− currents. Furthermore, overexpression of bestrophins in cell lines induces novel Cl− currents (Sun et al., 2002; Qu et al., 2003, 2004, 2006b; Tsunenari et al., 2003; Qu and Hartzell, 2004). hBest1 Cl− current is dependent on intracellular Ca2+ (EC50, ∼150 nm). Because virtually all disease-causing mutations examined affect hBest1 Cl− channel function (Sun et al., 2002; Yu et al., 2006, 2007), the simplest hypothesis is that macular degeneration is caused by Cl− channel dysfunction. However, experiments with knock-out mice suggest that this explanation is incomplete (for review, see Hartzell et al., 2008). Although humans with BVMD have a decreased electro-oculogram light peak (LP) that is thought to be mediated by Ca2+-activated hBest1 Cl− channels in the RPE, mice with the mouse Best1 (mBest1) gene disrupted (mBest1−/−) exhibit normal LP amplitudes at saturating light intensity (Marmorstein et al., 2006). Furthermore, Ca2+-dependent Cl− currents in the knock-out are the same as wild type (wt) (Marmorstein et al., 2006). This shows that mBest1 is not responsible for the Ca2+-activated Cl− current and does not generate the LP. However, mice with the Ca2+ channel CaVα1.3 or CaVβ4 subunits knocked out have significantly reduced LPs, suggesting that the LP relies on voltage-gated Ca2+ channels (Marmorstein et al., 2006; Wu et al., 2007). These results, coupled with the finding that hBest1 overexpression alters the kinetics of endogenous voltage-gated Ca2+ channels in RPE-J cells (Rosenthal et al., 2005), have led to the proposal that hBest1 is not a Cl− channel but rather a regulator of Ca2+ signaling (Marmorstein et al., 2006). This raises the question whether disease-causing mutations in hBest1 impair not only Cl− channel function but also affect voltage-gated Ca2+ channels. Freshly isolated human RPE cells express the L-type CaV channel α subunits α1.2 and α1.3 and β subunits β2 and β4 (Wimmers et al., 2008). Here, we examine the effect of hBest1 expression on CaV1.3 L-type channels and have found hBest1 regulates channel function via β subunits. Several disease-causing mutations that disrupt Cl− channel function do not abolish the inhibitory effect on CaV channels. However, some mutations, notably G222E and A146K, are less effective in inhibiting CaV channels than wild-type hBest1.
Materials and Methods
Constructs and molecular biology.
hBest1 tagged with the myc epitope at the C terminus in pRK5 was obtained from Dr. Jeremy Nathans (Johns Hopkins University, Baltimore, MD) (Sun et al., 2002). Site-specific mutations were generated using PCR-based mutagenesis (Quickchanger; Stratagene) (Qu et al., 2006b). Channel fragments were generated by PCR using Pfx polymerase and confirmed by sequencing. cDNAs encoding CaV subunits were as follows: rat α11.3 (GenBank accession number AF370010; provided by Dr. Diane Lipscombe, Brown University, Providence, RI) (Helton et al., 2005); FLAG-tagged α11.3 (Calin-Jageman et al., 2007); FLAG-tagged β2a (Zhou et al., 2004); β1b (GenBank accession number NM017346); β2a (GenBank accession number NM053581); β4 (GenBank accession number L02315; provided by Dr. E. Perez-Reyes, University of Virginia, Charlottesville, VA); and α2δ (GenBank accession number M21948). For pull-down assays, glutathione S-transferase (GST)-tagged β2a fusion protein was used (Roger Colbran, Vanderbilt University Medical Center, Nashville, TN) (Grueter et al., 2006).
Cell culture and transfection.
Human embryonic kidney (HEK) cells were cultured as described previously (Yu et al., 2006). For electrophysiology, low-passage HEK293 cells were transiently transfected using Fugene-6 (Roche Diagnostics) with ∼4 μg of total DNA consisting of a mixture of α11.3 (1.5 μg); β1b, β2a, or β4 (0.8 μg); and α2δ (0.8 μg), with or without hBest1 (0.5 μg) per 35 mm dish. Cells were also transfected with pEGFP (0.1 μg) for fluorescent detection of transfected cells. RPE-J cells (American Type Culture Collection) were cultured in DMEM supplemented with 4% fetal calf serum at 32°C and 5% CO2 in air. The 60–80% confluent cells were transfected with Lipofectamine (Invitrogen) according to manufacturer instructions. The total amount of DNA per dish (35 mm) was 2 μg. Cotransfection of wild type or mutant hBest1 with enhanced green fluorescent protein was performed at a ratio of 5:1. Transfected cells were plated at low density and used 24–72 h later.
Electrophysiology.
Cells were whole-cell patch clamped with a HEKA Elektronik EPC-7 patch-clamp amplifier. Clampfit Software (Molecular Devices) was used for data acquisition and leak subtraction using a P/−4 protocol. The standard pipette solution contained the following (in mm): 146 CsCl, 2 MgCl2, 2 Mg-ATP, 5 EGTA, 8 HEPES, pH 7.3, adjusted with N-methyl-d-glucamine. The “high” Ca2+ pipette solution used for recording Cl− current contained 5 mm (Ca2+)-EGTA instead of EGTA, which supplied free Ca2+ of ∼10 μm. The standard extracellular solution contained the following (in mm): 130 NaCl, 3 TEA-Cl, 10 BaCl2, 1 MgCl2, 10 glucose, 10 HEPES, pH 7.3 with Tris. Osmolarity was adjusted with sucrose to 305 mOsm for all solutions. Electrode resistances were typically 2–3 MΩ in the bath solution, and series resistance was ∼2–4 MΩ compensated up to 80%. For measuring IBa, a standard I–V protocol with 20 ms step depolarizations (−70 to +60 mV) was used to evoke currents from a −80 mV holding potential. Tail currents were measured at −70 mV. I–V relationships for each cell were fitted to the following equation: I = G(V − Vrev)/(1 + exp[(V1/2 − V)/k]), where I is the whole-cell current density, G is the specific conductance, Vrev is the reversal potential, V1/2 is the voltage of half-maximal activation, and k is a slope factor. Normalized tail current–voltage and steady-state inactivation data were fit with a single Boltzmann function as follows: I/Imax = A/(1 + exp[(V − V1/2)/k]) + b, where V is test or prepulse voltage, V1/2 is the midpoint of the activation or inactivation curve, k is a slope factor, A is the amplitude, and b is the baseline. For measuring ICl, the I–V protocol consisted of 750 ms pulses from a 0 mV holding potential. Data were analyzed with Clampfit 8.2 and Origin 7.0 software. Averaged data are mean ± SEM. Statistical significance was determined by two-way ANOVA or Student's t test.
Coimmunoprecipitation and Western blot.
HEK293 cells were transfected with FLAG- CaV subunits and myc-tagged hBest1 or hBest1-C terminus. Cell lysates were prepared by homogenization in 1 ml of ice-cold lysis buffer (10 mm HEPES, 50 mm NaCl, 1 mm benzamidine, and 0.5% Triton X-100, pH 7.4) above and incubated with 40 μl of anti-FLAG M2 affinity gel (Sigma-Aldrich) for 1 h, rotating at 4°C. After removing the beads by centrifugation and five washes with 1 ml of lysis buffer, proteins were eluted with sample buffer and subjected to SDS-PAGE. Proteins were detected by Western blotting with monoclonal anti-myc or anti-FLAG antibodies (1:2000; Sigma-Aldrich), followed by HRP-secondary antibodies and ECL detection.
Pull-down assay.
Fusion proteins were grown in BL21 Escherichia coli, induced by isopropyl-β-d-thiogalactopyranoside, and purified on glutathione agarose (β2a) or a Co2+ (hBest1) affinity column. For binding assays (Grueter et al., 2006), GST-tagged β2a or GST (control) were incubated at 4°C for 1 h with myc-tagged hBest1 or hBest1-C terminus in 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 0.1% Triton X-100. Glutathione-agarose was added, and the incubation was continued for 1 h. Resin was collected by centrifugation and washed four times in binding buffer. Bound proteins were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and then immunoblotted.
Results
hBest1 specifically reduces CaV1.3 current density
Different protocols were used to measure Ca2+ currents and Cl− currents in HEK cells transfected with hBest1 and CaV subunits (Fig. 1A). hBest1 Cl− currents were measured with high intracellular free Ca2+ and 0 mV holding potential. The depolarized holding potential (and possibly the high intracellular Ca2+) induced CaV channel inactivation so that the Cl− current was measured with little contamination from CaV channels. The Cl− currents in cells coexpressing CaV1.3 plus hBest1 and those expressing hBest1 alone were the same, showing that the Cl− currents were not contaminated with Ca2+ currents (Fig. 1C,D). CaV currents were measured with Ba2+ as charge carrier with nominally zero intracellular free Ca2+ (10 mm EGTA with no added Ca2+) from a holding potential of −80 mV. Under these conditions, hBest1 was not activated, and the CaV current could be measured with no contamination from hBest1 (Fig. 1C,D). Expression of wt hBest1 in HEK293 cells induced Ca2+-dependent Cl− currents with similar amplitudes and biophysical properties when expressed alone or with CaV channel subunits α11.3/β1b/α2δ (Fig. 1C,D). Cl− currents were not observed in cells transfected with CaV subunits alone (Fig. 1B).
Figure 1.

Inhibition of CaV1.3 currents by coexpression of hBest1. A, Voltage protocols used to record whole-cell Ca2+-activated Cl− current (a) and Ba2+ current (b) from transfected HEK293 cells. Ba2+ currents were measured by 50 ms voltage pulses from a holding potential of −80 mV to various depolarizing voltages in cells bathed with 10 mm [Ba2+]o and <20 nm [Ca2+]i. Cl− currents were measured by 750 ms voltage steps in 20 mV increments from −100 to +100 mV from a holding potential of 0 mV in cells bathed with 2 mm [Ca2+]o and >600 nm [Ca2+]i. B–D, I–V relationships for Ca2+-activated Cl− current (triangle) and Ba2+ current (circle) recorded from HEK293 cells transfected with CaV1.3 (α11.3/β1b/α2δ) alone (B), α11.3/β1b/α2δ plus hBest1 (C), or hBest1 alone (D). Representative current traces are shown above. The peak current densities at the end of the test pulse were plotted against test voltage. Each point represents the mean of 10–15 cells.
In cells cotransfected with α11.3/β1b/α2δ plus hBest1, IBa was significantly smaller in amplitude compared with IBa in cells transfected with α11.3/β1b/α2δ alone. The decrease in current amplitude occurred across the entire range of test pulse voltages. Furthermore, there was no change in reversal potential (Fig. 1B,C), indicating hBest1 did not influence the permeation properties of CaV1.3. The effects of hBest1 were not caused by a nonspecific decrease in CaV1.3 expression as a consequence, for example, of competition for translation machinery because mBest1, mBest2, or mBest3 did not decrease IBa (Fig. 2).
Figure 2.
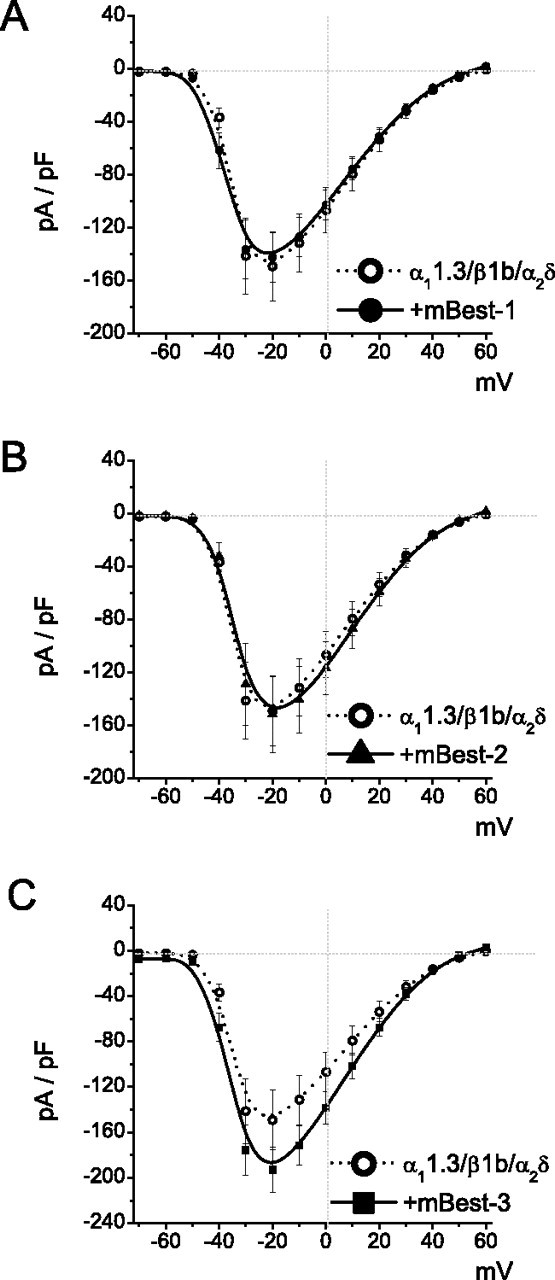
mBest1, mBest2, and mBest3 have no apparent effects on CaV1.3. I–V relationships for Ba2+ currents were recorded from HEK293 cells transfected with α11.3/β1b/α2δ plus mBest1 (A), α11.3/β1b/α2δ plus mBest2 (B), and α11.3/β1b/α2δ plus mBest3 (C). Data were from the same protocol as in Fig. 1Ab. Each point represents the mean of 10–15 cells.
Amino acids 330–370 in the hBest1 C terminus are critical for inhibition of CaV1.3
Bestrophins consist of four to six highly conserved transmembrane domains located in the first ∼295 amino acids and a highly variable cytoplasmic C-terminal domain (Fig. 3A) (Tsunenari et al., 2003; Milenkovic et al., 2007). To identify the region of hBest1 responsible for inhibition of CaV1.3, we cotransfected CaV1.3 subunits with cDNAs corresponding to either the C-terminal (CT292–586) or N-terminal (NT1–289) part of hBest1. Neither of these fragments generated Cl− currents when transfected alone (data not shown). Although hBest1-NT1–289 did not influence IBa, hBest1-CT292–586 caused a similar decrease in IBa amplitude to that produced by full-length hBest1 (Fig. 3A–C). These results demonstrate that inhibition of CaV1.3 does not require the Cl−-conducting properties of hBest1 but relies on determinants in hBest1-CT292–586.
Figure 3.

Effects of N-terminal and C-terminal hBest1 fragments on CaV1.3. A, The top panel shows a schematic of hBest1. The triangle marks amino acid 292 after the last transmembrane domain, which splits hBest1 into N- and C-terminal fragments, hB1-Nt1–289, and hB1-Ct292–586. Exemplar whole-cell Ba2+ currents evoked by 50 ms test pulses to the indicated potentials in HEK293 cells transfected with α11.3/β1b/α2δ alone, α11.3/β1b/α2δ plus hBest1, α11.3/β1b/α2δ plus hB1-Nt1–289, and α11.3/β1b/α2δ plus hB1-Ct292–586. Holding potential was −80 mV. B, I–V relationships for α11.3/β1b/α2δ alone (open circle), α11.3/β1b/α2δ plus hBest1 (filled circle), α11.3/β1b/α2δ plus hB1-Nt1–289 (orange triangle), and α11.3/β1b/α2δ plus hB1-Ct292–586 (green triangle). C, Averaged peak current densities at −10 mV test pulse were measured for each group in B. *p < 0.01 for each group compared with α11.3/β1b/α2δ alone by two-way ANOVA. Each point and averaged values mentioned above represent the mean of 10–15 cells.
To pinpoint these determinants, we tested the effects of truncated fragments of hBest1-CT292–586 (Fig. 4A). Peak IBa densities were significantly reduced by coexpression with C-terminal fragments CT292-X with X ≥ 400. Therefore, amino acids 292–400 of hBest1 contain the region involved in Cav1.3 regulation.
Figure 4.

Identification of critical region of hBest1 required for Cav1.3 inhibition. A, Averaged Ba2+ current densities recorded during 50 ms test pulse at −10 mV for α11.3/β1b/α2δ either alone or with hBest1 or six hBest1 C-terminal fragments. The percentage of inhibition compared with control (α11.3/β1b/α2δ alone) is also shown. B, Averaged Ba2+ current densities recorded during 50 ms test pulse at −10 mV for α11.3/β1b/α2δ either alone or with hBest1 and six hBest1 N-terminal fragments. The right panels show the design for hBest1 fragments. Each averaged value represents the mean of 10–15 cells. *p < 0.05 for each group compared with CaV1.3 alone by two-way ANOVA.
The critical region was further refined by expressing N-terminal constructs of hBest1 truncated at positions 380, 370, 360, 350, 340, 330, and 289 (Fig. 4B). Significant inhibition was induced by all hBest1-NT fragments truncated beyond 340 (NT1-X with X ≥ 340). However, the inhibition caused by NT1–340, NT1–350, and NT1–360 was less than that caused by NT1–370 or longer. NT1–370 or NT1–380 exerted the same inhibition as full-length hBest1. Together, these results suggest that the region from 330 to 370 is important in the regulation of CaV by hBest1.
CaVβ is required for CaV1.3 modulation by hBest1
Because CaVβ auxiliary subunits (CaVβ1-CaVβ4) strongly influence CaV current density and activation properties (Dolphin, 2003), we tested whether the effect of hBest1 on CaV1.3 depended on CaVβ. hBest1 inhibited CaV1.3 channels containing β1b, β2A, or β4 similarly (Figs. 1B,D, 5A,B). However, no inhibition of IBa by hBest1 was seen on the channel consisting of α11.3/α2δ without a β subunit (Fig. 5C). Furthermore, hBest1 had no effect on CaV3.1 expressed without a β subunit (Table 1). These results show that CaVβ subunits are required for CaV1.3 inhibition by hBest1.
Figure 5.
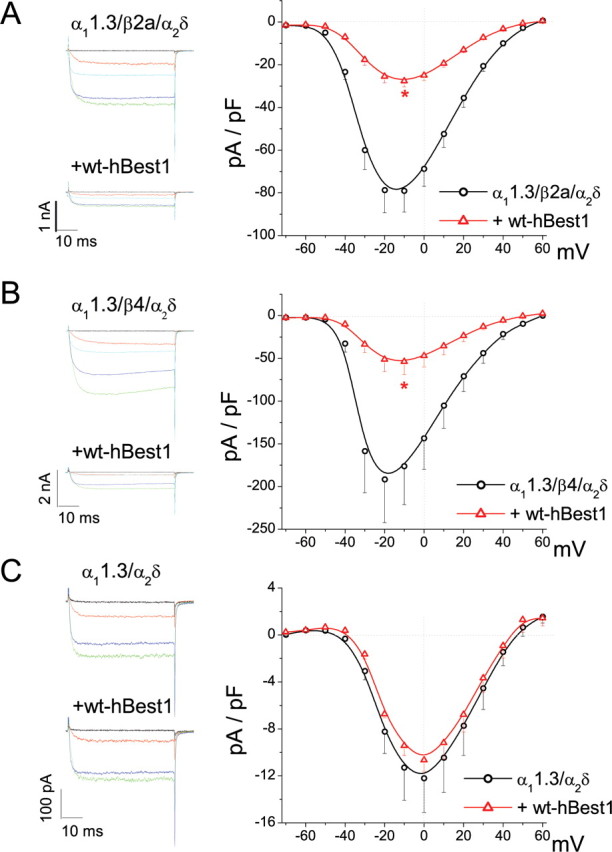
CaVβ is required for the inhibition caused by hBest1. A, I–V relationships for α11.3/β2a/α2δ alone (circle) and with hBest1 (triangle). B, I–V relationships for α11.3/β4/α2δ alone (circle) and with hBest1 (triangle). Representative current traces evoked by test pulses to voltages between −60 and +20 mV test pulses (20 mV steps) are shown for each group in the left panel. *p < 0.01 for CaV1.3 plus hBest1 compared with CaV1.3 alone by two-way ANOVA. C, I–V relationships for recombinant channel without CaVβ (α11.3/α2δ) alone (circle) and with hBest1 (triangle). Representative current traces evoked by test pulses to voltages between −40 and +20 mV (20 mV steps) are shown.
Table 1.
Effect of wt hBest1 on electrophysiological properties of recombinant voltage-gated Ca channels
| Transfected constructs | (±) hBest1 | G (pA · pF−1 · mV−1) | Activation V1/2 (mV) | Activation kslope | Inactivation V1/2 (mV) | Inactivation kslope |
|---|---|---|---|---|---|---|
| α11.3/α2δ | − | 0.26 ± 0.05 | −18.8 ± 1.3 | 6.0 ± 0.4 | ||
| n = 9 | n = 9 | n = 9 | ||||
| + | 0.25 ± 0.02 | −16.7 ± 0.9 | 6.2 ± 0.3 | |||
| n = 5 | n = 6 | n = 6 | ||||
| α11.3/β1b/α2δ | − | 1.7 ± 0.3 | −33.3 ± 0.7 | 4.1 ± 0.5 | −34.7 ± 1.1 | 6.9 ± 0.4 |
| n = 14 | n = 14 | n = 14 | n = 21 | n = 11 | ||
| + | 0.6 ± 0.1** | −24.6 ± 1.1** | 5.7 ± 0.6* | −33.5 ± 0.6 | 6.0 ± 0.3 | |
| n = 13 | n = 13 | n = 13 | n = 10 | n = 10 | ||
| α11.3/β2a/α2δ | − | 1.2 ± 0.1 | −31.7 ± 1.2 | 5.9 ± 0.3 | ||
| n = 11 | n = 11 | n = 11 | ||||
| + | 0.45 ± 0.04** | −29.0 ± 0.8 | 6.7 ± 0.2* | |||
| n = 9 | n = 9 | n = 9 | ||||
| α11.3/β4/α2δ | − | 3.0 ± 0.7 | −31.7 ± 0.7 | 4.3 ± 0.5 | −41.7 ± 1.9 | 8.5 ± 0.7 |
| n = 9 | n = 9 | n = 9 | n = 9 | n = 9 | ||
| + | 1.0 ± 0.3* | −28.8 ± 1.1 | 6.9 ± 0.5** | −36.1 ± 1.2* | 8.1 ± 0.8 | |
| n = 10 | n = 10 | n = 10 | n = 8 | n = 8 | ||
| α13.1 (stable expression) | − | 19.3 ± 2.9 | −56.9 ± 2.3 | 4.9 ± 0.5 | ||
| n = 4 | n = 4 | n = 4 | ||||
| + | 13.6 ± 2.4 | −55.5 ± 3.4 | 5.8 ± 0.7 | |||
| n = 6 | n = 6 | n = 6 |
*p < 0.05,
**p < 0.005, compared with (−)hBest1.
hBest1 inhibits the number of available CaV1.3 channels
To determine the mechanism of CaV1.3 inhibition by hBest1, activation and steady-state inactivation curves were measured by tail current analysis for α11.3/β1b/α2δ expressed with and without full-length hBest1 or hBest1-CT292–586 (Fig. 6, Table 1). The inactivation curve was not affected by either full-length hBest1 or its C-terminal fragment (Fig. 6B). Activation was not significantly affected by the C-terminal fragment but was shifted ∼8 mV in the depolarizing direction by the full-length hBest1 (Fig. 6A). This shift in the activation can also be seen in shift in the peak the I–V curves in Figure 1, B and C. These data suggest that although the inhibitory effects of hBest1 can be mostly accounted for by structures located in the C terminus, other regions of hBest1 may contribute to other effects. Although full-length hBest1 has small effects on the voltage dependence of channel activation (Table 1), these effects are not sufficient to explain the effect of hBest1 on IBa amplitude. In order for the shift in the activation curve to affect peak IBa amplitude significantly, inactivation would need to occur much more rapidly than it does (Fig. 1).
Figure 6.

Effects of hBest1 and hBest1-CT292–586 on CaV activation and inactivation. A, Normalized tail current activation curves for cells transfected with α11.3/β1b/α2δ alone (open circle) and α11.3/β1b/α2δ plus hB1-Ct292–586 (triangle) and α11.3/β1b/α2δ + hBest1 (filled circle). B, Steady-state inactivation curves for cells transfected with α11.3/β1b/α2δ alone (open circle), α11.3/β1b/α2δ plus hB1-Ct292–586 (triangle), and α11.3/β1b/α2δ plus hBest1 (filled circle). Peak Ba2+ currents were measured during 20 ms test step at −20 mV after 2 s conditioning pulses in the range of −120 to +40 mV (20 mV increments) and normalized to the largest in the series.
The inhibitory effects of hBest1 could be explained by negative regulation of gating or a decrease in the number of available CaV1.3 channels. To distinguish between these possibilities, we compared the effects of hBest1 on gating and ionic currents. The whole-cell ionic current (I) is described by the relationship I = N × i × Po, where N is the number of available channels at the membrane, Po is the channel open probability, and i is the unitary current amplitude. The on gating charge (Qon) is a useful index of N [Qon = Nq (Wei et al., 1994)]. We calculated Qon as the time integral of the on gating current isolated at the reversal potential of IBa (Erev = +60 mV) (Fig. 7A,B). hBest1-CT292–586 caused a similar inhibition of Qon as the whole-cell current (Itail). The slope of the line provided by a regression of Itail obtained at +60 mV (Erev) versus Qon provided a measure of relative Po (Fig. 7C). By this analysis, Po displayed no difference between CaV1.3 alone and CaV1.3 plus hB1-CT292–586. Because Qon is rather small for hBest1 expressing cells, this measurement is subject to some error and cannot definitively rule out the possibility that hBest1 produces a change in Po. However, the major effect of hBest1 is to reduce the number (N) of available CaV1.3 channels. To determine whether hBest1 interferes with CaV α11.3 channel expression, we measured the amount of CaV α11.3 protein in HEK cells transfected with FLAG-tagged α11.3, β1b, and α2δ with and without myc-tagged wt-hBest1 or hB1-CT292–586. Western blot with anti-FLAG antibody detected similar levels of expression of FLAG-α11.3 (∼210 kDa) in all three lysates (Fig. 7D, left panel). wt-hBest1 (68 kDa) and hB1-CT (50 kDa) detected with anti-myc antibody were observed only in lysates from cells transfected with wt-hBest1 or hB1-CT (Fig. 7D, right panel).
Figure 7.
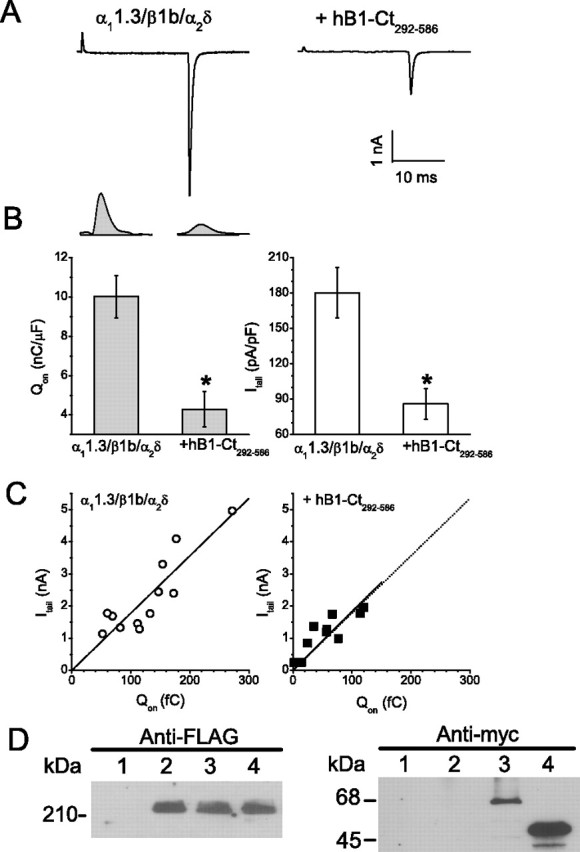
Effect of hBest1 on CaV1.3 gating currents. A, Exemplar currents from cells transfected with α11.3/β1b/α2δ alone and α11.3/β1b/α2δ plus hB1-Ct292–586. Currents were evoked with a 20 ms test pulse to +60 mV followed by a −60 mV repolarization. B, The left panel presents QON measurements for each group with exemplar gating current isolated at the reversal potential shown above. The right panel presents the averaged tail current densities recorded simultaneously for each group. *p < 0.01 for α11.3/β1b/α2δ plus hB1-Ct292–586 compared with α11.3/β1b/α2δ alone by two-way ANOVA. C, Scatter plot of tail current amplitude versus QON and regression line for cells transfected with α11.3/β1b/α2δ alone (left) and α11.3/β1b/α2δ plus hB1-Ct292–586 (right). The regression line for α11.3/β1b/α2δ alone is reproduced (gray) in the right panel to facilitate direct visual comparison. D, Cells were untransfected (lane 1) or transfected with CaV1.3 (FLAG-α11.3/β1b/α2δ) alone (lane 2), CaV1.3 plus wt-hBest1 (lane 3), and CaV1.3 plus hBest1-CT292–586 (lane 4) and subjected to lysis. An equal amount of supernatant proteins was detected by Western blotting with antibodies recognizing FLAG-epitopes (left) or myc-epitopes (right).
Mutation of a proline-rich motif in hBest1 eliminates effects of hBest1 on CaV1.3
The C-terminal domain of bestrophins is proline rich and is predicted to contain src-homology (SH3)-binding domains (Hartzell et al., 2008). Because SH3 domains of CaVβ subunits are essential for their effects on CaV channel gating and trafficking (Takahashi et al., 2005), we hypothesized that SH3-binding domains of bestrophins might be required for CaV modulation. We compared the sequences of several bestrophins and found a unique proline-rich motif (330PXXXP334) in hBest1, which is not present in mBest1, mBest2, or mBest3 (Fig. 8A). This region is predicted to be within or immediately flank an SH3-binding domain. To determine whether this domain is responsible for the effects of hBest1, we mutated hBest1 at Pro330 and Pro334 and tested the influence of the double mutation P330W/P334W on CaV1.3. As shown in Figure 8, B and C, the mutant had no effect on CaV1.3. However, this mutant is expressed at similar levels as wild type and generates normal Cl− currents (Fig. 8D,E).
Figure 8.
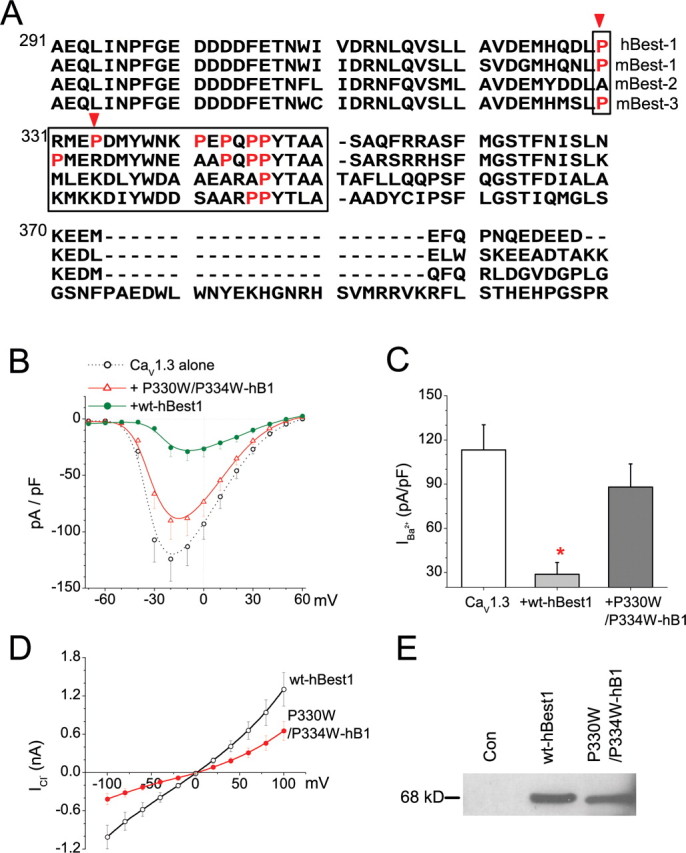
Critical prolines required for effect of hBest1 on CaV1.3. A, Alignment (ClustalW) of the C-terminal region of hBest1, mBest1, mBest2, and mBest3. Boxed amino acids represent the critical CT region involved in the regulation of Ca2+ channels. Prolines located at 330 and 334 (indicated by arrows) were mutated. B, I–V relationships for recombinant L-type CaV1.3 channel (α11.3/β1b/α2δ) alone (open circles), with wt-hBest1 (filled circles), or P330W/P334W-hBest1 (triangles). C, Averaged Ba2+ peak current densities at −10 mV for CaV1.3. *p < 0.01 for Ca channel plus hBest1 compared with Ca channel alone by two-way ANOVA. D, I–V relationship for Ca2+-activated Cl− currents recorded with the protocol described in Figure 1Aa from cells transfected with wt-hBest1 (open circles) or P330W/P334W-hBest1 (filled circles). E, Lysates from intact, wt-hBest1, or P330W/P334W mutant transfected HEK293 cells were detected with anti-myc antibodies.
CaVβ binds to hBest1
Based on our physiological evidence supporting an interaction between hBest1 and CaVβ, we tested whether hBest1 physically interacts with CaVβ subunits by coimmunoprecipitation. HEK293 cells were cotransfected with myc-tagged hBest1 and FLAG-tagged β2a or, as a negative control, untagged β2a. Anti-FLAG antibodies coimmunoprecipitated FLAG-β2a, and hBest1 from lysates of cells cotransfected with FLAG-β2a and myc-hBest1, but not from cells cotransfected with untagged β2a and myc-hBest1 (Fig. 9A).
Figure 9.

Binding of hbest1 to CaVβ2a. A, Coimmunoprecipitation (co-ip) of full-length myc-hBest1 with FLAG-tagged β2a. myc-hBest1 was cotransfected with FLAG-β2a (lanes 1, 3) or untagged β2a (lanes 2, 4) as a negative control in HEK293 cells. Cell lysates were incubated with FLAG antibodies, and immunoprecipitated proteins (left) were detected by Western blot with anti-FLAG or myc antibodies. Input lanes (right) represent ∼5% of cell lysate used for the assay. B, Pull-down of myc-tagged hBest1 C terminus (CT292–586) from transfected HEK293 cell lysates by GST-β2a but not GST. Bound protein was detected by Western blot with anti-myc antibodies (left). The blot with anti-GST antibodies (right) shows levels of GST-proteins used in assay. C, Pull-down of myc-tagged hBest1 mutation and CT fragments by GST-β2a (left). Ponceau staining shows levels of GST-β2a used for pull-down. Input lanes (right) represent ∼5% of cell lysate used for the assay.
To determine which regions of hBest1 were involved in the binding to CaVβ, the ability of immobilized GST-β2a to pull down C-terminal fragments of hBest1 from lysates of transfected HEK cells was measured. As shown in Figure 9B, immobilized GST-β2a, but not the GST control, bound to myc-tagged hBest1-CT292–586. Furthermore, hBest1-CT380–586 did not bind GST-β2a (Fig. 9C), as expected from the inability of hBest1-CT380–586 to inhibit Ca current (Fig. 4). Surprisingly, both hBest1-CT352–586 and P330W/P334W-hBest1, which had no inhibitory effect on Ca current, both bound to GST-β2a beads. These data suggest that, although the effect of hBest1 on Ca current requires amino acids 330–370 and prolines 330 and 334, binding of hBest1 to CaVβ does not require amino acids 330–352. However, it should be noted that this assay may not reflect quantitative changes in binding affinity.
Effects of hBest1 on endogenous Ca2+ currents in the RPE-J cell line
Rosenthal et al. (2005) reported that hBest1 can regulate endogenous Ca2+ channels in RPE-J cells. The molecular identity of the current is not absolutely certain, however. The cells express α11.3 by immunocytochemistry (Wollmann et al., 2006; Wimmers et al., 2008), but expression of other α and β subunits was not investigated. The current was not shown to exhibit negative activation threshold and limited dihydropyridine sensitivity that defines CaV1.3 currents. According to their finding, hBest1 plays a role as Ca2+ channel regulator by shifting the voltage dependence of Ca2+ channel activation to the left and accelerates activation without changing current amplitude. In contrast to the results of Rosenthal et al. (2005), we found that hBest1 caused a decrease in endogenous Ca2+ current in RPE-J cells with no significant effects on channel voltage dependence (Fig. 10). These results are very similar to what we have reported above for the effects of hBest1 on CaV1.3 expressed in HEK cells. The inhibitory effect is partly eliminated by the disease-causing hBest1 mutant G299R-hBest1 (Fig. 10).
Figure 10.
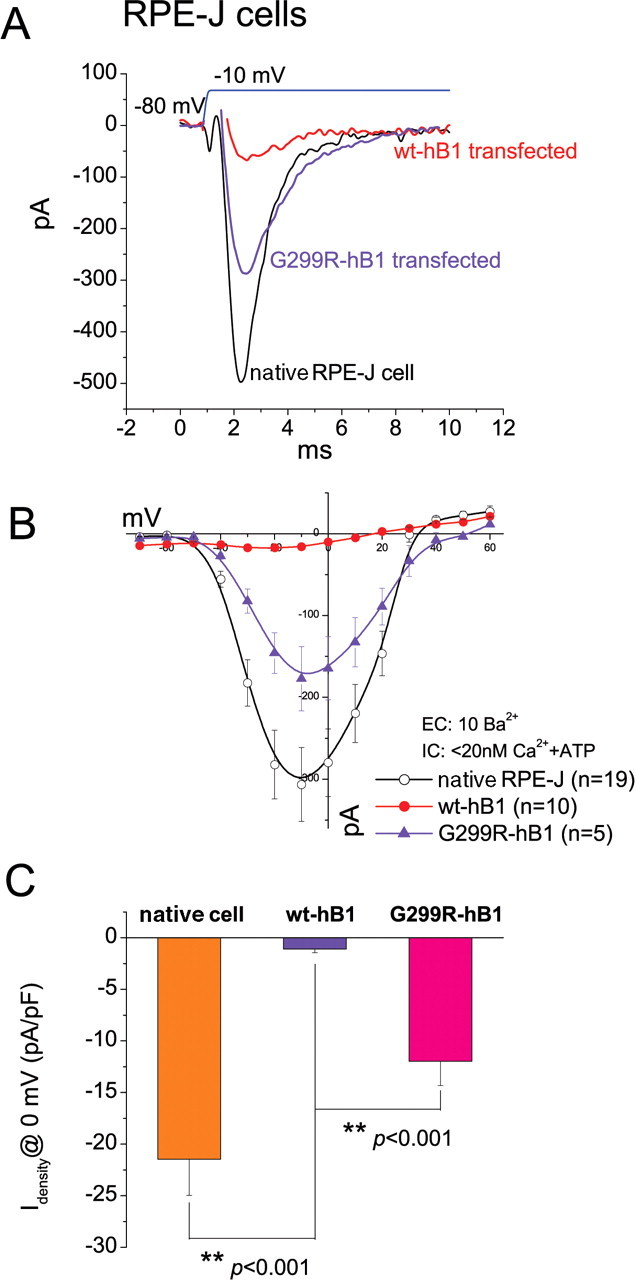
Effects of wt and G299R hBest1 on native CaV currents in RPE-J cells. A, Representative current traces evoked by −10 mV test pulse from native, hBest1 transfected, and G299R hBest1 transfected RPE-J cells. B, I–V relationships for native CaV channel (open circles), with wt-hBest1 (filled circles), or G299R-hBest1 (triangle). C, Averaged Ba2+ current densities at 0 mV for native CaV either alone or with additional wt-hBest1 and G299R mutant. **p < 0.001 for Ca channel plus hBest1 compared with Ca channel alone by two-way ANOVA.
Effects of hBest1 disease-causing mutations on expressed CaV currents
To investigate the effect of disease-causing mutations on the regulation of Ca2+ currents by hBest1, we studied the effects of mutants on currents generated by the defined CaV 1.3 channel (α11.3/β1b/α2δ) expressed in HEK cells (Fig. 11). We tested six different mutants: D312N, R92S, G299E, G299R, G222E, and A146K. Tested by Western blot with anti-myc antibody, these mutants are all expressed, although G299R and G299E may be reduced compared with wild-type hBest1 (Fig. 11A, top). All of these mutants were either nonfunctional as Cl− channels or produced Cl− currents that were significantly smaller than wild type (Fig. 11A, bottom). Wild-type hBest1 and the six mutants reduced CaV current (Fig. 11B,C). Although the Ca2+ current density was significantly smaller than control when coexpressed with mutant or wild-type hBest1 (p < 0.01), the percentage of inhibition ranged from 85% for wild-type hBest1 to 50% for G222E and A146K. The amount of inhibition produced by the G222E and A146K mutants was significantly less than the inhibition produced by wild-type hBest1 (p < 0.05). Because the effect of some of the mutants was quite variable, we quantified the amount of inhibition not only by mean current density but also by the fraction of cells that had current densities smaller than control current amplitude minus three SEMs (Fig. 11C, horizontal line). Whereas <7% of control cells had currents smaller than this value, 96% of hBest1 transfected cells had currents below this threshold (Fig. 11D). However, for G222E and A146K, only 46% of the cells had currents below this level. These results show that the G222E and A146K mutants were less effective in inhibiting CaV currents than wild type or the D312N or R92S mutants.
Figure 11.

Effects of hBest1 mutants on CaV 1.3 currents. Cells were transfected with the CaV subunits α11.3, β1b, and α2δ with and without hBest1 wild type or mutants. A, Lysates from varied mutant hBest1 transfected cells were detected by Western blot with anti-myc antibodies (top). Cl− currents generated by hBest1 mutants are shown in the bottom panel. Cl− current data for D312N, G299E, and A146K have been published previously (Yu et al., 2007). B, Effects of mutant bestrophins on CaV1.3 current density. Error bars are means ± SEM. Asterisks show values of individual cells. Horizontal line is control mean minus three SEM. Statistical significance (ANOVA, both Tukey's and Bonferroni's test) is shown in the box above the data. +p < 0.01, significantly different from control; op < 0.05, indicates significantly different from wild-type hBest1. C, Percentage of inhibition of CaV current relative to mean control value. D, Percentage of cells having CaV currents smaller than the three SEM threshold in B. The numbers at the bottom of the bars are numbers of cells. Con, Control.
Discussion
The main finding of this study is that hBest1 modulates CaV1.3 L-type Ca channels. The inhibitory effect of hBest1 requires the presence of β subunits: the effect is not seen with CaV1.3 or CaV3.1 expressed in the absence of β subunits. The C-terminal domain of hBest1, especially a proline-rich region located within amino acids 330–370, is critical for the modulation. The physiological findings are supported by the finding that CaVβ directly binds the C terminus of bestrophin. Several bestrophin homologs, including mBest1, mBest2, and mBest3, do not exert any obvious effect on Ca channel properties, possibly because they lack the proline-rich domain 330PxxxP334. We hypothesize that hBest1 specifically regulates Ca2+ channels by associating with SH3 domains in CaVβ subunits. The data have important implications for not only understanding how bestrophins function but also for describing new mechanistic paradigms for SH3-binding partners controlling Ca channels through CaVβ.
Role of hBest1 in RPE
Although these experiments use cells that heterologously express CaV1.3 and hBest1, it is very likely that our results are physiologically relevant to RPE cell function. Human RPE cells and the RPE cell line ARPE-19 express CaV α1.3 and β2 subunits (Wimmers et al., 2008). Furthermore, it has been shown recently that hBest1 coimmunoprecipitates with CaVβ subunits from freshly isolated human RPE cells (Strauss et al., 2008). These results suggest that it is likely that the interaction between hBest1 and CaV subunits has an important physiological function and may help resolve the present controversy of whether Best1 is a Cl− channel (Hartzell et al., 2008).
As reviewed in the Introduction, there is strong evidence that bestrophins are Cl− channels (Hartzell et al., 2008), but this idea has been questioned (Marmorstein et al., 2004a,b, 2006; Rosenthal et al., 2005; Marmorstein and Kinnick, 2007). These authors have proposed that hBest1 is not a Cl− channel but rather is a regulator of Ca2+ signaling (Rosenthal et al., 2005; Marmorstein et al., 2006). This suggestion is based on the finding that hBest1 overexpression shifts the voltage dependence of Ca2+ channel activation to the left and accelerates activation of endogenous voltage-gated Ca2+ channels in RPE-J cells (Rosenthal et al., 2005). The W93C mutant shifts voltage dependence of activation to the left and slows inactivation. In contrast, the R218C mutation accelerates and augments Ca2+ current inactivation. The LP of the electrooculogram, which is a hallmark diagnostic feature of BVMD, is apparently dependent on voltage-gated Ca2+ channels in mouse, because the LP is reduced by the Ca2+ channel blocker nimodipine and is abolished in β4 (Marmorstein et al., 2006) and CaVα1.3 knock-out (Wu et al., 2007) mice. Our results strengthen the suggestion that hBest1 is a multifunctional protein, both a Cl− channel and a channel regulator. However, it is puzzling that mouse Best1 does not regulate CaV1.3, because the study by Marmorstein et al. (2006) in mouse was the motivation for this paper.
Regardless of whether hBest1 functions as a Cl− channel and/or a Ca2+-channel regulator, it seems clear that bestrophin participates in epithelial transport across the RPE. How might disrupted epithelial transport be linked to macular degeneration? RPE has a variety of essential functions in retina, including paracrine hormone secretion, participation in the blood-eye barrier, epithelial transport, regeneration of visual pigment, and phagocytosis of photoreceptor outer segments (Strauss, 2005).
It is well established that RPE Cl− channels are activated (indirectly) by light (Gallemore et al., 1998), and it seems likely that these channels correspond to hBest1 (Hartzell et al., 2008). We propose hBest1 inhibits CaV channels only when hBest1 is in the closed conformation and that CaV channels are activated by the conformational change that occurs when hBest1 is activated by light. That is, we imagine that when hBest1 channels are tuned on, hBest1 inhibition of CaV channels is relieved. The efflux of Cl− through hBest1 will depolarize the cell to facilitate CaV channel opening. Ca2+ influx will then augment hBest1 Cl− current by a positive feedback. Ca2+ influx provides Ca2+ for the many Ca2+-dependent processes that occur in RPE in light, such as phagocytosis of photoreceptor outer segments and secretion of various paracrine hormones.
Our finding that six hBest1 disease-causing mutants have different abilities to inhibit CaV1.3 currents suggests that CaV channel regulation may not be coupled tightly to Cl− channel activity. Four of these mutants, D312N, R92S, G299E, and G222E, are also dominant negative on wild-type hBest1 Cl− channel function: coexpression of these mutants inhibits the currents generated by hBest1 (Yu et al., 2007). Although D312N, R92S, and G299E have statistically the same effect on CaV currents as wild-type hBest1, G222E has a significantly smaller effect. The topological location of G222 in hBest1 is controversial. Tsunenari et al. (2003) place it extracellularly, whereas Milenkovic et al. (2007) place it in the same intracellular loop as A146. The A146K mutation produces measurable Cl− currents, although they are smaller than wild type, and is not dominant negative (Yu et al., 2007). This mutant also has smaller effects on CaV channels. Although quantitative differences exist between different mutations in their ability to inhibit CaV currents, our data would be consistent with a model in which BVMD is caused primarily by a defect in Cl− channel function and not a defect in the ability of hBest1 to regulate CaV channels.
SH3-binding partners as modulators of voltage-gated Ca2+ channels
L-type voltage-gated Ca channels are composed of multiple subunits, the pore-forming α1 subunit and the accessory β and α2δ subunits (Catterall, 2000). It is well established that CaVβ subunits modulate CaV α1 channel function in multiple ways (Birnbaumer et al., 1998; Dolphin, 2003). β subunits act as chaperones that are essential for the proper assembly and trafficking of CaV channels to the plasma membrane. However, in addition, β subunits play a direct modulatory role on the pore-forming α1 subunit voltage sensitivity and gating. At least some of these direct modulatory effects are mediated by interactions between an SH3 domain linked to a guanylate kinase (GK) domain in the β subunit (Chen et al., 2004; Opatowsky et al., 2004; Van et al., 2004). These two domains are required for both the trafficking and gating effects of β subunits, and interactions between the two domains are important in mediating these functions (Takahashi et al., 2005). Here, we show that the bestrophin SH3-binding domain is crucial for the regulation of CaV currents and that CaVβ subunits are required for the effect. Both gating currents and kinetics are altered in a way that is consistent with altered CaVβ function by bestrophin. The simplest model that one might imagine could involve hBest1 disrupting the interaction between the SH3 and GK domains of the β subunit (Takahashi et al., 2004, 2005).
It is worth noting that all bestrophins have a proline-rich C terminus with several predicted SH3-binding domains. However, the SH3 binding domain that is predicted in hBest1 between amino acids 336 and 350 is not found in the other bestrophins. It is particularly interesting that mouse Best1 lacks this domain and does not regulate the Ca2+ currents. Whether this reflects some mismatch caused by species-specific sequences or reflects a fundamental difference between human and mouse Best1 remains to be determined. It also raises questions about the role of Best1 in mouse and human as discussed above. It is also intriguing that this predicted SH3-binding domain is adjacent to the 358SFxGS362 domain that we have shown previously to have important regulatory functions in Best2 and Best3 (Qu et al., 2006a, 2007).
Footnotes
This work was supported by National Institutes of Health Grants GM60448 (H.C.H.), EY014852 (H.C.H.), and NS044922 (A.L.). We thank Dr. Dorothy Hanck (University of Chicago) for providing the data on the effect of hBest1 on CaV3.1; Jeremy Nathans, Dianne Lipscombe, and Eduardo Perez-Reyes for cDNA constructs; and Roger Colbran for donating the FLAG- and GST-β2a constructs.
References
- Barro Soria R, Spitzner M, Schreiber R, Kunzelmann K. Bestrophin 1 enables Ca2+ activated Cl− conductance in epithelia. J Biol Chem. 2008 doi: 10.1074/jbc.M605716200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaumer L, Qin N, Olcese R, Tareilus E, Platano D, Costantin J, Stefani E. Structures and functions of calcium channel beta subunits. J Bioenerg Biomembr. 1998;30:357–375. doi: 10.1023/a:1021989622656. [DOI] [PubMed] [Google Scholar]
- Calin-Jageman I, Yu K, Hall RA, Mei L, Lee A. Erbin enhances voltage-dependent facilitation of Cav1.3 Ca2+ channels through relief of an autoinhibitory domain in the Cav1.3 α1 subunit. J Neurosci. 2007;27:1374–1385. doi: 10.1523/JNEUROSCI.5191-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L, Yang J. Structural basis of the alpha1-beta subunit interaction of voltage-gated Ca2+ channels. Nature. 2004;429:675–680. doi: 10.1038/nature02641. [DOI] [PubMed] [Google Scholar]
- Chien LT, Hartzell HC. Drosophila bestrophin-1 chloride current is dually regulated by calcium and cell volume. J Gen Physiol. 2007;130:513–534. doi: 10.1085/jgp.200709795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien LT, Zhang ZR, Hartzell HC. Single Cl− channels activated by Ca2+ in Drosophila S2 cells are mediated by bestrophins. J Gen Physiol. 2006;128:247–259. doi: 10.1085/jgp.200609581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. Beta subunits of voltage-gated calcium channels. J Bioenerg Biomembr. 2003;35:599–620. doi: 10.1023/b:jobb.0000008026.37790.5a. [DOI] [PubMed] [Google Scholar]
- Gallemore RP, Hughes BA, Miller SS. Light-induced responses of the retinal pigment epithelium. In: Marmor MF, Wolfensberger TJ, editors. The retinal pigment epithelium. Oxford: Oxford UP; 1998. pp. 175–198. [Google Scholar]
- Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, Anderson ME, Colbran RJ. L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol Cell. 2006;23:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Guziewicz KE, Zangerl B, Lindauer SJ, Mullins RF, Sandmeyer LS, Grahn BH, Stone EM, Acland GM, Aguirre GD. Bestrophin gene mutations cause canine multifocal retinopathy: a novel animal model for best disease. Invest Ophthalmol Vis Sci. 2007;48:1959–1967. doi: 10.1167/iovs.06-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzell HC, Qu Z, Yu K, Xiao Q, Chien LT. Molecular physiology of bestrophins: multifunctional membrane proteins linked to Best disease and other retinopathies. Physiol Rev. 2008;88:639–672. doi: 10.1152/physrev.00022.2007. [DOI] [PubMed] [Google Scholar]
- Helton TD, Xu W, Lipscombe D. Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci. 2005;25:10247–10251. doi: 10.1523/JNEUROSCI.1089-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer F, White K, Pauleikhoff D, Gehrig A, Passmore L, Rivera A, Rudolph G, Kellner U, Andrassi M, Lorenz B, Rohrschneider K, Blankenagel A, Jurklies B, Schilling H, Schutt F, Holz FG, Weber BH. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. Eur J Hum Genet. 2000;8:286–292. doi: 10.1038/sj.ejhg.5200447. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Milenkovic VM, Spitzner M, Soria RB, Schreiber R. Calcium-dependent chloride conductance in epithelia: is there a contribution by Bestrophin? Pflügers Arch. 2007;454:879–889. doi: 10.1007/s00424-007-0245-z. [DOI] [PubMed] [Google Scholar]
- Marmorstein AD, Kinnick TR. Focus on molecules: bestrophin (Best-1) Exptl Eye Res. 2007;2007:423–424. doi: 10.1016/j.exer.2006.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein AD, Marmorstein LY, Rayborn M, Wang X, Hollyfield JG, Petrukhin K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral membrane of the retinal pigment epithelium. Proc Natl Acad Sci USA. 2000;97:12758–12763. doi: 10.1073/pnas.220402097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein AD, Rosenthal R, Stanton JB, Bakall B, Wadelius C, Marmorstein LY, Goldberg AFX, Peachey N, Strauss O. Bestrophin is not a chloride channel. ARVO Meet Abstr. 2004a;45:1759. [Google Scholar]
- Marmorstein AD, Stanton JB, Yocom J, Bakall B, Schiavone MT, Wadelius C, Marmorstein LY, Peachey NS. A model of best vitelliform macular dystrophy in rats. Invest Ophthalmol Vis Sci. 2004b;45:3733–3739. doi: 10.1167/iovs.04-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein LY, Wu J, McLaughlin P, Yocom J, Karl MO, Neussert R, Wimmers S, Stanton JB, Gregg RG, Strauss O, Peachey NS, Marmorstein AD. The light peak of the electroretinogram is dependent on voltage-gated calcium channels and antagonized by bestrophin (Best-1) J Gen Physiol. 2006;127:577–589. doi: 10.1085/jgp.200509473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt A, Stohr H, Passmore LA, Kramer F, Rivera A, Weber BH. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best's disease) Hum Mol Genet. 1998;7:1517–1525. doi: 10.1093/hmg/7.9.1517. [DOI] [PubMed] [Google Scholar]
- Milenkovic VM, Rivera A, Horling F, Weber BH. Insertion and topology of normal and mutant bestrophin-1 in the endoplasmic reticulum membrane. J Biol Chem. 2007;282:1313–1321. doi: 10.1074/jbc.M607383200. [DOI] [PubMed] [Google Scholar]
- Opatowsky Y, Chen CC, Campbell KP, Hirsch JA. Structural analysis of the voltage-dependent calcium channel beta subunit functional core and its complex with the alpha 1 interaction domain. Neuron. 2004;42:387–399. doi: 10.1016/s0896-6273(04)00250-8. [DOI] [PubMed] [Google Scholar]
- Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T, Sandgren O, Forsman K, Holmgren G, Andreasson S, Vujic M, Bergen AAB, McGarty-Dugan V, Figueroa D, Austin CP, Metzker ML, Caskey CT, Wadelius C. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998;19:241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- Qu Z, Hartzell HC. Determinants of anion permeation in the second transmembrane domain of the mouse bestrophin-2 chloride channel. J Gen Physiol. 2004;124:371–382. doi: 10.1085/jgp.200409108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Wei RW, Mann W, Hartzell HC. Two bestrophins cloned from Xenopus laevis oocytes express Ca-activated Cl currents. J Biol Chem. 2003;278:49563–49572. doi: 10.1074/jbc.M308414200. [DOI] [PubMed] [Google Scholar]
- Qu Z, Fischmeister R, Hartzell HC. Mouse bestrophin-2 is a bona fide Cl− channel: identification of a residue important in anion binding and conduction. J Gen Physiol. 2004;123:327–340. doi: 10.1085/jgp.200409031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Cui Y, Hartzell C. A short motif in the C-terminus of mouse bestrophin 4 inhibits its activation as a Cl channel. FEBS Lett. 2006a;580:2141–2146. doi: 10.1016/j.febslet.2006.03.025. [DOI] [PubMed] [Google Scholar]
- Qu Z, Chien LT, Cui Y, Hartzell HC. The anion-selective pore of the bestrophins, a family of chloride channels associated with retinal degeneration. J Neurosci. 2006b;26:5411–5419. doi: 10.1523/JNEUROSCI.5500-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Yu K, Cui Y, Ying C, Hartzell C. Activation of bestrophin Cl− channels is regulated by C-terminal domains. J Biol Chem. 2007;282:17460–17467. doi: 10.1074/jbc.M701043200. [DOI] [PubMed] [Google Scholar]
- Rosenthal R, Bakall B, Kinnick T, Peachey N, Wimmers S, Wadelius C, Marmorstein A, Strauss O. Expression of bestrophin-1, the product of the VMD2 gene, modulates voltage-dependent Ca2+ channels in retinal pigment epithelial cells. FASEB J. 2005;20:178–180. doi: 10.1096/fj.05-4495fje. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Afshari MA, Sharma S, Bernstein PS, Chong S, Hutchinson A, Petrukhin K, Allikmets R. Assessment of mutations in the best macular dystrophy (VMD2) gene in patients with adult-onset foveomacular vitelliform dystrophy, age-related maculopathy, and bull's-eye maculopathy. Ophthalmol. 2001;108:2060–2067. doi: 10.1016/s0161-6420(01)00777-1. [DOI] [PubMed] [Google Scholar]
- Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- Strauss O, Milenkovic V, Striessnig J, Rejcova S. Direct interaction of bestrophin-1 and beta-subunits of voltage-dependent calcium channels. Invest Ophthalmol Vis Sci. 2008 in press. [Google Scholar]
- Sun H, Tsunenari T, Yau K-W, Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci USA. 2002;99:4008–4013. doi: 10.1073/pnas.052692999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi SX, Miriyala J, Colecraft HM. Membrane-associated guanylate kinase-like properties of beta-subunits required for modulation of voltage-dependent Ca2+ channels. Proc Natl Acad Sci USA. 2004;101:7193–7198. doi: 10.1073/pnas.0306665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi SX, Miriyala J, Tay LH, Yue DT, Colecraft HM. A CaV{beta} SH3/guanylate kinase domain interaction regulates multiple properties of voltage-gated Ca2+ channels. J Gen Physiol. 2005;126:365–377. doi: 10.1085/jgp.200509354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunenari T, Sun H, Williams J, Cahill H, Smallwood P, Yau KW, Nathans J. Structure-function analysis of the bestrophin family of anion channels. J Biol Chem. 2003;278:41114–41125. doi: 10.1074/jbc.M306150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van PF, Clark KA, Chatelain FC, Minor DL., Jr Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature. 2004;429:671–675. doi: 10.1038/nature02588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Neely A, Lacerda AE, Olcese R, Stefani E, Perez-Reyes E, Birnbaumer L. Modification of Ca2+ channel activity by deletions at the carboxyl terminus of the cardiac α1 subunit. J Biol Chem. 1994;269:1635–1640. [PubMed] [Google Scholar]
- Wimmers S, Coeppicus L, Rosenthal R, Strauss O. Expression profile of voltage-dependent Ca2+ channel subunits in the human retinal pigment epithelium. Graefes Arch Clin Exp Ophthalmol. 2008;246:685–692. doi: 10.1007/s00417-008-0778-7. [DOI] [PubMed] [Google Scholar]
- Wollmann G, Lenzner S, Berger W, Rosenthal R, Karl MO, Strauss O. Voltage-dependent ion channels in the mouse RPE: comparison with Norrie disease mice. Vision Res. 2006;46:688–698. doi: 10.1016/j.visres.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Wu J, Marmorstein AD, Striessnig J, Peachey NS. Voltage-dependent calcium channel CaV1.3 subunits regulate the light peak of the electroretinogram. J Neurophysiol. 2007;97:3731–3735. doi: 10.1152/jn.00146.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardley J, Leroy BP, Hart-Holden N, Lafaut BA, Loeys B, Messiaen LM, Perveen R, Reddy MA, Bhattacharya SS, Traboulsi E, Baralle D, De Laey JJ, Puech B, Kestelyn P, Moore AT, Manson FD, Black GC. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC) Invest Ophthalmol Vis Sci. 2004;45:3683–3689. doi: 10.1167/iovs.04-0550. [DOI] [PubMed] [Google Scholar]
- Yu K, Cui Y, Hartzell HC. The bestrophin mutation A243V, linked to adult-onset vitelliform macular dystrophy, impairs its chloride channel function. Invest Ophthalmol Vis Sci. 2006;47:4956–4961. doi: 10.1167/iovs.06-0524. [DOI] [PubMed] [Google Scholar]
- Yu K, Cui Y, Hartzell HC. Chloride channel activity of bestrophin mutants associated with mild or late-onset macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:4694–4705. doi: 10.1167/iovs.07-0301. [DOI] [PubMed] [Google Scholar]
- Zhou H, Kim SA, Kirk EA, Tippens AL, Sun H, Haeseleer F, Lee A. Ca2+-binding protein-1 facilitates and forms a postsynaptic complex with Cav1.2 (L-type) Ca2+ channels. J Neurosci. 2004;24:4698–4708. doi: 10.1523/JNEUROSCI.5523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]